Compound Heterozygous Protein C Deficiency Presenting With Splenic Infarction After COVID-19: A Case Report
Saki Imai, Soichiro Ishimaru, Masayuki Hirai, Makito Tanaka, Tetsushi Yoshikawa

TL;DR
A 17-year-old with a rare protein C deficiency developed severe blood clots and a splenic infarction after recovering from COVID-19, highlighting the need for close monitoring in such patients.
Contribution
This case report highlights the risk of thrombosis in protein C deficiency patients during and after COVID-19, even after initial recovery.
Findings
A patient with compound heterozygous protein C deficiency developed VTE and splenic infarction after COVID-19.
Thrombotic events occurred despite anticoagulant therapy and initial clinical improvement.
The case underscores the need for individualized anticoagulation strategies in such patients during and after infection.
Abstract
Congenital protein C deficiency is a rare autosomal recessive disorder that predisposes patients to severe thrombosis due to markedly reduced protein C activity. Although primarily associated with venous events, an increased risk of arterial thrombosis is also recognized, particularly in severe cases. We report the case of a 17-year-old male patient with compound heterozygous protein C deficiency who developed venous thromboembolism (VTE) after coronavirus disease 2019 (COVID-19) and subsequently experienced splenic infarction despite anticoagulant therapy. Diagnosed shortly after birth following neonatal purpura fulminans, he had been maintained on long-term warfarin therapy. The patient presented with fever and cough and was diagnosed with COVID-19 by antigen testing. Soon after symptom onset, he developed right thigh pain with rapidly expanding ecchymosis, leading to a diagnosis of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
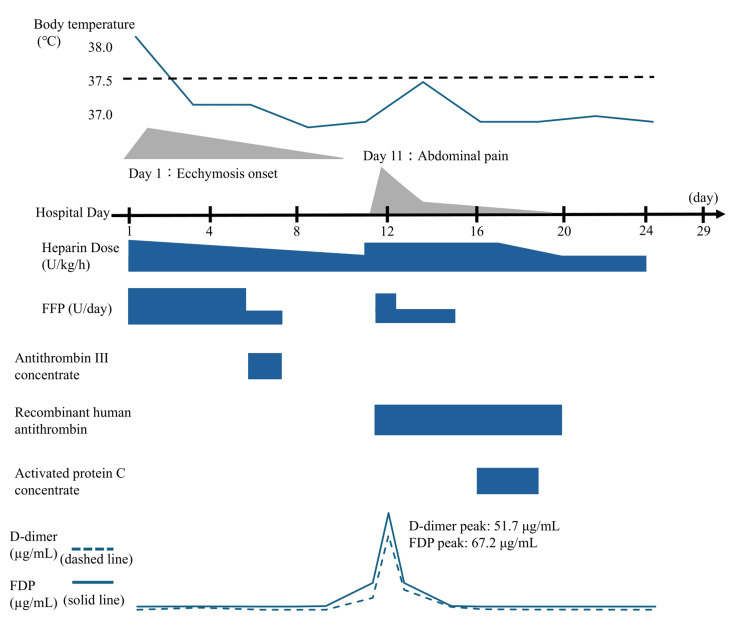 Figure 1
Figure 1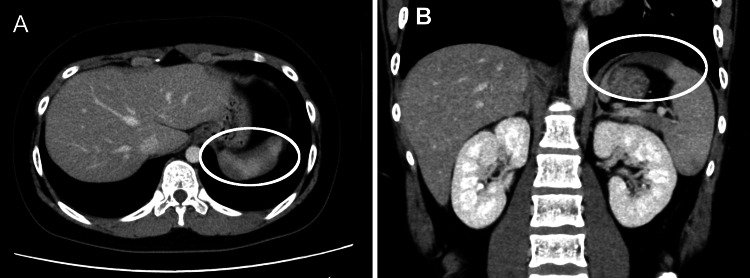 Figure 2
Figure 2| Parameters | Patient Value | Reference Range |
| WBC (/µL) | 5900 | 4000–11000 |
| Neut (%) | 52.2 | 40–70 |
| Lym (%) | 29.1 | 20–50 |
| RBC (×10⁶/µL) | 4.98 | 4.3–5.7 |
| Hb (g/dL) | 12.4 | 13–18 |
| Hct (%) | 40.1 | 40–52 |
| Plt (×10⁵/µL) | 3.06 | 1.5–4.0 |
| PT-INR | 2.13 | 0.9–1.1 |
| APTT (s) | 53 | 25–35 |
| D-dimer (µg/mL) | 1.7 | <1.0 |
| TAT (ng/mL) | 7 | 0–4 |
| PIC (µg/mL) | 0.76 | 0.2–0.8 |
| PAI-1 (ng/mL) | 20.6 | 5–40 |
| Sodium (mEq/L) | 141 | 135–145 |
