2Pipe starts with a question: matching you with the correct pipeline for MAG reconstruction
Jeferyd Yepes-García, Laurent Falquet

TL;DR
This paper introduces 2Pipe, a web tool that helps researchers choose the best pipeline for reconstructing microbial genomes from environmental DNA data.
Contribution
The novel contribution is the development of 2Pipe, an interactive decision-support tool for selecting MAG reconstruction pipelines.
Findings
2Pipe assists users in selecting suitable MAG reconstruction pipelines based on input data and computational constraints.
The paper provides a technical overview of 41 publicly available MAG reconstruction pipelines.
The tool includes a pipeline comparison and a mechanism for developers to add new workflows.
Abstract
Whole-genome sequencing has boosted our ability to explore microbial diversity by enabling the recovery of metagenome-assembled genomes (MAGs) directly from environmental DNA. As a result, the vast availability of sequencing data has prompted the development of numerous bioinformatics pipelines for MAG reconstruction, along with challenges to identify the most suitable pipeline to perform the analysis according to the user needs. This report briefly discusses the computational requirements of these pipelines; presents the variety of interfaces, workflow managers, and package managers they feature; and describes the typical modular structure. Also, it provides a compacted technical overview of 41 publicly available pipelines or platforms to build MAGs starting from short and/or long sequences. Moreover, recognizing the overwhelming number of factors to consider when selecting an…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
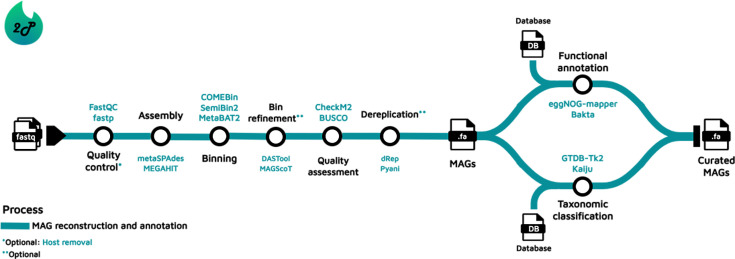 Fig 1
Fig 1| No. | Pipeline/platform | Quality control preprocessing | Assembly | Binning | Quality | Bin | Taxonomic | Functional | Other |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Ancient DNA ( | FastQC ( | Bowtie2 and MEGAHIT ( | CONCOCT ( | CheckM ( | DASTool ( | GTDB-Tk ( | mapDamage2 ( | |
| 2 | Anvi'o | Illumina-utils ( | metaSPAdes ( | MetaBAT2 ( | DASTool | KrakenUniq ( | DIAMOND ( | ||
| 3 | Aviary ( | FastQC, Filtlong ( | metaSPAdes, MEGAHIT, metaFlye ( | MetaBAT2, MetaBAT, MaxBin2, VAMB
( | CheckM, metaQUAST ( | DASTool | GTDB-Tk | Prodigal ( | Lorikeet ( |
| 4 | BugBuster ( | fastp and Bowtie2 ( | MEGAHIT | METABAT2, SemiBin2 ( | CheckM2 ( | MetaWRAP-native module ( | GTDB-Tk2 ( | Prodigal and MetaCerberus ( | Kraken2 ( |
| 5 | BV-BRC | TrimGalore ( | metaSPAdes and MEGAHIT | PATRIC metagenome binning service
( | EvalG and EvalCon ( | RASTtk ( | VIGOR4 ( | ||
| 6 | DATMA ( | Trimmomatic ( | metaSPAdes, Velvet ( | CLAME ( | CheckM | BLAST and Kaiju ( | Prodigal and GeneMark ( | Krona ( | |
| 7 | EasyMetagenome ( | KneadData ( | metaSPAdes and MEGAHIT | MetaWRAP-native ( | CoverM and CheckM2 | MetaWRAP-native module | GTDB-Tk2 | MetaProdigal ( | dRep ( |
| 8 | EasyNanoMeta ( | fastp, Minimap2 ( | metaFlye, OPERA-MS ( | SemiBin2, MetaBAT2, MaxBin2, CONCOCT, and VAMB | CheckM2 | GTDB-Tk2 and PhyloPhlAn ( | Prokka ( | Kraken2 and Centrifuge | |
| 9 | Eukfinder ( | Bowtie2 and Trimmomatic | metaSPAdes | MyCC ( | Centrifuge and PLAST ( | ||||
| 10 | EURYALE (MEDUSA) ( | FastQC, fastp, Bowtie2, and MultiQC
( | MEGAHIT | Kaiju and Kraken2 | DIAMOND | Krona | |||
| 11 | Galaxy | FastQC, Seqtk ( | metaSPAdes | MaxBin2 | GTDB-Tk2 and Contig Annotation Tool
(CAT) ( | Prokka | Kraken ( | ||
| 12 | GEN-ERA ( | fastp and FastQC | SPAdes ( | MetaBAT2 and CONCOCT | CheckM, GUNC ( | AMAW ( | Prodigal, Mantis ( | OrthoFinder ( | |
| 13 | HiFi-MAG ( | MetaBAT2 and SemiBin2 | CheckM2 | DASTool | GTDB-Tk2 | ||||
| 14 | IDseq | Trimmomatic, STAR ( | SPAdes and Bowtie2 | GSNAPL ( | |||||
| 15 | IMG/M | SemiBin2 | CheckM | GTDB-Tk | Prodigal, GeneMarkS-2 ( | EukCC, SignalP ( | |||
| 16 | JAMS ( | Trimmomatic and Bowtie2 | MEGAHIT and SPAdes | Kraken2 | Prokka and | Samtools and | |||
| 17 | KBase | FastQC, Trimmomatic, | metaSPAdes, MEGAHIT, and IBDA-UD | MetaBAT2, CONCOCT, and MaxBin2 | CheckM | DASTool | RASTtk and GTDB-Tk | Prokka, dbCAN3 ( | OMEGGA ( |
| 18 | MAGNETO ( | fastp, Bowtie2, and FastQscreen ( | MEGAHIT and Simka ( | MetaBAT2 | CheckM | GTDB-Tk, | Prodigal, | mOTUs ( | |
| 19 | MAGO ( | FastQC and fastp | metaSPAdes, MEGAHIT, and IBDA-UD | MaxBin2, MetaBAT, CONCOCT, and BinSanity | CheckM | GTDB-Tk | Prokka | Roary ( | |
| 20 | Mapler ( | FastQC | metaMDBG ( | MetaBAT2 | CheckM2 and metaQUAST | GTDB-Tk2 and Kraken2 | KAT ( | ||
| 21 | MetaGEM ( | fastp | MEGAHIT and BWA | MetaBAT2, CONCOCT, and MaxBin2 | MetaWRAP–native module | GTDB-Tk | Prokka | Roary, CarveMe ( | |
| 22 | MetaGenePipe ( | Trimmomatic, TrimGalore, and FastQC | MEGAHIT | DIAMOND (SwissProt [ | Prodigal and HMMER ( | BLAST | |||
| 23 | Metagenome-Atlas ( | BBTools | MEGAHIT and metaSPAdes | MetaBAT2, MaxBin2, and VAMB | BUSCO, CheckM, and CheckM2 | DASTool | GTDB-Tk | Prodigal, eggNOG, | dRep |
| 24 | Metagenomics- | fastp, Porechop, Filtlong, NanoPack2,
KMC ( | metaFlye, metaSPAdes, MEGAHIT, and
Assembler Resource Estimator ( | MetaBAT2, MetaCoAG ( | CheckM | MAGScoT ( | MMSeqs2 taxonomy ( | Prodigal, Prokka, and RGI ( | CarveMe, SMETANA, MEMOTE, gapseq
( |
| 25 | Metaphor ( | FastQC, fastp, and MultiQC | MEGAHIT | VAMB, MetaBAT2, and CONCOCT | metaQUAST | DASTool | DIAMOND (NCBI COG) | Prodigal and Prokka | |
| 26 | metagWGS ( | FastQC, Cutadapt, Sickle ( | metaSPAdes, MEGAHIT, hifiasm, and metaFlye | MetaBAT2, CONCOCT, and MaxBin2 | metaQUAST | Binette ( | GTDB-Tk2 | Prodigal and
