An increase in environmental temperature within the growth range suppresses phage resistance in Escherichia coli
Satoshi Takayama, Yoshimitsu Masuda, Ken-ichi Honjoh, Takahisa Miyamoto

TL;DR
This study shows that increasing temperature can suppress phage-resistant Escherichia coli, offering a potential non-antibiotic method for controlling these bacteria.
Contribution
The study identifies a novel temperature-dependent vulnerability in phage-resistant E. coli that could be used for biocontrol without antibiotics.
Findings
Phage-resistant E. coli strains exhibit increased membrane permeability and sensitivity to monocaprin.
Elevated temperature (46°C) significantly reduces viability and regrowth of phage-resistant E. coli.
Phage-resistant strains show increased Congo Red binding and autoaggregation at higher temperatures.
Abstract
To develop countermeasures against phage-resistant bacteria without antibiotics, a detailed phenotypic characterization of phage-resistant Escherichia coli BW25113 was performed. Phage susceptibility testing of E. coli BW25113 deletion mutants involved in lipopolysaccharide (LPS) synthesis revealed that the first glucose residue of the LPS outer core was essential for infection by phage S127. From E. coli BW25113 cells that survived S127 exposure, four phage-resistant strains were isolated and characterized. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis showed that the phage-resistant strains had LPS with a smaller molecular mass compared with that of the E. coli BW25113 parental strain. Fluorescence microscopy after BacLight staining, along with comparisons of viable counts on non-selective versus selective media, indicated increased membrane permeability in the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
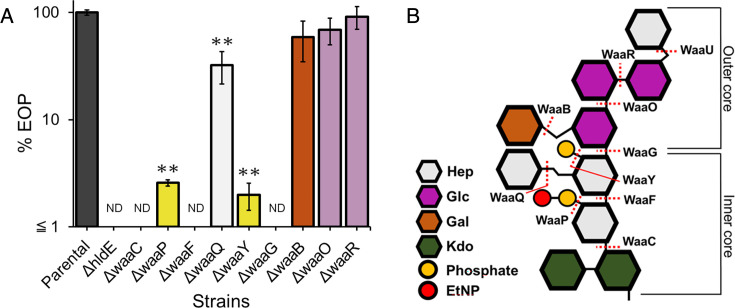 Fig 1
Fig 1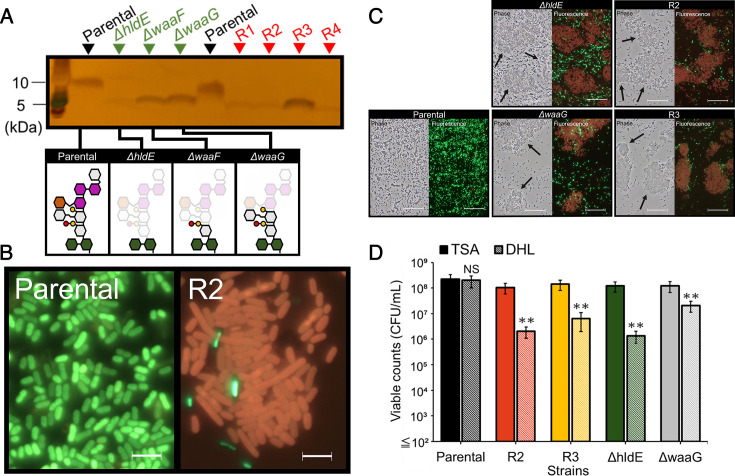 Fig 2
Fig 2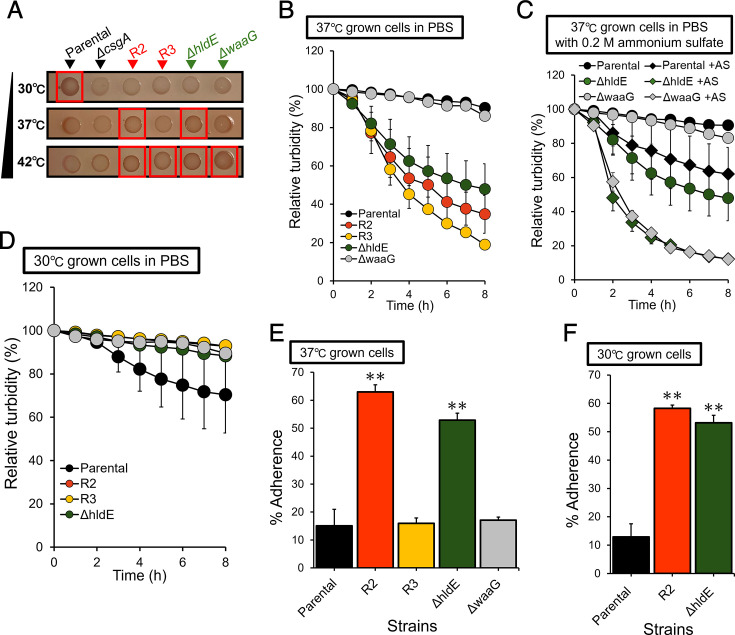 Fig 3
Fig 3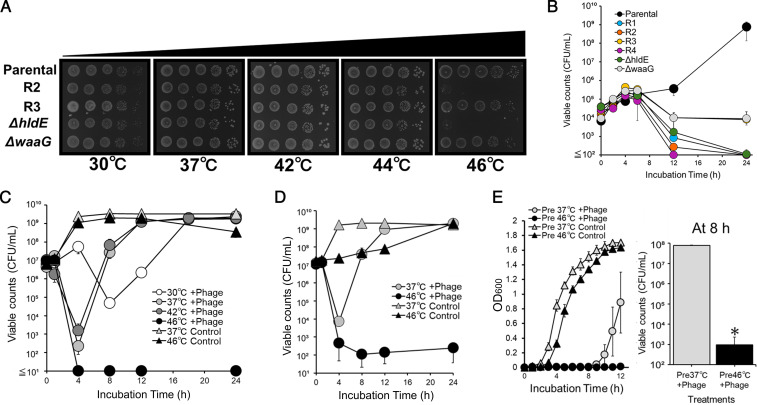 Fig 4
Fig 4| Strain | MIC (µM) |
|---|---|
| Parental (BW25113) | >2,000 |
| R2 | 250 |
| R3 | 1,000 |
| 250 | |
| 1,000 |
- —Japan Society for the Promotion of Sciencehttp://dx.doi.org/10.13039/501100001691
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacteriophages and microbial interactions · Bacterial Genetics and Biotechnology · Vibrio bacteria research studies
INTRODUCTION
The widespread use of conventional antimicrobial agents has led to the emergence of antimicrobial resistance (AMR), which is now recognized as a major global public health concern and a critical food safety issue (1, 2). A comprehensive global assessment of AMR in 2022 estimated that bacterial AMR was associated with 4.95 million deaths in 2019, with projections reaching 10 million deaths annually by 2050 if no effective countermeasures are implemented (3, 4). Any environment where antimicrobials are administered can be a risk factor for the selection and dissemination of AMR bacteria. In particular, the use of antimicrobials in animals/crops production facilitates the transmission of foodborne AMR bacteria to humans through the consumption of contaminated food or drinking water (5, 6). The foodborne AMR bacteria have made it challenging to treat patients with foodborne illness; thus, approaches to tackle AMR bacteria and means to prevent the emergence of AMR are in urgent need (7) throughout the process of “farm to folk” (8–10).