| Potassium (mEq/L) | 4 | 3.5–5.0 |
| Chloride (mEq/L) | 106 | 98–108 |
| BUN (mg/dL) | 15.7 | 7–20 |
| Cr (mg/dL) | 0.55 | 0.5–1.1 |
| TP (g/dL) | 7.1 | 6.5–8.0 |
| Alb (g/dL) | 4.2 | 3.5–5.2 |
| T-bil (mg/dL) | 0.2 | 0.2–1.2 |
| AST (U/L) | 20 | 13–33 |
| ALT (U/L) | 16 | 8–42 |
| LDH (U/L) | 225 | 120–245 |
| AMY (U/L) | 66 | 40–120 |
| GLU (mg/dL) | 105 | 70–110 |
| CRP (mg/dL) | 0.17 | <0.3 |
| Hospital Day | PT-INR | APTT (seconds) | D-dimer (µg/mL) | FDP (µg/mL) | Clinical notes |
| 1 | 2.13 | 53 | 1.7 | 2.6 | Admission, diagnosis of VTE |
| 4 | 1.25 | 41 | 0.3 | 2.5 | Improvement under therapy |
| 8 | 1.71 | 90.8 | 0.3 | 2.5 | Purpura resolved |
| 11 | 2.13 | 200a | 28.8 | 48.7 | Onset of splenic infarction |
| 12 | 2.19 | 200 | 51.7 | 67.2 | - |
| 13 | 2.18 | 200 | 14.1 | 18.8 | - |
| 15 | 2.27 | 200 | 3.6 | 4.7 | - |
| 17 | 2.69 | 200 | 1.2 | 2.5 | - |
| 24 | 4.06 | 63 | 0.4 | 2.5 | Heparin discontinued |
| 29 | 5.76 | 86.8 | 0.3 | 2.5 | Discharge |
| Reference Range | 0.9~1.1 | 25~35 | <1.0 | <5.0 | - |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBlood Coagulation and Thrombosis Mechanisms · Vitamin K Research Studies · Retinopathy of Prematurity Studies
Introduction
Congenital protein C deficiency is a rare autosomal recessive disorder that leads to severe thrombotic events due to markedly reduced protein C activity, with an estimated incidence of one in 500,000-750,000 live births among homozygous or compound heterozygous patients [1]. Coronavirus disease 2019 (COVID-19) causes vascular endothelial injury and systemic inflammation, increasing the risk of venous and arterial thrombosis [2,3]. Because protein C also exerts endothelial-protective effects, its reduced activity may further amplify COVID-19-associated coagulopathy [4,5]. There is, however, only limited evidence to describe how congenital protein C deficiency interacts with post-COVID vascular endothelial dysfunction, particularly regarding arterial thrombosis in the recovery phase.
Here, we report the case of a patient with compound heterozygous protein C deficiency who developed venous thromboembolism (VTE) after COVID-19 and subsequently experienced splenic infarction despite apparent biochemical improvement and tapering of anticoagulation.
Case presentation
A 17-year-old male patient presented with fever and cough and was diagnosed with COVID-19 by antigen testing two days after symptom onset.