eggNOG- | dRep and Kaiju |
| 27 | MetaWRAP ( | FastQC and TrimGalore | metaSPAdes and MEGAHIT | MetaBAT2, CONCOCT, and MaxBin2 | CheckM | MetaWRAP-native module | Kraken and BLAST | Prokka | Kraken and Blobology ( |
| 28 | MG-TK ( | Trimmomatic, Porechop, Kraken,
Kraken2, and SDM ( | SPAdes, MEGAHIT, Flye ( | MetaBAT2, SemiBin2, and MetaDecoder
( | CheckM and CheckM2 | GTDB-Tk | Prodigal and DIAMOND (KEGG
Carbohydrate-Active enZYmes, CAZy [ | mOTUs2 ( | |
| 29 | MGnify | Trimmomatic and Biopython ( | metaSPAdes | DIAMOND (UniRef90 [ | Prodigal, FragGeneScan ( | mOTUs2 and antiSMASH ( | |||
| 30 | MOSHPIT | Cutadapt and Bowtie2 | SPAdes and MEGAHIT | MetaBAT2 | QUAST and BUSCO | Sourmash | Kraken2 and Kaiju | eggNOG- | |
| 31 | MUFFIN ( | fastp and Filtlong | SPAdes, Flye, and Unicycler | MetaBAT2, CONCOCT, and MaxBin2 | CheckM | MetaWRAP-native module | Sourmash (Genome Taxonomy Database,
GTDB [ | eggNOG- | Salmon ( |
| 32 | NanoPhase ( | Filtlong | metaFlye, Racon ( | MetaBAT2 and MaxBin2 | CheckM and QUAST | MetaWRAP-native module | GTDB-Tk | Prodigal and DIAMOND (UniProtKB [ | |
| 33 | nf-core/mag ( | fastp, AdapterRemoval ( | MEGAHIT, metaSPAdes, Flye, metaMDBG,
and hybridSPAdes ( | MetaBAT2, CONCOCT, and MaxBin2 | BUSCO, CheckM, CheckM2, GUNC, and QUAST | DASTool | GTDB-Tk2 and CAT | Prodigal, Prokka, and MetaEuk ( | Kraken2, MultiQC,
Centrifuge, |
| 34 | ngs-preprocess | Porechop, Nanopack2, pycoQC ( | SPAdes, Flye, Canu, Unicycler, Shovill
( | Prokka, antiSMASH, KofamScan ( | AMRFinderPlus ( | ||||
| 35 | nIMP3 ( | BWA, Samtools, BBTools, FastQC,
Kraken2, | MEGAHIT | mOTUs, MultiQC, MetaPhlAn4 ( | |||||
| 36 | SnakeMAGs ( | Illumina-utils, Trimmomatic, and Bowtie2 | MEGAHIT | MetaBAT2 | CheckM, GUNC, and CoverM | GTDB-Tk2 | |||
| 37 | SPIRE ( | NGLess ( | MEGAHIT, BWA, and Samtools | MetaBAT2 | CheckM2 and GUNC | GTDB-Tk2 | Prodigal and eggNOG-mapper | Barrnap, RGI ( | |
| 38 | SqueezeMeta ( | PRINSEQ | MEGAHIT, SPAdes, Canu, and Flye | MetaBAT2, CONCOCT, and MaxBin2 | CheckM, CheckM2, and CompareM | DASTool | GTDB-Tk2 | Prodigal, MUMmer | DIAMOND (NCBI COG,
KEGG), |
| 39 | Sunbeam ( | Trimmomatic, Cutadapt, Komplexity
( | MEGAHIT | Prodigal, | Kraken | ||||
| 40 | VEBA ( | KneadData, fastp, BBTools, Bowtie2, NanoPack2, and Minimap2 | metaSPAdes, SPAdes, rnaSPAdes ( | MetaBAT2, CONCOCT, MaxBin2, and SemiBin2 | CheckM2, Tiara, CheckV ( | Binette | GTDB-Tk2, MetaEuk, geNomad, and
VirFinder ( | Prodigal, DIAMOND (UniRef50/90, MIBiG
[ | antiSMASH, Muscle5, FastTree2,
FastANI, sylph ( |
| 41 | WGSA2+/LoRA | KneadData, fastp, and Kraken2 | metaSPAdes, metaFlye, MiniMap2, and Samtools | MetaBAT2 | CheckM and CheckM2 | GTDB-Tk2 | Prodigal,
eggNOG- | SortMeRNA, Krona, Trinity, |
| No. | Pipeline/ | Category | Short reads | Long reads | Hybrid assembly | Multiple samples | Co-assembly
and/or | Bin refinement | Infrastructure | Interface | Workflow manager | Software execution | Special features | Last update | Number of citations | License |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ancient DNA ( | Special | Yes | No | No | No | No | Yes | Local and HPC | CLI | Local | Ancient DNA identification | 2024 | 0 | Not specified | |
| 2 | Anvi'o ( | Short-read-centered | Yes | No | No | Yes | Yes | Yes | Local and HPC | CLI/graphical user interface (GUI) | Conda | Visualization module | 2025 | 678 | GNU GPL v3 | |
| 3 | Aviary ( | Hybrid | Yes | Yes | Yes | Yes | No | Yes | Local, HPC, and CC | CLI | Snakemake | Conda | Genotype recovery | 2025 | Not found | GNU GPL v3 |
| 4 | BugBuster ( | Short-read-centered | Yes | No | No | Yes | No | Yes | Local, HPC, and CC | CLI | Nextflow | Docker | Taxonomic profiling and antimicrobial resistance gene prediction | 2025 | 0 | Not specified |
| 5 | BV-BRC ( | Web-based | Yes | No | No | Yes | No | No | External | GUI | External | Taxonomic profiling and viral MAGs | 2024 | 783 | MIT License | |
| 6 | DATMA ( | Short-read-centered | Yes | No | No | No | No | No | Local and HPC | CLI | COMP Superscalar ( | Local | Reads first grouped (binning) and assembled in batches | 2020 | 4 | GNU GPL v3 |
| 7 | EasyMetagenome ( | Short-read-centered | Yes | No | No | Yes | Yes | Yes | Local and HPC | CLI | Conda | Taxonomic profiling | 2024 | 14 | GNU GPL v3 | |
| 8 | EasyNanoMeta ( | Long-read-focused | No | Yes (ONT) | Yes | Yes | No | No | Local and HPC | CLI | Conda, Singularity | Taxonomic profiling | 2024 | 0 | GNU GPL v3 | |
| 9 | Eukfinder ( | Special | Yes | Yes | No | No | No | No | Local and HPC | CLI | Conda | Eukaryotic MAGs | 2025 | 1 | MIT License | |
| 10 | EURYALE (MEDUSA) ( | Short-read-centered | Yes | No | No | Yes | No | No | Local, HPC, and CC | CLI | Nextflow | Conda, Singularity, Docker | 2024 | 7 | MIT License | |
| 11 | Galaxy ( | Web-based | Yes | Yes | Yes | No | No | Yes | External | GUI | External | Taxonomic profiling | 2024 | 1168 | Academic Free License v3 | |
| 12 | GEN-ERA ( | Dual | Yes | Yes (ONT) | No | Yes | No | No | Local, HPC, and CC | CLI | Nextflow | Singularity | Metabolic modeling | 2024 | 7 | GNU GPL v3 |
| 13 | HiFi-MAG ( | Long-read-focused | No | Yes (PacBio) | No | Yes | No | Yes | Local, HPC, and CC | CLI | Snakemake | Conda | 2025 | 8 | BSD-3-Clause-Clear License | |
| 14 | IDseq ( | Web-based | Yes | Yes (ONT) | No | No | No | No | External | GUI | External | Viral MAGs | 2025 | 347 | MIT License | |