An alternative strategy to mitigate the emergence of AMR bacteria is the use of bacteriophages (phages) as antibacterial agents (11, 12). Phages are bacterial viruses that exist in every environment of the biosphere inhabited by host bacteria. Foods and food-producing environments are no exception to this inherent existence of phages. Exploiting their nature as bacterial predators, phages have been applied to control foodborne pathogens (13–15). From the perspective of AMR, the key advantages of phages are their ability to kill AMR bacteria, their sustainability as naturally abundant antimicrobial resources (16, 17), without contributing to the emergence or spread of AMR. Phages also offer distinct benefits as food additives. First, being naturally occurring, they do not alter the flavor or texture of foods, enabling effective decontamination without synthetic antimicrobials while maintaining the original quality of food or feed (14, 18, 19). Second, harmless microflora native to the environment may not be eliminated because phages selectively infect and lyse their target bacterium, which differentiates them from existing antimicrobials with broad spectra (20, 21). Taken together, these attributes highlight phages as promising antimicrobial agents that can be applied in multiple stages of the food chain.
Despite their potential, it is typically observed that a certain population of the host bacterium survives phage treatment and regrows. This is “phage resistance” of the host, escaping from infection by the same phage. For phages to be practically applied to control foodborne pathogens, this resistant population must be successfully suppressed. Several studies have explored the mechanisms of phage resistance (22), which are generally classified into three categories: modification of phage receptors, host phage defense systems, and phage-derived phage defense systems (23). Among these, receptor modification frequently alters cell-surface properties, leading to a reduction in stress tolerance or pathogenicity of phage-resistant bacteria (24, 25). This phenomenon highlights the trade-off associated with acquiring phage resistance (26). To date, numerous studies have attempted to exploit this trade-off between phage resistance and reduced stress tolerance to control phage-resistant populations, although they have often focused on the efficacy of phage-antibiotic combinations or decreased pathogenicity, particularly in clinical settings (27–30). Therefore, there have been few reports on control methods based on the characteristics of phage-resistant bacteria that do not use antibiotics. From the One Health perspective, it is crucial to explore the possibility of controlling phage resistance in a different way.
In the present study, we aimed to establish a scientific basis for developing novel strategies to control phage-resistant bacteria without the use of antibiotics by characterizing the unique phenotypes of phage-resistant strains. Escherichia coli and the lytic phage S127BCL3 (S127) were used as model organisms to investigate phage-host interactions and to analyze phage-resistant bacteria. Phage S127, a lytic phage of the genus Vequintavirus, was originally isolated from chicken livers using E. coli O157:H7 as an original host and possesses a 135.5 kbp genomic DNA (31).
Building on these findings, we discuss the thermosensitivity of phage-resistant strains, which leads to distinctive phenotypes in phage-resistant strains, and the possibility of controlling phage resistance by applying mild heat treatment to target bacterial populations.