The patient had been followed for compound heterozygous protein C deficiency at our hospital since birth. He presented with purpura fulminans shortly after birth and was diagnosed with protein C deficiency (protein C activity <10%, protein C antigen <5%), maintaining a prothrombin time-international normalized ratio (PT-INR) of approximately 4. Genetic analysis had revealed compound heterozygous mutations in the protein C gene (PROC), the inactivator of coagulation factors Va and VIIIa (c.631C>T and c.1268delG), inherited from each parent. These variants have been previously reported in Japanese patients with severe congenital protein C deficiency [6]. The patient had recurrent episodes of VTE triggered by infection or trauma and had been treated with heparin and periodic fresh frozen plasma (FFP) supplementation.
At the current presentation, he had no history of COVID-19 vaccination and had not received antiviral therapy. The warfarin dose was increased from 6.0 to 6.5 mg/day because his PT-INR had decreased during the early phase of COVID-19 infection. Shortly thereafter, he developed right thigh pain with rapidly expanding ecchymosis and was admitted the same day for evaluation and treatment of suspected VTE. On admission (defined as day 1), laboratory tests showed elevated D-dimer (1.7 μg/mL) and thrombin-antithrombin complex (TAT) (7.0 ng/mL), consistent with VTE (Table 1).
Given his history of multiple VTE episodes with identical presentations, recurrent VTE was clinically diagnosed based on physical findings and corroborative laboratory data without additional imaging at that time, to expedite treatment initiation. Continuous intravenous unfractionated heparin at 18 units/kg/hour and FFP at 16 units/day were immediately initiated, while warfarin was continued throughout the treatment period, in accordance with his long-term management protocol for severe protein C deficiency.
Antithrombin supplementation with antithrombin III concentrate (Nonthron; Takeda Pharmaceutical Co., Ltd., Tokyo, Japan) at 1,500 units/day was initiated on day 6 and continued for two days. Heparin and FFP were gradually tapered as shown in Figure 1 while monitoring D-dimer levels and reduction in size and tenderness of the right thigh ecchymosis.
Clinical course during hospitalizationTrends in body temperature, D-dimer, and fibrin degradation products (FDP) levels are shown, along with the progression of right thigh ecchymosis and the onset of abdominal pain. The timeline displays hospital days along with the corresponding doses of unfractionated heparin (U/kg/h) and daily fresh frozen plasma (FFP) administration (U/day). Ecchymosis gradually improved following the initiation of anticoagulation therapy. Abdominal pain emerged on hospital day 11, coinciding with a sharp rise in coagulation biomarkers and the diagnosis of splenic infarction. Adjustments in heparin and FFP dosing, including temporary discontinuation of FFP on day 8 and reinitiation from days 12 to 15, illustrate the therapeutic decision-making throughout the hospital course. Antithrombin III concentrate (Nonthron; 1,500 units/day for two days), recombinant human antithrombin (Acoalan; 1,800 units/day from days 11 to 20), and activated protein C concentrate (Anact C; administered for three days) are also indicated on the timeline.
On day 7, the FFP dose was reduced to four units/day, and FFP was discontinued on day 8. By day 9, unfractionated heparin had been reduced to 10 units/kg/hour, and the purpura on the right thigh had nearly resolved by day 10. This reduction in anticoagulation intensity coincided with a temporary improvement in coagulation biomarkers (Table 2).
While anticoagulation was being tapered, around day 11, the patient suddenly developed abdominal pain. Laboratory tests performed at that time showed a marked increase in D-dimer (28.8 μg/mL) and TAT (59.6 ng/mL), indicating a sudden exacerbation of the coagulation pathway (Table 2). A contrast-enhanced CT scan revealed a partial splenic infarction (Figure 2).
Axial (A) and coronal (B) contrast-enhanced CT images showing splenic infarctionA well-demarcated, wedge-shaped hypoenhancing area is seen in the spleen (white circles), consistent with splenic infarction.