| 15 | IM | Web-based | NA | NA | NA | No | No | No | External | GUI | External | Eukaryotic MAGs | 2025 | 268 | IMG Expert Review Submission Agreement | |
| 16 | JAMS ( | Short-read-centered | Yes | No | No | No | No | No | Local and HPC | CLI | Conda | Direct sample comparison | 2025 | 7 | GNU GPL v3 | |
| 17 | KBase ( | Web-based | Yes | Yes | Yes | Yes | Yes | Yes | External | GUI | External | Taxonomic profiling and metabolic modeling | 2024 | 63 | MIT License | |
| 18 | MAGNETO ( | Short-read-centered | Yes | No | No | Yes | Yes | No | Local, HPC, and CC | CLI | Snakemake | Conda | Taxonomic profiling | 2025 | 13 | GNU GPL v3 |
| 19 | MAGO ( | Short-read-centered | Yes | No | No | No | No | Yes | Local and HPC | CLI | Singularity, Docker | Phylogenetic tree generation and pangenome analysis | 2020 | 21 | Creative Commons BY 4.0 | |
| 20 | Mapler ( | Long-read-focused | No | Yes (PacBio) | No | Yes | No | No | Local, HPC, and CC | CLI | Snakemake | Conda | Visualization module | 2025 | 0 | GNU AGPL v3 |
| 21 | MetaGEM ( | Short-read-centered | Yes | No | No | Yes | No | Yes | Local, HPC, and CC | CLI | Snakemake | Conda | Eukaryotic MAGs and metabolic modeling | 2023 | 99 | MIT License |
| 22 | MetaGenePipe ( | Short-read-centered | Yes | No | No | Yes | Yes | No | Local, HPC, and CC | CLI | WDL ( | Singularity | 2023 | 1 | Apache License 2.0 | |
| 23 | Metagenome-Atlas ( | Short-read-centered | Yes | No | Yes | Yes | Yes | Yes | Local, HPC, and CC | CLI | Snakemake | Conda | 2024 | 159 | BSD-3-Clause-Clear | |
| 24 | Metagenomics- | Dual | Yes | Yes (ONT) | No | Yes | No | Yes | Local, HPC, and CC | CLI | Nextflow | Docker | Plasmid assembly, metabolic modeling and controlled resource allocation | 2025 | 0 | GNU AGPL v3 |
| 25 | Metaphor ( | Short-read-centered | Yes | No | No | Yes | Yes | Yes | Local, HPC, and CC | CLI | Snakemake | Conda | Visualization module | 2024 | 13 | MIT License |
| 26 | metagWGS ( | Dual | Yes | Yes (PacBio) | No | Yes | Yes | Yes | Local, HPC, and CC | CLI | Nextflow | Singularity | Taxonomic profiling | 2025 | 2 | GNU GPL v3 |
| 27 | MetaWRAP ( | Short-read-centered | Yes | No | No | Yes | Yes | Yes | Local and HPC | CLI | Conda and Docker | Taxonomic profiling | 2020 | 1917 | MIT License | |
| 28 | MG-TK ( | Dual | Yes | No | No | Yes | Yes | No | Local and HPC | CLI | Conda | Taxonomic profiling and strain delineation | 2025 | 99 | GNU GPL v2 | |
| 29 | MGnify ( | Web-based | Yes | Yes | Yes | Yes | Yes | No | External | GUI | External | Taxonomic profiling | 2025 | 286 | Apache License 2.0 | |
| 30 | MOSHPIT ( | Short-read-centered | Yes | No | No | Yes | No | Yes | Local and HPC | CLI | Conda | Taxonomic profiling | 2025 | 1 | BSD-3-Clause-Clear | |
| 31 | MUFFIN ( | Hybrid pipelines | No | Yes (ONT) | Yes | Yes | No | Yes | Local, HPC, and CC | CLI | Nextflow | Conda, Docker, | Metatranscriptome support | 2022 | 34 | GNU GPL v3 |
| 32 | NanoPhase ( | Long-read-focused | No | Yes (ONT) | Yes | No | No | Yes | Local and HPC | CLI | Conda | 2023 | 73 | MIT License | ||
| 33 | nf-core/mag ( | Hybrid | Yes | Yes (ONT or PacBio) | Yes | Yes | Yes | Yes | Local, HPC, and CC | CLI | Nextflow | Conda, Docker, Singularity | Ancient DNA identification | 2025 | 57 | MIT License |
| 34 | ngs-preprocess | Hybrid | Yes | Yes | Yes | Yes | No | No | Local, HPC, and CC | CLI | Nextflow | Conda, Docker, Singularity | Antimicrobial resistance gene prediction, virulence factor annotation, and plasmid assembly | 2025 | 2 | GNU GPL v3 |
| 35 | nIMP3 ( | Short-read-centered | Yes | No | No | Yes | No | No | Local, HPC, and CC | CLI | Nextflow | Docker, Singularity | Metatranscriptome support and taxonomic profiling | 2024 | 150 | MIT License |
| 36 | SnakeMAGs ( | Short-read-centered | Yes | No | No | Yes | No | No | Local, HPC, and CC | CLI | Snakemake | Conda | 2024 | 6 | CeCILL Free Software License Agreement v2.1 | |
| 37 | SPIRE ( | Short-read centered | Yes | No | No | Yes | No | No | Local, HPC, and CC | CLI | Nextflow | Antimicrobial resistance gene prediction and virulence factor annotation | 2025 | 41 | MIT License | |
| 38 | SqueezeMeta ( | Hybrid | Yes | Yes | Yes | Yes | Yes | Yes | Local and HPC | CLI | Conda | Taxonomic profiling, metatranscriptome support, and visualization module | 2025 | 400 | GNU GPL v3 | |
| 39 | Sunbeam ( | Short-read-centered | Yes | No | No | Yes | No | No | Local and HPC | CLI | Snakemake | Conda and Docker | Taxonomic profiling | 2025 | 184 | GNU GPL v3 |
| 40 | VEBA ( | Dual | Yes | Yes (ONT or PacBio) | No | Yes | Yes and pseudo- coassembly | Yes | Local and HPC | CLI | GenoPype ( | Conda and Docker | Eukaryotic or viral MAGs, antimicrobial resistance gene prediction, and virulence factor annotation | 2025 | 23 | GNU AGPL v3 |
| 41 | WGSA2+/LoRA ( | Web-based | Yes | Yes (ONT or PacBio) | No | Yes | No | No | External and CC | GUI | AWS environment | External | Visualization module, metatranscriptome support, and antimicrobial resistance gene prediction | 2025 | 138 | CC0 1.0 Universal |
- —Federal Commission for Scholarships for Foreign Students
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Microbial Community Ecology and Physiology · Scientific Computing and Data Management
INTRODUCTION