RESULTS
The first glucose residue of lipopolysaccharide outer core for efficient infection by phage
We identified the putative receptor required for phage S127 to infect E. coli BW25113. Since lipopolysaccharide (LPS) commonly serves as a receptor for phage infection in gram-negative bacteria (32), the efficiency of plating (EOP) of the phage was evaluated against a series of E. coli BW25113 mutants, each lacking a single gene involved in LPS synthesis. In several mutants, the EOP of S127 was significantly decreased (Fig. 1A). These included deletions in genes responsible for the modification of the LPS inner core region (hldE, waaC, waaP, waaF, waaQ, waaY, and waaG). The degree of LPS truncation was determined based on the role of each deleted gene product in LPS core biosynthesis, as illustrated in the structure of E. coli BW25113 (Fig. 1B). The hldE gene product, HldE, is involved in the biosynthesis of heptose in the inner core of LPS. Of these seven genes, the deletion of hldE, waaC, waaF, and waaG resulted in completely abolished S127 infection, whereas the deletion of waaO had little effect on EOP, suggesting that the first glucose residue of the LPS outer core was critical for S127 infection. Additionally, deletion of waaP and waaY, both of which are involved in phosphorylation of heptose residues in the LPS inner core, also caused a significant decrease in EOP. Collectively, these results indicated that the boundary region between the outer and inner core of LPS, as well as the phosphate modifications of heptose, could play a role in maintaining the surface structure for phage S127 to initiate its infection.
*EOP of phage S127 on mutants with various lengths of LPS. (A) EOP of S127 on parental (black) and a series of single-gene deletion mutants lacking genes responsible for modifications of the LPS core region (other colors). Error bars show standard deviations of the mean for three biological replicates. The data were analyzed using one-way analysis of variance with Dunnett’s multiple comparison to parental. *P < 0.01. ND, not detected. (B) Structure of the LPS core from E. coli BW25113. Abbreviations are as follows: Hep, L-glycero-D-manno-heptose; Glc, D-glucose; Gal, D-galactose; Kdo, 3-deoxy-D-mannno-oct-2-ulosonic acid; P, phosphate; and EtNP, 2-aminoethyl phosphate. The assignment of function to genes encoding core glycosyltransferases and phosphotransferases has been previously reported (33, 34).
Characterization of S127-resistant strains
Spontaneous phage S127-resistant strains were isolated from single colonies formed by bacterial cells that survived overnight phage treatment in Luria-Bertani (LB) broth at a multiplicity of infection (MOI) of 0.1. The phage resistance of the four isolates (designated as R1, R2, R3, and R4) was confirmed by plaque assays, in which no plaques were detected on their lawns. To characterize the unique phenotypes of these strains, the LPS of the phage-resistant strains was analyzed. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis of purified LPS, visualized by silver staining, revealed that all phage-resistant strains produced LPS chains with a smaller molecular mass (MM) than that of the parental strain (Fig. 2A). The MM of LPS from R1, R2, and R4 was similar to that of ΔhldE, while that from R3 resembled ΔwaaG. Overall, the LPS profiles in the phage-resistant strains were comparable to those of ΔhldE, ΔwaaF, and ΔwaaG mutants, suggesting that phage-resistant strains possessed truncated LPS chains relative to the parental strain. To investigate the phenotypes of strains with distinct types of LPS, R2 and R3 were selected as representative strains for further characterization.
*Characterization of phage-resistant strains of E. coli BW25113. (A) Silver staining of LPS extracted from phage-resistant strains. (B) Baclight staining of parental and a phage-resistant strain, R2, as representative images. Scale bars indicate 5 µm. (C) Wider view of the result of Baclight staining. Black arrows point out cellular aggregates observed in phage-resistant strains. Scale bars indicate 50 μm. (D) Comparison of viable cell counts of parental (black) and R2 (orange), R3 (yellow), ΔhldE (green), and ΔwaaG (gray) between tryptic soy agar (TSA; solid) and deoxycholate hydrogen sulfide lactose (DHL) agar (dashed). Error bars show standard deviations of the mean for three biological replicates. The data were analyzed using a two-tailed Student’s t-test. *P < 0.01. NS, not significant.
To investigate the effect of LPS truncation on membrane property, changes in the outer membrane permeability (35) of phage-resistant strains were assessed. Fluorescence microscopy following BacLight staining revealed stronger red fluorescence derived from propidium iodide (PI) in phage-resistant strains such as R2, compared with the parental strain (Fig. 2B). Moreover, aggregates formed by PI-stained cells were frequently observed in the phage-resistant strains (Fig. 2C, black arrows). Viability assays on tryptic soy agar (TSA) and deoxycholate hydrogen sulfide lactose (DHL) agar plates also indicated that most of the cells of phage-resistant strains were viable but had an incomplete envelope function (Fig. 2D). These results suggested that the integrity of the outer membrane was compromised, leading to increased outer membrane permeability in the cells of phage-resistant strains, which exhibited enhanced aggregation.
The susceptibility of phage-resistant and mutant strains to monocaprin, a hydrophobic antimicrobial agent, was investigated to verify increased outer membrane permeability. As shown in Table 1, the phage-resistant strains exhibited increased susceptibility to monocaprin, with minimum inhibitory concentration (MIC) values decreasing to 1/8 in R2 and ΔhldE and 1/2 in R3 and ΔwaaG compared with that of E. coli BW25113 parental strain (Table 1).
Effects of culture temperature on curli production and autoaggregation
The involvement of curli fimbriae in the autoaggregation of phage-resistant strains was examined. The red coloration of the colony formed by the parental strain on YESCA agar containing Congo Red (CR) decreased as the culture temperature increased (Fig. 3A). Pale white colonies were observed in ΔcsgA, which is deficient in the synthesis of curli fimbriae, indicating the dependency of red colony phenotype on curli production. The red coloration of the colony by phage-resistant strains, as well as ΔhldE and ΔwaaG, increased at 37°C or 42°C and decreased at 30°C. This observation was the opposite of that observed in the parental strain, where curli production was prominent at a lower temperature.