The patient was treated conservatively with intensified anticoagulation, including an increased heparin dose to 20 units/kg/hour and supplementation with FFP at 8 units/day.
Following the diagnosis of splenic infarction, anticoagulation was intensified. Recombinant human antithrombin (Acoalan; Japan Blood Products Organization, Tokyo, Japan) was administered at 1,800 units/day from days 11 to 20. Beginning on day 16, activated protein C concentrate (Anact C; KM Biologics Co., Ltd., Kumamoto, Japan) was administered for three days during the subacute phase, with dose adjustments detailed in Figure 1. Heparin was gradually tapered thereafter and continued until day 23, while FFP was reduced to 4 units/day from days 12 to 15 and then discontinued. No further thrombotic events occurred after discontinuation of heparin, and the patient was discharged without complications on day 29 of hospitalization. A summary of the clinical course is shown in Figure 1. Follow-up imaging performed on day 39 confirmed resolution of the splenic infarction. Investigations, including echocardiography and pulmonary scintigraphy, ruled out cardiac shunt-related embolism or other systemic causes of thrombosis.
Summary of the clinical timeline
To clarify the temporal relationship between COVID-19 infection and thrombotic events, the key clinical events are summarized as follows: (i) Two days prior to admission: Onset of fever and cough; (ii) Day 1 (Admission): Diagnosis of COVID-19 via antigen testing; onset of right thigh pain and ecchymosis; admission for recurrent VTE; (iii) Days 4-10: Improvement of ecchymosis and normalization of coagulation markers (D-dimer/FDP) under anticoagulant therapy; (iv) Day 11 (13 days post-symptom onset): Sudden onset of abdominal pain; diagnosis of splenic infarction via contrast-enhanced CT; (v) Day 29: Discharged without sequelae.
Discussion
Severe congenital protein C deficiency poses substantial challenges in anticoagulation management. In this patient, splenic infarction occurred during heparin tapering in the setting of COVID-19, underscoring the difficulty of maintaining adequate anticoagulation in high-risk situations. Protein C deficiency confers an increased risk of both venous and arterial thrombosis, as demonstrated in previous cohort studies [7,8], and the presence of recurrent VTE and severely reduced protein C antigen levels further complicated management in this case. Protein C supplementation was considered necessary at the time of admission. However, because concentrate was initially unavailable, FFP was used as an alternative. Although the initial VTE improved with anticoagulation, the patient subsequently developed a splenic infarction. COVID-19-associated coagulopathy, characterized by endothelial injury and sustained inflammation, may have contributed to an elevated background thrombotic tendency [2,3].
Splenic infarction has been documented during both the acute and recovery phases of COVID-19 [9,10]. In this case, the event occurred 13 days after symptom onset, consistent with reports that arterial thrombosis can arise from persistent or delayed endothelial dysfunction even after clinical symptoms have resolved [2,3]. Notably, the infarction developed during a period when D-dimer and FDP levels had normalized (day 4-10), and the purpura had resolved. Such biochemical improvement may not necessarily indicate true resolution of hypercoagulability, particularly in patients with severe congenital thrombophilia. In this patient, the normalization of fibrinolytic markers likely reflected pharmacologic suppression of thrombin generation by heparin rather than genuine stabilization of the underlying prothrombotic state. Furthermore, echocardiography revealed no evidence of intracardiac shunt, making paradoxical embolism unlikely. Therefore, the splenic infarction in this case was most consistent with in situ arterial thrombosis triggered by COVID-19-related vascular endothelial injury, rather than embolic disease. This mechanism aligns with emerging evidence that COVID-19 can induce localized arterial thrombosis even in patients whose underlying thrombophilia predominantly affects the venous system.
In patients with congenital protein C deficiency who develop COVID-19, endothelial dysfunction and inflammation may persist longer than clinically apparent, creating a vulnerable period in which thrombotic events can occur despite apparent recovery. Although heparin initially controlled coagulation biomarkers, the patient’s intrinsic thrombotic potential was likely influenced by the combined effects of congenital thrombophilia and COVID-19-related endotheliopathy. Consequently, tapering anticoagulation solely on the basis of biomarker normalization may insufficiently reflect ongoing thrombotic risk, potentially allowing residual hypercoagulability to manifest as arterial thrombosis.