Metagenomics has advanced the study of microbial communities by diminishing the need for cultivation and enabling direct DNA sequencing from complex environments such as the human body, soil, or aquatic ecosystems (1). This has been possible thanks to the combination of high-quality and high-throughput sequencing technologies and recent advances in bioinformatics tools, increasing the scope and resolution at which the microbiota can be explored (2). Moreover, reconstructing metagenome-assembled genomes (MAGs) has enabled the genomic characterization of uncultured microorganisms, the discovery of previously unknown species, the inference of the community’s metabolic and functional potential, the establishing of ecological interactions, and the detection of evolutionary mechanisms (2, 3).
Considering the ecological importance of the MAGs, genomic criteria have been designed to determine whether a recovered bin (draft genome) truly represents a MAG or not. For instance, the Minimum Information about MAGs guidelines establish that MAGs can be classified into three quality tiers: high-quality drafts (HQ), medium-quality drafts (MQ), and low-quality drafts; the specific details regarding the genomic quality metrics used for this classification were introduced by Bowers et al. (4). MAGs can also be divided into species-assigned MAGs (SMAGs), that is, MAGs for which a species can be assigned, and hypothetical MAGs (HMAGs), that is, MAGs that are supposedly genomes of novel species, according to the genome heterogeneity spectrum proposed by Setubal (5).
In a simplified manner, MAGs are obtained through bioinformatics pipelines that include quality control, assembling and binning the sequences, and the annotation of each recovered genome (6) (Fig. 1). These pipelines are then responsible for the correct MAG assembly and have a key role at extracting meaningful information about the structure and function of microbial communities (1). Through their orchestrated workflow, they simplify and standardize the common tasks that are required to achieve HQ MAGs, reducing the occurrence of manual errors by improving reproducibility (7). Nonetheless, pipeline choice may not be a trivial decision, given that it should be based on the alignment between user needs and workflow key factors such as the type of sequencing data they handle (short or long reads, or both), analytical functions (i.e., co-assembly, sequential co-assembly, taxonomic profiling, and eukaryotic recovery), and computational environment (e.g., availability of local resources, high-performance computing [HPC] infrastructure, or web-based tools). Therefore, pipeline selection can quickly become an overwhelming process and challenge researchers with a vast landscape of options, delaying the start of the analysis or even not obtaining the expected results since the incorrect workflow was chosen.
Usual bioinformatics workflow followed to perform MAG recovery, classification, and annotation. Some common tools incorporated by the pipelines are highlighted.
Here, we describe the general workflow followed by bioinformatics pipelines to recover MAGs directly from metagenomics data, discussing important aspects the pipelines feature, such as the tools they encompass and the type of data they can handle. We also succinctly highlight major considerations regarding pipeline execution, storage needs, and computational infrastructure. Likewise, we provide a compact overview of 41 publicly available pipelines, suites, or platforms that enable MAG reconstruction and/or annotation starting from short and/or long sequences. Finally, considering the main practical features of each pipeline and aiming at aiding researchers in navigating the ecosystem of workflows, we also introduce 2Pipe, a decision-support web application designed to match metagenomics community users with the most suitable MAG pipeline based on their input data, technical requirements, bioinformatics experience, and preferred interface.
PIPELINE WORKFLOW, TOOLS, AND BENCHMARKS
The traditional computational workflow to build and annotate MAGs involves several steps (6); Fig. 1 introduces the general series of steps to potentially achieve MQ or HQ MAGs, along with some common software integrated by the pipelines. In brief, it begins with quality control, where low-quality reads and contaminants are removed (8, 9); when required, some pipelines include the option to discard host organism sequences (10). This is followed by the assembly step, where reads are extended to create contiguous sequences, also called contigs. The contigs are then grouped into bins that ideally represent individual genomes, based on the sequence composition and coverage patterns, among other genomic features (11). Optionally, the bins are subjected to a process of refinement when researchers consider it necessary (12, 13). Afterward, these bins are evaluated for common metrics such as completeness and contamination to assess their quality and hence determine whether they constitute MAGs or not, using the criteria previously mentioned (14). In some cases, the workflows can encompass dereplication tools or modules that attempt to curate the MAG set by clustering them according to their genomic similarity and thus selecting a representative MAG from each cluster (15). To conclude with the workflow, the MAGs are then taxonomically affiliated and functionally annotated to assign biological meaning, extracting insights related to their identity and potential roles within their microbial communities (16, 17). A detailed description of the tools for each step of the workflow is provided by Yang et al. (6), and Wajid et al. (18) present an overview of the typical analysis pipeline and software using an interesting music analogy.