*Effects of culture temperatures on curli fimbriae production, autoaggregation, and cell-surface hydrophobicity of E. coli strains. (A) CR binding of E. coli BW25113 parental, ΔcsgA, R2, R3, ΔhldE, and ΔwaaG. Each strain was cultured on a YESCA plate in the presence of CR at different temperatures. (B) Changes in OD600 of the cell suspensions in phosphate-buffered saline (PBS) of E. coli BW25113 parental (black), R2 (orange), R3 (yellow), ΔhldE (green), and ΔwaaG (gray) strains cultured at 37°C were determined. (C) Changes in OD600 of the cell suspensions in PBS supplemented with 0.2 M ammonium sulfate of E. coli BW25113 parental (black), ΔhldE (green), and ΔwaaG (gray) strains cultured at 37°C were determined. Ammonium sulfate-supplemented groups (+AS) are shown with diamonds. (D) Changes in OD600 of the cell suspensions in PBS of E. coli BW25113 parental (black), R2 (orange), R3 (yellow), ΔhldE (green), and ΔwaaG (gray) strains cultured at 30°C were determined. (E) Cell-surface hydrophobicity of parental (black), R2 (orange), R3 (yellow), and ΔhldE (green) cultured at 37°C or (F) 30°C determined by the degree of adherence of bacterial cells to n-octane. Error bars show standard deviations of the mean for three biological replicates, and data were analyzed using one-way analysis of variance followed by Dunnett’s multiple comparisons to parental. *P < 0.01.
In the autoaggregation assay, the isolated phage-resistant strains and ΔhldE cultured at 37°C showed a rapid decrease in turbidity of bacterial suspensions in phosphate-buffered saline (PBS), whereas the parental strain and ΔwaaG did not (Fig. 3B). The addition of ammonium sulfate to PBS promoted a decrease in turbidity of bacterial suspensions from ΔhldE and ΔwaaG (Fig. 3C), indicating that autoaggregation in these strains was salt concentration dependent. Autoaggregation was canceled in all bacterial cell suspensions prepared from the cells cultured at 30°C (Fig. 3D), which was consistent with the temperature dependency observed in the CR binding of phage-resistant strains (Fig. 3A).
To examine the involvement of hydrophobic interactions in the autoaggregation of phage-resistant cells, cell-surface hydrophobicity was assessed using the bacterial adhesion to hydrocarbons (BATH) assay, which measures adherence of bacterial cells to *n-*octane. When cultured at 37°C, the percent adherence of R2 and ΔhldE strains was significantly higher than that of the parental, R3, and ΔwaaG strains (Fig. 3E). To evaluate the temperature dependency of the cell-surface hydrophobicity, parental, R2, and ΔhldE strains were also cultured at 30°C. Cells of R2 and ΔhldE strains showed higher percent adherence than that of the parental strain (Fig. 3F), indicating that these strains consistently exhibited higher cell-surface hydrophobicity than the parental strain, irrespective of culture temperature.
Effects of culture temperature on the growth of phage resistance of the isolated strains and deletion mutants
To examine the thermosensitivity of the phage-resistant and mutant strains, their colony-forming abilities were determined at different culture temperatures on LB agar. Deep rough mutants, including ΔhldE and ΔwaaG, were examined in this study owing to their inability to grow at 46°C (36). All strains grew on LB agar at 44°C; however, only parental, R3, and ΔwaaG strains grew at 46°C, whereas R2 and ΔhldE did not (Fig. 4A). A time-kill assay in LB broth at 46°C further supported these observations. All strains increased their viable counts for the first 4 h, but thereafter, all except the parental strain showed a decline. By 24 h, the viable counts of R1, R2, R4, and ΔhldE decreased to the detection limit, whereas R3 and ΔwaaG, which retained LPS inner core structures, still maintained 10^4^ CFU/mL (Fig. 4B). Parental cells grown at 37°C were treated with phage S127 at different temperatures. In the presence of the phage, viable counts initially decreased but eventually recovered along with regrowth of the phage-resistant population at 30°C, 37°C, and 42°C. In contrast, at 46°C, viable counts dropped to the detection limit within 4 h and showed no recovery for 20 h at 46°C (Fig. 4C). A similar effect was observed in EHEC strain E. coli No. 127, where the phage-resistant population grew significantly slower when treated with phage S127 at 46°C (Fig. 4D). Finally, the effect of the incubation temperature before phage treatment on the survival of phage-resistant populations was investigated. To this end, the parental strain precultured at 37°C or 46°C was treated with phage S127 at 37°C. Regrowth of E. coli BW25113 precultured at 46°C was significantly delayed in the presence of phage compared with that of the cells precultured at 37°C, as shown by reduced turbidity (Fig. 4E, left). After 8 h of phage treatment, viable cell counts differed by 10^5^-fold between the cells precultured at 37°C and 46°C (Fig. 4E, right).