This case highlights that in high-risk patients with severe congenital thrombophilia and COVID-19, anticoagulation management, particularly tapering, should not be guided solely by the normalization of biomarkers like D-dimer. Decisions must integrate the clinical context, the known severity of the underlying thrombophilia, and an awareness of the potential for delayed endothelial injury from COVID-19, which may outlast symptomatic and biochemical improvement. These findings underscore the need to develop clearer anticoagulation management strategies, particularly regarding the safe tapering of therapy, for patients with congenital coagulation disorders during and after COVID-19 infection [11]. The lack of imaging confirmation for the initial recurrent VTE, while clinically justified to prioritize rapid intervention, is a minor limitation of this report. The spleen can be a target organ in various hematologic and thrombotic diseases. While infarctions may occur in hypercoagulable states, other severe complications like spontaneous rupture are documented in the context of hematologic malignancies [12]. Reporting such atypical splenic manifestations is therefore essential to enhance clinician awareness, facilitate early recognition, and prevent fatal outcomes.
Conclusions
This case underscores the elevated risk of arterial thrombosis in patients with protein C deficiency who develop COVID-19, even during periods of apparent biochemical improvement. The occurrence of splenic infarction illustrates the combined effect of congenital thrombophilia and COVID-19-associated endotheliopathy. Close monitoring and carefully tailored anticoagulation strategies are essential to prevent thrombotic complications in such patients during and after COVID-19, regardless of disease severity. Clinicians should exercise caution when tapering anticoagulation based solely on biomarker normalization, as underlying hypercoagulability may persist despite apparent recovery.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Protein C deficiency Arch Pathol Lab Med Dinarvand P Moser KA 1281128514320193070233410.5858/arpa.2017-0403-RS · doi ↗ · pubmed ↗
- 2The coagulopathy, endotheliopathy, and vasculitis of COVID-19Inflamm Res Iba T Connors JM Levy JH 118111896920203291856710.1007/s 00011-020-01401-6PMC 7486586 · doi ↗ · pubmed ↗
- 3Understanding COVID-19-associated coagulopathy Nat Rev Immunol Conway EM Mackman N Warren RQ 6396492220223593181810.1038/s 41577-022-00762-9PMC 9362465 · doi ↗ · pubmed ↗
- 4Protein C and S activities in COVID-19: a systematic review and meta-analysis J Thromb Thrombolysis Khoshnegah Z Siyadat P Rostami M Sheikhi M Ghorbani M Mansouritorghabeh H 101810305720243872252110.1007/s 11239-024-02971-6 · doi ↗ · pubmed ↗
- 5Protein C pretreatment protects endothelial cells from SARS-Co V-2-induced activation Viruses Silva BR Sidarta-Oliveira D Morari J 10491620243906621210.3390/v 16071049 PMC 11281670 · doi ↗ · pubmed ↗
- 6Definite diagnosis in Japanese patients with protein C deficiency by identification of causative PROC mutations Int J Hematol Takagi A Tanaka R Nakashima D 5555578920091937352210.1007/s 12185-009-0312-7 · doi ↗ · pubmed ↗
- 7Analysis of 45 episodes of arterial occlusive disease in Japanese patients with congenital protein C deficiency Thromb Res Sakata T Kario K Katayama Y 69789419991023089110.1016/s 0049-3848(98)00194-7 · doi ↗ · pubmed ↗
- 8Hereditary deficiency of protein C or protein S confers increased risk of arterial thromboembolic events at a young age: results from a large family cohort study Circulation Mahmoodi BK Brouwer JL Veeger NJ van der Meer J 1659166711820081882464210.1161/CIRCULATIONAHA.108.780759 · doi ↗ · pubmed ↗