We present on Table 1 the tools and third-party software for quality control, assembly, binning, refinement, taxonomic classification, and functional annotation that each of the pipeline documented here encompasses. Additionally, a detailed description of the main workflow for each of them can be found in File S1, where important technical considerations such as the type of input (short reads, long sequences, or both), tools employed at each step, advantages, limitations, and/or special features they depict are presented.
As previously mentioned, the MAG reconstruction workflow is triggered with the quality control of the raw reads to ensure the accuracy and integrity of downstream analyses. Usually, the reads received from the sequencing facility contain sequencing errors, low-quality bases, adapters, and contaminant sequences (e.g., host or environment DNA) that can lead to fragmented assemblies or chimeric bins if not properly removed (6, 10). These issues are addressed by filtering and trimming, if required, the raw reads using tools like Trimmomatic (9), fastp (8), Cutadapt (139), or BBTools (21). In the case of contamination removal, tools such as KneadData (82), Bowtie2 (55), Minimap2 (88), BWA (75), or Kraken (either v1 or v2) (62, 108) are commonly used to screen and remove host-derived or non-target reads. For long-read data (Oxford Nanopore known as ONT or Pacific Biosciences known as PacBio), Filtlong (42), Nanofilt (43), and Porechop (90) are used for length filtering, quality trimming, and adapter removal. The pipeline quality control and contamination removal modules are often complemented by FastQC (20) or MultiQC (103), the standard methods to evaluate the overall quality and report it; NanoPack2 and pycoQC (215) provide detailed quality summaries for long reads. In a recent report, Gao et al. (10) compared many available tools for removing host contamination, namely, KneadData, Bowtie2, KMCP (260), BWA, KrakenUniq (35), and Kraken2, highlighting the superior performance depicted by Bowtie2 in terms of resource usage, while Kraken2 demonstrated the shortest execution times; the accuracy of Bowtie2, KneadData, and BWA outperformed the rest of the tools.
Furthermore, the assembly step represents the core of the process since it reconstructs longer contiguous sequences from the high-quality reads. Notably, assembling metagenomics data sets faces complex challenges due to varying species abundance, uneven coverage, and the presence of closely related organisms (261). The short-read assemblers rely mainly on two strategies: overlap-layout-consensus, which aligns overlapping reads to build contigs, and the more widely used De Bruijn graph method, which decomposes reads into k-mers and represents them as nodes and edges in a graph (261). MEGAHIT (22), metaSPAdes (30), and IDBA-UD (31) are examples of tools that implement the De Bruijn graph approach, incorporating heuristics to address the coverage variation and strain complexity. In contrast, assemblers for long-read data such as metaFlye (45), Canu (111), and hifiasm (157) are designed to apply graph-based algorithms optimized for higher error rates and uneven depth. In some cases, hybrid strategies are employed, combining long reads for structural resolution with accurate short reads for initial graph assembly, as implemented in tools like OPERA-MS (92) and hybridSPAdes (209).
To this date, some authors have attempted to provide a comprehensive and unbiased benchmark of the most popular assemblers using different data sets that vary in complexity. For instance, Goussarov et al. (262) developed a comparison among short, long, and hybrid assemblers using a complex mock metagenome with more than 200 bacterial strains, demonstrating that metaSPAdes can achieve superior performance in terms of assembly fragmentation and chimerism when using Illumina reads, while Canu depicted the best metrics (chimerism and fragmentation) for ONT data. A similar conclusion regarding short-read assemblers was presented by Meyer et al. (263), where although MEGAHIT and metaSPAdes showed similar performance, metaSPAdes delivers fewer fragmented assemblies using simulated mouse gut sequences that enclosed more than 540 species. During the analysis of data sets enclosing mixed real metagenomic reads and reads from known genomes, Wang et al. (264) reported MEGAHIT as the most efficient assembler, while metaSPAdes outperformed MEGAHIT, IDBA-UD, and Faucet (265) in terms of integrity and continuity at the species level, and it showed the overall best performance at the strain level.
In the case of hybrid assembly, Brown et al. (266) showed boosted contiguity and reduced assembly errors with either hybridSPAdes or OPERA-MS, although yielding frequent misassemblies during in silico spike-in experiments using real and simulated reads. Nevertheless, assemblies obtained with these hybrid same tools were less complete and more fragmented than long-read only assemblies using the same data set of more than 200 bacterial strains mentioned above (262). As a result, Goussarov et al. suggest constructing the assembly using long reads complemented with short-read polishing, when the coverage is sufficient.
Accompanying the core of the pipelines, binning tools also represent an important step to reconstruct as accurately as possible the genomes present in the microbial communities. Classical binning strategies can be divided into different categories: (i) algorithms based on the genomic composition (mainly k-mer frequencies and GC content), (ii) approaches using read depth (coverage) profiles across multiple samples to link contigs with similar abundance patterns, and (iii) combined strategies that integrate both sequence composition and coverage signals (6). Classical tools based on these strategies such as MetaBAT2 (32), MaxBin2 (33), and CONCOCT (23) have been widely incorporated into the workflows given their efficiency and robustness. Nevertheless, more recent methods leverage machine learning and semi-supervised approaches to improve the resolution in more complex environments such as soil or ocean (267). SemiBin2 (56) represents an example of these recent strategies as it uses deep learning with semi-supervised contrastive learning to incorporate both intrinsic sequence information and external reference genomes. Another example is represented by COMEBin (57), which employs graph neural networks to integrate contrastive multiview representation learning, coverage, and a clustering algorithm.
Similar to the assembly case, there have been efforts to benchmark the performance of the available binning tools. In a recent report, Han et al. (11) used different combinations of short, long, and hybrid data to compare the outcomes from 10 binners, finding that deep-learning-based tools (COMEBin and SemiBin2) were almost always among the top three high-performance binners regardless of the combination of the contig provenance. Through comparisons among less tools, Cansdale & Chong (268) showed that CONCOCT generated more high-quality bins than MetaBAT2 using a simple gut metagenome, while Meyer et al. (263) reported homogeneous results among CONCOCT, MetaBAT2, and MaxBin2, with MAG completeness slightly increased by CONCOCT at the expense of genome purity. Contrastingly, Groopm2 (269) and MetaBAT2 provided the best performance metrics in recall, purity, and the number of high-quality genome bins at recovering MAGs from Critical Assessment of Metagenome Interpretation (CAMI) data sets (270). In addition, Yepes-García and Falquet (271) used environmental metagenomics samples (rice soil) to show how MetaBinner stands out for the greater number of bins recovered as compared with MetaBAT2 and SemiBin2, albeit only 10% of these were at least MQ MAGs.