*Effects of culture temperatures on regrowth and viability of E. coli strains in the absence and presence of phage S127. (A) Colony formation of E. coli BW25113 parental and R2, R3, ΔhldE, and ΔwaaG strains was examined on LB agar plates at 30°C, 37°C, 42°C, 44°C, and 46°C. (B) Changes in viability at 46°C in LB broth of parental (black), R1 (blue), R2 (orange), R3 (yellow), R4 (purple), ΔhldE (green), and ΔwaaG (gray) strains precultured at 37°C. (C) E. coli BW25113 parental strain was incubated with phage S127 (circles) at 30°C (white), 37°C (light gray), 42°C (dark gray), and 46°C (black) or without phage at 37°C and 46°C (triangles) at MOI = 0.1. (D) EHEC strain E. coli No. 127 was incubated with phage (circles) at 37°C (light gray) and 46°C (black) or without phage (triangles). (E) Effect of preculture at 46°C on the susceptibility to phage S127 in BW25113 parental strain. E. coli BW25113 parental strain was precultured at 37°C (light gray) or 46°C (black) and then incubated with phage (circles) or without phage (triangles) at MOI = 100. The OD600 of the culture was measured at designated time points (left graph), and viable cell counts were measured after 8 h of incubation (right). Error bars show the standard deviation of the mean for three biological replicates. Data were analyzed using the Mann-Whitney U test for the comparison of two treatments at 8 h. P < 0.05.
DISCUSSION
Bacterial phage resistance associated with phenotypic trade-offs has been of growing interest because it is valuable for optimizing bacterial control strategies (25, 37). In this study, E. coli isolates resistant to phage S127 were characterized to develop novel strategies for controlling phage-resistant populations without antibiotics. The requirement of the first glucose residue in the LPS outer core for S127 infection (Fig. 1A), together with the lower MM of the LPS chain in all isolated phage-resistant strains (Fig. 2A), indicated that deletion of the outer core of the LPS chain conferred resistance to S127 in E. coli BW25113. Fluorescence microscopy and viability assays on TSA and DHL agar further demonstrated that the truncation of the LPS chain compromised membrane integrity, resulting in an increase in viable cells with increased outer membrane permeability (Fig. 2B through D). Since the LPS core oligosaccharide portion confers hydrophilicity to the cell surface (38), the truncation of the LPS chain resulted in increased cell-surface hydrophobicity in R2 and ΔhldE strains (Fig. 3E) and heightened their susceptibility to monocaprin (Table 1). Monocaprin, a non-ionic surfactant and fatty acid monoglyceride, is generally recognized as safe and widely used in food as both an emulsifier and antimicrobial (39–41). These findings suggest that further evaluation of monocaprin’s efficacy against phage-resistant bacteria is warranted. The susceptibility to monocaprin and cell-surface hydrophobicity of the R2 closely resembled those of ΔhldE, whereas R3 resembled ΔwaaG (Table 1; Fig. 3E). Consistent with this phenotypic variation, the strains also differed in autoaggregation and thermosensitivity. Collectively, the degree of LPS truncation produced distinct phenotypic outcomes among the phage-resistant isolates ([Fig. 3A through D and 4B](#F3 F4)). The physiological mechanisms underlying these phenotypes, along with the potential applications of increased thermosensitivity in phage-resistant bacteria, are discussed below.
Autoaggregation is a hallmark of E. coli deep rough mutants (38, 42). In the present study, autoaggregation was investigated in relation to potential mediators, including curli fimbriae, cell-surface hydrophobicity, environmental salt concentration, and culture temperature. Previous studies on curli production in deep rough mutants have yielded conflicting conclusions, likely due to differences in culture temperatures (43, 44). The CR binding assay suggested that curli production in the phage-resistant strains increased at higher culture temperatures, in contrast to the parental strain, where curli expression was greater at lower temperatures (Fig. 3A). Because curli expression is regulated by σ^S^ (45), which can be indirectly induced by σ^E^ (46), it is plausible that the shorter LPS chains in resistant strains trigger envelope stress at elevated temperatures, thereby increasing σ^E^ activity and, in turn, σ^S^-dependent curli expression. In line with these CR binding patterns, autoaggregation in phage-resistant strains was also culture temperature dependent. However, the BATH assay indicated that the inherent increase in cell-surface hydrophobicity in R2 and ΔhldE did not correlate with autoaggregation behavior (Fig. 3E and F). The R3 strain, which likely retains an inner core heptose in its LPS, exhibited strong autoaggregation despite having hydrophobicity levels similar to the parental strain (Fig. 3B). These findings suggest that factors beyond hydrophobicity are involved in autoaggregation. Ag43 and Type I fimbriae, encoded by flu and fimH, respectively, are well-known mediators of autoaggregation in E. coli (47, 48), whereas Nakao et al. have shown that even a double deletion of flu and hldE did not abolish enhanced autoaggregation in E. coli BW25113 (38). Similarly, overnight cultures of ΔcsgA and ΔfimH treated with phage S127 still showed a rapid decrease in turbidity (data not shown), indicating that loss of single aggregation-mediating factors is insufficient to abolish autoaggregation in deep rough populations. For ΔwaaG, previous studies reported that autoaggregation was comparable to the parental strain when cells were suspended in PBS (38), whereas enhanced autoaggregation of ΔwaaG was observed when cells were suspended in LB broth (42, 49), which contains a larger amount of organic compounds compared with PBS, suggesting that autoaggregation of ΔwaaG depends on the substances in the cell suspension. Based on these facts, it was shown in this study that the autoaggregation of ΔwaaG and ΔhldE appeared to be solute concentration dependent (Fig. 3C). Taken together, these results highlight the multifactorial nature of autoaggregation, which is influenced by multiple factors, including culture temperature and salt concentration. Given the diversity of curli production and autoaggregation with changes in culture temperature in phage-resistant strains, further analysis is warranted to elucidate the underlying mechanisms and potential impact of these behaviors on the outcome of phage therapy.