Moreover, the inclusion (or enabling) of tools within the workflows to recover a non-redundant and high-quality MAG set is determinant. Several pipelines incorporate bin refinement modules or tools to improve the quality of the bin set as they reduce contamination, increase completeness, and may recover mis-binned contigs (12, 13, 85). The tools in charge of this task take as input the bins from different binning software to provide the best possible version of each bin and potential MAG. Among the existing tools for bin refinement, MAGScoT (13) is claimed by the developers as the piece of software with the best performance, as compared to DASTool (12) and the MetaWRAP-binning module (59), in terms of MAG quantity and quality using simulated marine and human gut data sets. Nonetheless, Han et al. (11) showed how MetaWRAP achieved the highest rank score (custom ranking score developed for the study) followed closely by MAGScoT, although this former tool demanded 10 times less memory and carried the bin refinement in one-tenth of a fraction of the time required by MetaWRAP.
Contamination estimation tools aid in the main goal of ensuring the reliability of the MAGs, with representative tools such as CheckM (26), BUSCO (116), and CheckM2 (58) that infer completeness and contamination based on single-copy marker genes from specific lineages or deep learning models. Notwithstanding, a benchmarking study (14) showed that CheckM may underestimate contamination, mainly if sequences from distantly related taxa are present, as it reported contamination values between 1% and 2% when the true contamination introduced by the researchers was 11%. In contrast, in the same study, the authors found that tools integrating phylogenomic signals or read classification strategies like GUNC (114), Kraken2 (62), Physeter (117), and Forty-Two (272) achieved contamination estimations closer to the true values and performed overall better at detecting inter-domain contamination. Further, within the CheckM2 paper itself, the developers demonstrated its greater accuracy to detect genome contamination conferred by unusual lineages and to predict genome completeness.
Similarly, some pipelines could include dereplication strategies after quality assessment, typically based on Average Nucleotide Identity with the aim of curating the MAG set and selecting the best representative MAG in each cluster of MAGs. Nonetheless, enabling the execution of these dereplication tools (85, 144, 177), as well as the parameter configuration, should always be thought thoroughly as discussed by Evans and Denef (15), who analyzed the advantages and drawbacks of running de-replication procedures. Briefly, these authors highlighted how dereplication maintains high quality of genomic databases and enhances coverage pattern estimations; however, dereplication may lead to a loss of information on variability in the auxiliary gene content among representatives from the same species.
One of the final stages when building MAGs is represented by reporting the taxonomic affiliation of each genome. The most common tool included within the workflows (Table 1) is GTDB-Tk (17) since it demonstrated that its phylogeny-based approach achieves high agreement (around 90%) with manually curated classifications in the GTDB, while GTDB-Tk v2 (GTDB-Tk2) is further optimized to reduce memory requirements without compromising the accuracy. Beyond this, the report describing the capabilities of CAT and BAT (107) included a benchmark against GTDB-Tk that demonstrated very similar performance as BAT and GTDB-Tk provided the same final MAG annotations.
Other classifiers not particularly designed to annotate MAGs can be included within the workflows such as MetaPhlAn4 (190), Kraken (108), Kraken2 (62), Centrifuge (36), and Kaiju (78) through the re-formatting of the draft genomes to make them suitable as input for these tools. There have been several efforts to benchmark taxonomic classifiers in a wide variety of scenarios and using different types of data (10, 273–279); however, these studies contrasting their performance and precision have shown variable results. For instance, Kraken2 in combination with Bracken exhibited superior precision, sensitivity, F1 score, and overall sequence classification of a custom in silico mock community within a comparison against MetaPhlAn and Kaiju (273); similar results were described by Timilsina et al. (274), who reported the highest accuracy and broad sensitivity achieved by Kraken2/Bracken (86) in simulated microbial communities as compared against MetaPhlAn4 and Centrifuge. Meanwhile, Irankhah et al. (275) observed how MetaPhlAn4 exhibited higher precision in identifying species in a simulated data set, outperforming Kraken2, Bracken, and Centrifuge. In contrast, when attempting to classify long reads (ONT), Kraken2 and Centrifuge demonstrated low to very low precision for all defined mock communities considered in the study (276). Similarly, Centrifuge depicted the worst performance at classifying sequences belonging to a mock community built from human fecal samples, within the study that introduced the tool DeepMicrobes (277).
To complete the final stages of the MAG reconstruction, functional annotation serves to reveal metabolic potential and ecological roles of microbial communities, with a remarkably high number of options available (280). The selection of these tools depends on the study goal, and it is usually a conscious decision made by the researchers. For more than 10 years, Prokka (96) has remained as standard for rapid genome annotation, predicting coding sequences, rRNAs, and tRNAs and assigning functions through curated databases. Nevertheless, more elaborated tools like eggNOG-mapper (84) have emerged to provide large-scale functional annotation, and the DRAM pipeline (141) offers detailed metabolic summaries. Web-based systems like RASTtk (70) (implemented within the Bacterial and Viral Bioinformatics Resource Center, BV-BRC [65]) and MGnify (193) can achieve quick and reliable annotations, while for specialized functional insights, tools like antiSMASH (197), KOfamKOALA (167), and dbCAN3 (140) are often incorporated into the workflows.
As shown on Table 1, taxonomic and functional annotation steps heavily rely on existing databases, highlighting the importance of these information resources. In the case of taxonomic classification, the GTDB (200) provides a phylogenetically consistent framework for prokaryotic and archaeal taxonomy, while nucleotide and protein repositories like UniRef (195) and Swiss-Prot (165) offer curated sequences that serve reliable standards for accurate assignments. On the functional prediction side, the KEGG (122) and its ortholog collection (KOfam [167]) enables the reconstruction of metabolic pathways, while Pfam (132) catalogs protein domains and families that help identify conserved protein functions. In the same sense, the database for evolutionary genealogy of genes: non-supervised Orthologous Groups (eggNOG) (52) covers orthologous groups linked to functional categories including COG (38), KEGG, and Gene Ontology terms (281). Other specialized databases are represented by the CAZy (188) and the database of proteolytic enzymes, their substrates, and inhibitors (MEROPS) (282). Please note that this is not a comprehensive review, and hence we suggest further reading of the works by Zeller and Huson (283) and Lin et al. (280), who explored and compared computational methods and classification systems, including databases, for protein function prediction.