In the present study, the elevated temperature dependence of CR binding and autoaggregation in phage-resistant strains prompted a detailed investigation of their thermosensitivity. Membrane integrity has been reported to play a critical role in bacterial thermotolerance (50, 51). Reduced membrane integrity can induce oxidative stress caused by the disruption of the electron transport chain and impair the ability to maintain cell structure due to increased membrane fluidity, which affects adaptation to external temperature changes (51, 52). Intact LPS structure has been reported to maintain outer membrane integrity by enabling divalent cations to crosslink adjacent LPS chains via phosphate groups and by contributing to the balanced composition of phospholipids and proteins in the outer membrane (35, 53, 54). Consistent with the increased membrane permeability observed in phage-resistant strains (Fig. 2B through D), the truncation of the LPS chain likely reduces membrane stability by compromising these critical envelop factors (55, 56), which impaired their thermotolerance, resulting in a decreased acclimatization ability and growth suppression at 46°C (Fig. 4A and B). Previous studies have shown that the extent of LPS chain truncation resulted in the difference in outer membrane properties in E. coli (38, 57), and the difference in thermosensitivity among the strains investigated here could be explained by the varied magnitude of destabilization of the outer membrane in each strain, which corresponded with their susceptibility to monocaprin (Fig. 2D). Specifically, R2 and ΔhldE, which had a lower MIC value for monocaprin, were more thermosensitive than R3 and ΔwaaG. Increased thermosensitivity in phage-resistant bacteria may also occur in other gram-negative bacteria infected by phages that require LPS as a receptor, similar to S127.
In addition to suppressing the growth of phage-resistant strains by elevating the culture temperature (Fig. 4C and D), we observed increased susceptibility to phage S127 in the parental E. coli BW25113 when elevating the preculture temperature (Fig. 4E). Based on previous reports on phenotypic heterogeneity in bacterial populations (58, 59), cells harboring truncated LPS chains may naturally occur within the parental population of E. coli, representing potential phage-resistant subpopulations that display increased thermosensitivity. Accordingly, preculture at 46°C may have reduced the subpopulation of these thermosensitive, potential phage-resistant cells, thereby explaining the delayed regrowth of phage-resistant cells following phage treatment of the parental cells precultured at 46°C. Alternatively, the observed effect may be partly attributable to impaired adaptation of cells shifted from elevated to lower temperatures, as indicated by the prolonged lag phase of the parental strain precultured at 46°C (Fig. 4E, left). As the experiment was performed based on the hypothesis that potential phage-resistant cells preexist prior to phage treatment, further analysis to confirm their presence in the parental population is required to better understand this phenomenon. Nevertheless, the results suggest that a high-temperature history within the normal growth range significantly decreases the population of phage-resistant cells, leading to delayed regrowth of phage-resistant bacteria.
Although it remains to be confirmed whether the increased thermosensitivity of E. coli phage-resistant strains demonstrated in this study is also observed in other gram-negative bacteria, exploring the feasibility of applying mild heat treatment combined with phage treatment is worthwhile. Such an approach could be particularly useful for controlling phage-resistant bacterial populations in food-manufacturing environments as well as veterinary and clinical settings. For example, maintaining food at elevated temperatures above 46°C after phage treatment may prevent potential regrowth of the phage-resistant population of the target bacterium, while the growth of off-target bacteria should be taken into account at the same time. In addition, the combined use of phage therapy and hyperthermia could be evaluated as a therapeutic option for wounds resulting from bacterial infections in the veterinary and clinical contexts. Further elucidation to ascertain the practical exploitation of increased thermosensitivity is anticipated.
MATERIALS AND METHODS
Bacterial strains, phage, and culture conditions
E. coli BW25113 parental and single-gene knockout mutants from the Keio collection were obtained from the National BioResource Project (NBRP, Shizuoka, Japan). E. coli O157: H7 strain No. 127 was kindly provided by the Fukuoka City Institute of Health and Environment (Fukuoka, Japan). E. coli strains were cultured in LB Miller broth (Kanto Chemical, Japan) or on LB agar plates at 37°C unless otherwise specified. The BW25113 single-gene knockout mutants used in this study included ΔhldE (JW3024), ΔwaaC (JW3596), ΔwaaP (JW3605), ΔwaaF (JW3595), ΔwaaQ (JW3607), ΔwaaY (JW3600), ΔwaaG (JW3606), ΔwaaB (JW3603), ΔwaaO (JW3602), ΔwaaR (JW3601), and ΔcsgA (JW1025). Each mutant harbored a kanamycin-resistant cassette, as previously described (60), and was precultured in LB containing 20 µg/mL kanamycin. Phage S127BCL3 (S127) was originally isolated from chicken liver in a previous study, and the phage S127 solution was prepared as described elsewhere (31).
Isolation of phage-resistant strains
Phage-resistant strains were isolated from bacterial cultures that were treated overnight with phage S127. Briefly, an overnight culture of E. coli BW25113 parental strain was diluted with LB broth to get a 10⁸ CFU/mL suspension. Fifty microliter of phage S127 solution was added to 4.95 mL of the diluted E. coli suspension in a test tube to attain a MOI of 0.1. The mixture in the test tube was incubated overnight at 37°C with shaking. The resulting culture was then streaked onto LB agar plates, and four single colonies that formed on the agar were randomly selected. Each colony was purified by two successive re-streakings on a fresh LB agar plate, each accompanied by cultivation in LB broth. The phage-resistant phenotype was confirmed by plaque assays, in which no plaque formation was observed after spotting the phage solution onto bacterial lawns of the isolates. Finally, the purified resistant cultures were stored in 20% glycerol at −80°C for subsequent characterization.