Finally, benchmarking entire pipelines can be more challenging as they include many pieces of software which difficults setting a groundline for comparisons. Notwithstanding, there are a few works where the whole pipeline execution has been benchmarked, for instance, Churcheward et al. (147), who tested their pipeline performance (MAGNETO) against similar workflows such as nf-core/mag, Metagenome-Atlas, and MetaWRAP. These authors recovered a superior number of HQ MAGs from human gut microbiomes (Integrative Human Microbiome Project) through MetaWRAP operated in either single-assembly with single binning or co-assembly with a co-binning approach (see the next section for a detailed explanation of these approaches). Meanwhile, Yepes-García and Falquet (271), starting from sequences belonging to a mock community, depicted slight differences in terms of genome completeness, contamination, and number of MAGs taxonomically annotated at species level among MetaWRAP, nf-core/mag, SnakeMAGs, and Metagenome-Atlas. nf-core/mag reached the highest percentages of MQ and HQ MAGs, whilst DATMA, also included in this study, performed poorly as only 40% of the MAGs were assigned a proper taxonomic classification and not a single MQ or HQ MAG was recovered.
PRACTICAL AND TECHNICAL CONSIDERATIONS FOR PIPELINE EXECUTION
As high-throughput sequencing technologies have grown in the past years, the availability of MAG-centered pipelines has been quickly expanded to handle and integrate different data types and computational strategies (169, 180, 250). Specifically, recent pipelines have been designed or have evolved to assemble and bin short reads (normally Illumina), long reads (mainly ONT and PacBio), or a blend of both technologies to maximize base calling, depth, contiguity, and structural information (180, 250). Short reads synthesized through DNA nanoball sequencing (284) or long reads derived from CycloneSEQ (285) can be eventually processed by some pipelines (207, 214). Differences or similarities among these MAG-reconstruction approaches based on the type of sequence used as input have been studied by Goussarov et al. (262), and Kim et al. (286) analyzed the variations in terms of genome recovery between Illumina and MGI platforms.
Among the several tools that compose a pipeline (Fig. 1), assembly and binning tools are mainly responsible for the scaling up in the hardware demands, especially when handling data sets with several samples encompassing millions of short-read sequences (6). Moreover, these tools can be executed in different configurations such as co-assembly and co-binning, as these strategies can increase the overall MAG recovery rate and quality (287). Briefly, co-assembly refers to the possibility of performing the metagenome assembly after merging user-specified samples to enhance the coverage, capturing a higher fraction of the diversity (287), while co-binning establishes the possibility of binning contigs using coverage information across multiple samples simultaneously after single or co-assembly (11). Co-binning is advantageous at exploring coverage across samples and improving separation of closely related genomes (47). Despite the desirable benefits co-assembly can bring to the analysis, it is computationally intensive and increases the probability of generating fragmented assemblies (147), although sequential co-assembly has emerged recently as an efficient alternative that enhances both time and memory requirements by the assembler (288). Similarly, co-binning can be sensitive to uneven sequencing depth, requires high-quality coverage profiles, and can be affected by low diversity among samples (147). Vosloo et al. (287) and Han et al. (11) have demonstrated how superior performance can be achieved by applying co-assembly and/or co-binning.
On the other hand, the workflow execution varies in terms of computational demands, where small-scale data sets can be processed on high-end workstations, while large or complex metagenomes often require access to HPC clusters or cloud-based environments (Azure, Amazon Web Services or AWS, Google Cloud, and Terra, among others). Beyond sample-specific computational requirements, and as mentioned before, most metagenomics pipelines rely on external reference databases to perform taxonomic classification, functional annotation, and quality assessment of MAGs. Commonly used databases, namely, RefSeq (289), GTDB, UniProt (206), KEGG, and eggNOG, are large and require substantial local storage that ranges from tens to hundreds of gigabytes. For instance, the latest GTDB release (R226) exceeds 140 GB, while comprehensive functional annotation pipelines like DRAM can demand up to 500 GB to exploit its full potential. Being so, MAG building is a demanding process that needs adequate disk space, CPU capacity, and memory availability.
For researchers without access to HPC resources, web-based platforms such as KBase (290), MGnify (193), Galaxy (105), and BV-BRC (65), among others, can assist them by carrying out analysis execution in their servers. In addition, these platforms aid users without a strong experience in command line interface (CLI) interaction since they provide user-friendly interfaces where users can upload raw reads and run predefined workflows. As a result, these platforms eliminate the need for CLI proficiency and offer built-in visualization applications and databases for downstream interpretation; a complete landscape of web-based applications is compiled by Achudhan et al. (291) and Chivian et al. (138).
Furthermore, given the MAG pipeline evolution in complexity, involving multiple tools, dependencies, and steps, the use of workflow managers has become the standard to ensure reproducibility, scalability, and portability (292). Specifically, workflow managers ease pipeline step definition in a modular and automated architecture to orchestrate entire analyses, tracking software versions, managing intermediate files, restarting the process if interrupted, handling multiple samples as input, and enabling parallel processing in a reproducible manner. Some representatives of these helpful orchestrators are Snakemake (293), Nextflow (294), and Workflow Definition Language (WDL) (295) whose design, implementation, benefits, and scope have been reviewed in some reports (292–294, 296); also, important guidelines for pipeline design based on workflow managers have been published by Roach et al. (297), Reiter et al. (298), and Ahmed et al. (7). Advantageously, containerization platforms such as Docker, Singularity, and Seqera Containers, or package managers like Conda or the Python Package Index complement workflow orchestrators by offering a flexible and reproducible solution for software and dependency management (299). As a result, this combination allows users to run the analysis without system conflicts, specific versions of the software, and libraries.
In contrast, beyond the MAG assembly and annotation, some pipelines feature interesting options that complement the analysis and provide a wider understanding of the microbial community. The range of these special options is wide, and therefore they must be carefully selected. In this sense, read-based taxonomic profiling (1) is one of the most common offerings by the pipelines as this process does not rely on the main workflow and can be executed in parallel. Furthermore, some pipelines can incorporate tools or modules to recover viral or eukaryotic MAGs (250), and it is even possible to find pipelines mostly focused on this type of MAGs (97). Another popular extra option is represented by the possibility of establishing genome-scale metabolic models among the built MAGs (159, 169). However, in many cases, some workflows can be considered unique since they include options that no other pipeline encompasses. Examples of these rare features are the possibility to assemble plasmids (169), genotype recovery (41) , controlled resource allocation (169), and an alternative assembly and binning order, where the reads are first grouped (binning) and then assembled in batches (73).
On Table 2, we present a summarized overview of the technical features and methodological factors each workflow presents, and hence these same pipeline aspects are also the basis for the questionnaire presented on 2Pipe. Methodological factors include the ability to assemble short reads, long sequences, or both in a hybrid approach; the possibility to request a co-assembly and/or co-binning natively; whether the user can input multiple samples or not; if the pipeline includes a bin refinement tool; and special functionalities they may incorporate. In the same sense, technical features are described through factors like which kind of resources the user is planning to use for the pipeline execution, the interface they feel more comfortable working with, the workflow manager they expect to orchestrate the data flow, and the software/package technology management available within each workflow. We assigned one of the following (non-mutually exclusive) labels in order to classify them: short-read-centered or long-read-focused (if their main input is short or long reads), dual (if they can handle both long and short reads, but they do not perform hybrid assembly), hybrid (pipelines able to assemble short and long reads together), web-based (pipelines offered by online platforms or suites), or special (pipelines designed for a specific purpose).