EOP test
EOP of phage S127 was evaluated against the E. coli BW25113 parental strain and its single-gene deletion derivatives with various lengths of the LPS core. Ten microliter of serially diluted phage solutions was spotted onto bacterial lawns prepared from each strain and incubated overnight at 37°C. The phage titer against each strain was calculated based on the plaque numbers. The EOP was determined by dividing the phage titer obtained for each mutant strain by that of the parental strain.
LPS extraction and silver staining
LPS was extracted from overnight bacterial cultures using the hot aqueous phenol extraction (61). Purified LPS was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 1.2% acrylamide gel containing 4 M urea. Silver staining (62) was used to visualize the LPS bands in the gel.
Membrane permeability assay
The membrane permeability of the test strains was assessed using fluorescence microscopy with BacLight Live/Dead Bacterial Viability Kit (Invitrogen). Overnight cultures were diluted approximately 1:100 in 3 mL of fresh LB broth in a test tube and incubated at 37°C until the OD_600_ reached 0.4. Cells were then collected by centrifugation at 8,000 × g, 4°C for 5 min, washed twice with 0.1 M HEPES buffer (pH 7.1), and suspended in 100 µL of the same buffer. Staining was performed following the manufacturer’s instructions, except that the bacterial suspension was incubated for 2 h. Microscopic observation was performed using a BX53 fluorescence microscope (OLYMPUS) equipped with a U-FBW dichroic mirror (OLYMPUS), which provided excitation at 460–495 nm and emission at 510 nm or higher. In this assay, intact cells appear green due to SYTO9 staining, whereas cells with increased membrane permeability are stained red as they allow the penetration of PI.
Counting viable cells and cells with incomplete envelope function
To quantify cells with incomplete envelope function, viable and intact cells were assessed using spot tests on TSA and DHL agar with modifications to previously described methods (63, 64). Bacterial suspensions of each strain in 0.1 M HEPES buffer (pH 7.1) were prepared as described above for the membrane permeability assay and serially diluted in PBS. Ten microliter of each diluent was spotted onto both TSA (Oxoid, UK) and DHL agar (Nissui Pharmaceutical, Japan) plates. After overnight incubation at 37°C, the colonies were enumerated. Because sodium deoxycholate in DHL agar inhibits the growth of cells with incomplete envelope function, only intact cells form colonies on DHL agar, whereas both intact cells and envelope-compromised cells form colonies on TSA. The difference in the viable counts between TSA and DHL agar thus indicates the number of viable cells with incomplete envelope function.
MIC measurements
The MIC of monocaprin was determined by evaluating the growth of the test strains in the presence of various concentrations of monocaprin. Monocaprin (Taiyo Kagaku, Japan) was dissolved in 100% dimethyl sulfoxide and serially diluted in LB broth containing 1% dimethyl sulfoxide in 96-well plates. For inoculum preparation, overnight cultures were adjusted to an OD_600_ = 0.1 in LB broth and further diluted 1:100. Equal volumes (100 µL) of bacterial suspension were added to wells containing 100 µL of the broth with varying monocaprin concentrations. The plates were incubated at 37°C for 24 h with shaking, and OD_600_ of each well was measured using a spectrophotometer (Infinite F50, TECAN) after the incubation. The MIC of monocaprin against different strains was defined as the lowest concentration at which the OD_600_ of the well remained less than 0.01 after 24 h of cultivation.
BATH assay
Cell-surface hydrophobicity was assessed by examining bacterial adherence to n-octane using the BATH assay (65) with some modifications. Briefly, cells were suspended in potassium phosphate buffer containing 3.5 M ammonium sulfate (pH 7.1) to attain an OD_600_ of 1.3–1.4 using a spectrophotometer (UV-1800, SHIMADZU). Two hundred microliters of n-octane (Nacalai Tesque, Japan) was added to 1.5 mL of the cell suspension in a test tube (φ 10 mm), followed by incubation at 30°C for 10 min. The suspension was then vigorously vortexed for 2 min and incubated at room temperature for 20 min to allow phase separation. The bottom aqueous phase was transferred to a 1 mL disposable cuvette (Dsp Semi 1.5 ML, Fisher Scientific) using a Pasteur pipette, and the OD_600_ of the aqueous phase was measured. Adherence to *n-*octane was expressed as the percentage decrease in OD_600_ after vortexing, relative to the initial OD_600_ of the aqueous phase before vortexing, using the following formula: percent adherence = [1 – (OD_600_ after vortex/OD_600_ before vortex)] × 100.
CR binding assay
CR is an anionic diazo dye that binds specifically to amyloid fibers such as curli fimbriae in E. coli and to polysaccharides such as cellulose and chitin (66). CR binding was evaluated as previously described, with minor modifications (67). Overnight cultures of test strains were adjusted to equal turbidity and were spotted onto YESCA agar containing CR (1% casamino acids, 0.1% yeast extract, 2% agar, 20 µg/mL CR [SIGMA], and 10 µg/mL Coomassie Brilliant Blue R-250 [Nacalai Tesque]). The plates were incubated statically overnight at 30°C, 37°C, and 42°C for 24 h. CR binding was assessed based on the intensity of the red coloration of colonies after incubation. Note that the cellulose production by E. coli BW25113 was abolished owing to the mutation of bcsQ, which is involved in cellulose synthesis (68). Accordingly, the red color of the colony can be attributed to curli production in BW25113 (69).
Autoaggregation assay
The autoaggregation assay was performed by measuring the reduction in turbidity of bacterial cell suspensions due to cell precipitation, as previously described (38, 48). Briefly, overnight-cultured cells were harvested by centrifugation at 6,000 × g, 25°C for 5 min, and the resulting pellets were resuspended in PBS (pH 7.4) to an OD_600_ of 1.2–1.4. The suspensions were then statically incubated at room temperature in the test tubes. When required, ammonium sulfate (Nacalai Tesque) was supplemented to PBS to a final concentration of 0.2 M. At designated time points, the OD_600_ of the phase above the sediment formed by aggregation was measured using a spectrophotometer (Miniphoto 518R, TAITEC). Autoaggregation was expressed as relative turbidity, defined as the ratio of turbidity at a given time point to the initial turbidity.
Thermosensitivity test
The thermosensitivity of the strains was evaluated using viability assays at different temperatures. Overnight cultures grown at 37°C of test strains were diluted in LB broth to an OD_600_ = 0.1 and serially diluted in PBS, and 10 µL of each dilution was spotted onto LB agar. Plates were incubated overnight at 30°C, 37°C, 42°C, 44°C, and 46°C, and colony formation was assessed. A time-kill assay was also performed in a broth at 46°C. Briefly, overnight cultures of the test strains were diluted in 5 mL of LB broth and incubated with shaking at designated temperatures. At each specified time point, 20 µL of an aliquot was taken and serially diluted in PBS, and 10 µL of each dilution was spotted on LB agar. Viable cell counts were determined based on the number of colonies formed on the plate after incubation for 24 h at 37°C and the dilution factor.
Phage treatment at different temperatures
E. coli BW25113 parental strain and E. coli O157:H7 No. 127, an EHEC strain, were used as test strains. Phage-treated bacterial cultures were prepared as described previously. For the control, the same volume of SM buffer (50 μL) was added in place of the phage solution. The cultures were incubated with or without phage S127 at different temperatures with shaking, and viable cell counts were determined at designated time points using a spot test. For undiluted samples, 100 µL of culture was directly plated onto LB agar.
Phage treatment of bacteria with different temperature histories
The effect of a high-temperature history prior to phage treatment was evaluated using the E. coli BW25113 parental strain precultured at different temperatures. Briefly, 10 µL of an overnight culture of parental strain was transferred into 5 mL of fresh LB broth and incubated overnight at either 37°C or 46°C. The cultures were then diluted to an OD_600_ = 0.1, and 50 µL of the diluted suspension was inoculated into 4.95 mL of LB containing S127 at 10^8^ PFU/mL, yielding approximately 100 of MOI. Following incubation at 37°C, OD_600_ was measured at designated time points using a spectrophotometer (Miniphoto 518R, TAITEC), and viable counts were determined using the spot test or direct plating.
Statistical analysis
All statistical analyses were performed using Statcel4 (70) implemented in Microsoft Excel. Specific details regarding the statistical methods are provided in the corresponding figure legends.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Okeke IN, de Kraker MEA, Van Boeckel TP, Kumar CK, Schmitt H, Gales AC, Bertagnolio S, Sharland M, Laxminarayan R. 2024. The scope of the antimicrobial resistance challenge. Lancet 403:2426–2438. doi:10.1016/S 0140-6736(24)00876-638797176 · doi ↗ · pubmed ↗
- 2Djordjevic SP, Jarocki VM, Seemann T, Cummins ML, Watt AE, Drigo B, Wyrsch ER, Reid CJ, Donner E, Howden BP. 2024. Genomic surveillance for antimicrobial resistance — a One Health perspective. Nat Rev Genet 25:142–157. doi:10.1038/s 41576-023-00649-y 37749210 · doi ↗ · pubmed ↗
- 3O’Neill J. 2016. Tackling drug-resistant infections globally: final report and recommendations. The Review on Antimicrobial Resistance, chaired by Jim O’Neill. London HM Government and Wellcome Trust
- 4Murray CJL, Ikuta KS, Sharara F, Swetschinski L, Robles Aguilar G, Gray A, Han C, Bisignano C, Rao P, Wool E, et al.. 2022. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399:629–655. doi:10.1016/S 0140-6736(21)02724-035065702 PMC 8841637 · doi ↗ · pubmed ↗
- 5FAO, WHO. 2023. Foodborne antimicrobial resistance – Compendium of Codex standards. First revision. Codex Alimentarius Commission, Rome.
- 6Kniel KE, Kumar D, Thakur S. 2018. Understanding the complexities of food safety using a “one health” approach. Microbiol Spectr 6. doi:10.1128/microbiolspec.pfs-0021-2017 PMC 1163355329451115 · doi ↗ · pubmed ↗
- 7Ghosh C, Sarkar P, Issa R, Haldar J. 2019. Alternatives to conventional antibiotics in the era of antimicrobial resistance. Trends Microbiol 27:323–338. doi:10.1016/j.tim.2018.12.01030683453 · doi ↗ · pubmed ↗
- 8Allen HK, Levine UY, Looft T, Bandrick M, Casey TA. 2013. Treatment, promotion, commotion: antibiotic alternatives in food-producing animals. Trends Microbiol 21:114–119. doi:10.1016/j.tim.2012.11.00123473629 · doi ↗ · pubmed ↗