TABLE 2: Technical and operational features for each pipeline or web-based platform
2PIPE: IT STARTS WITH A QUESTION
Considering the pipeline landscape identified in this review, we have developed a decision-support application that concatenates most of the features described for each workflow. 2Pipe is an interactive web application designed to help researchers identify the most suitable metagenomics pipeline for reconstructing and annotating MAGs. 2Pipe can be used by users with different expertise levels and computational access, simplifying the often-complex selection process by mapping user needs to a curated database of available pipelines.
At the core of 2Pipe, there is a dynamic and question-driven interface that guides users step by step through a personalized questionnaire. This adaptive form collects information related to the methodological factors and technical features detailed on Table 2. Therefore, every response is used to assign a score to each pipeline based on the presence or absence of specific features that align with the user’s input. The recommendation system will then suggest the pipeline with the highest score, as well as the second “best hit” for the user to check in case the first option does not fulfill their requirements; these suggestions can be as well the starting point for the user to dig into the other sections of 2Pipe. It is worth mentioning that the scoring is weighted, and some features have prevalence as they are definitive for the pipeline suggestion. Specifically, all matching features presented in the questions add one point to the final score, excepting type of reads to analyze (2 points), the need for a GUI (3 points), and the requirement for external computational resources (3 points). These features are prioritized then, and the recommendation must reflect them as they cannot simply be bypassed with any other pipeline. The system also includes a protection for cases when the users do not provide at least three answers, asking them to restart the questionnaire. Likewise, in case of a tie among more than two pipelines, the recommendation system will show all of them with the respective matching features.
Aside from the accession to the questionnaire and the response-based recommendation at the end of this, 2Pipe as well encompasses a pipeline gallery, where a visual catalog is displayed, offering individual summaries of each pipeline and a direct access to the source code or to the publication that documents the pipeline. Additionally, 2Pipe makes available an interactive view of Table 2 that includes the possibility of filtering by each feature or by a combination of them, allowing users to directly tailor the search for the pipeline that best suits their needs; the displayed categories are the same key attributes the question-based suggestion system relies on. 2Pipe also incorporates the features presented in Table 1, assisting the user when comparing the pipelines beyond technical aspects. Also, these tools and external software are organized in a gallery that allows the user to match pipelines that use them, which is useful if the user is looking for a specific software combination that a specific pipeline can offer.
On the other hand, given the importance pipeline and tool benchmarking represents, 2Pipe provides an exclusive page where the reports cited in this work comparing performance and/or technical features are introduced. This page is divided into sections according to the tools benchmarked in the papers, namely, assemblers, binners, bin-refinement tools, contamination-estimation software, complete pipelines, workflow managers, and taxonomic classifiers. Moreover, we include sections for reviews, tutorials, and protocols for manual MAG reconstruction and key papers that set interesting discussions around MAG recovery.
The source code for 2Pipe is available at the repository https://github.com/jeffe107/2pipe, and foreseeing the possibility of new pipelines being released in the near future, we provide a quick form for developers to include their workflow into 2Pipe’s recommendation system, pipeline gallery, and table comparison. Also, at the GitHub repository, developers can find a simple template and detailed instructions for the inclusion of their pipeline through a pull request.
CONCLUSION
The rapid evolution of sequencing technologies has broadened the availability of metagenomics data sets that demand bioinformatics tools adjusted to the user requirements to achieve cutting-edge analysis, including MAG reconstruction. As a result, in the past 10 years, a rise in the number of MAG reconstruction pipelines available has been observed, and the selection of the proper pipeline for the analysis has become an essential step during the execution of metagenomics projects. This review offers a compact description of 41 publicly available pipelines or platforms, with special focus on their capabilities and distinctive features to serve as a valuable resource for researchers navigating this overwhelming landscape. Beyond the scope of a classical review, we streamlined the selection process by introducing 2Pipe, an interactive decision-support web application that aligns the user needs with the most convenient workflow for their analysis and allows a general overview of the pipeline universe with its gallery and pipeline-comparison sections. Finally, this review and its accompanying application provide a unified framework that simplifies the decision-making process, releasing part of the burden and uncertainty when setting a metagenomics data analysis project.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Navgire GS, Goel N, Sawhney G, Sharma M, Kaushik P, Mohanta YK, Mohanta TK, Al-Harrasi A. 2022. Analysis and Interpretation of metagenomics data: an approach. Biol Proced Online 24:18. doi:10.1186/s 12575-022-00179-736402995 PMC 9675974 · doi ↗ · pubmed ↗
- 2Kim N, Ma J, Kim W, Kim J, Belenky P, Lee I. 2024. Genome-resolved metagenomics: a game changer for microbiome medicine. Exp Mol Med 56:1501–1512. doi:10.1038/s 12276-024-01262-738945961 PMC 11297344 · doi ↗ · pubmed ↗
- 3Lemos LN, Mendes LW, Baldrian P, Pylro VS. 2021. Genome-resolved metagenomics is essential for unlocking the microbial black box of the soil. Trends Microbiol 29:279–282. doi:10.1016/j.tim.2021.01.01333551270 · doi ↗ · pubmed ↗
- 4Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, et al.. 2017. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol 35:725–731. doi:10.1038/nbt.389328787424 PMC 6436528 · doi ↗ · pubmed ↗
- 5Setubal JC. 2021. Metagenome-assembled genomes: concepts, analogies, and challenges. Biophys Rev 13:905–909. doi:10.1007/s 12551-021-00865-y 35059016 PMC 8724365 · doi ↗ · pubmed ↗
- 6Yang C, Chowdhury D, Zhang Z, Cheung WK, Lu A, Bian Z, Zhang L. 2021. A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data. Comput Struct Biotechnol J 19:6301–6314. doi:10.1016/j.csbj.2021.11.02834900140 PMC 8640167 · doi ↗ · pubmed ↗
- 7Ahmed AE, Allen JM, Bhat T, Burra P, Fliege CE, Hart SN, Heldenbrand JR, Hudson ME, Istanto DD, Kalmbach MT, Kapraun GD, Kendig KI, Kendzior MC, Klee EW, Mattson N, Ross CA, Sharif SM, Venkatakrishnan R, Fadlelmola FM, Mainzer LS. 2021. Design considerations for workflow management systems use in production genomics research and the clinic. Sci Rep 11:1–18. doi:10.1038/s 41598-021-99288-834737383 PMC 8569008 · doi ↗ · pubmed ↗
- 8Chen S, Zhou Y, Chen Y, Gu J. 2018. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:i 884–i 890. doi:10.1093/bioinformatics/bty 56030423086 PMC 6129281 · doi ↗ · pubmed ↗