Carriage of hypervirulent and ESBL-producing Klebsiella pneumoniae complex among community-dwelling individuals in Japan
Akiko Watanabe, Yukio Tawada, Makoto Moriyama, Yohei Doi, Masahiro Suzuki

TL;DR
This study found that Klebsiella bacteria are common in the gut of healthy people in Japan, but dangerous antibiotic-resistant or highly virulent strains are rare in the community.
Contribution
The study provides new data on the community carriage rates of Klebsiella pneumoniae complex in Japan, including insights into hypervirulent and ESBL-producing strains.
Findings
Klebsiella pneumoniae complex was detected in 58.7% of community-dwelling individuals in Japan.
Hypervirulent and ESBL-producing strains were rare, each accounting for less than 1% of isolates.
Klebsiella variicola was more frequently detected than previously reported in Asian studies.
Abstract
Despite the increasing number of reports on hypervirulent and extended-spectrum β-lactamase (ESBL)-producing Klebsiella pneumoniae infections, data on the distribution of these pathogens in the community are limited. To address this knowledge gap, we investigated the carriage rates of K. pneumoniae complex in the stools of community-dwelling individuals in Japan. From 627 stool samples submitted to a commercial diagnostic laboratory, 407 Klebsiella strains were identified from 368 samples, corresponding to a colonization rate of 58.7%. Based on whole-genome sequencing, K. pneumoniae was the most prevalent species (n = 218, 53.6%), followed by Klebsiella variicola (n = 137, 33.7%). The detection rate of K. variicola was higher than previously reported in studies from other Asian countries. The overall distribution of sequence types (STs) was similar to those observed in previous studies…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
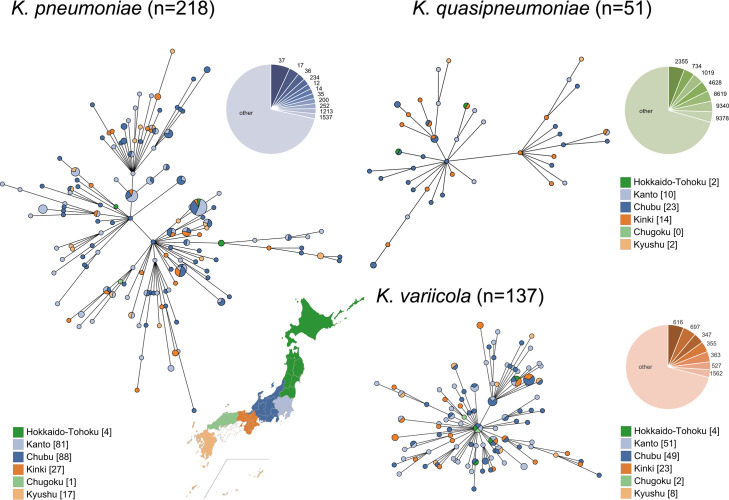 Fig 1
Fig 1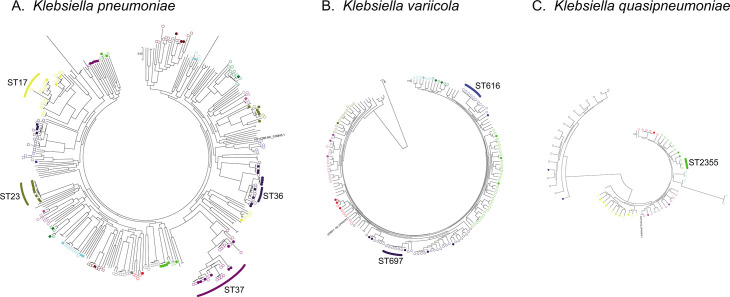 Fig 2
Fig 2| Species detected | Number of samples (%) | Adjusted proportion (%) |
|---|---|---|
|
| 180 (48.9) | 36 |
|
| 37 (10.1) | 5 |
|
| 112 (30.4) | 19 |
|
| 13 (3.5) | NA |
|
| 25 (6.8) | NA |
|
| 1 (0.27) | NA |
| Total | 368 | 60 |
| Strain no. | ST | β-Lactamase gene | MIC (μg/mL) of: | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AMC | TZP | CEZ | CMZ | FMOX | CTX | CAZ | FEP | C/T | GEN | LVX | |||
| FUJA0800 | 9372 |
| 4/2 | 4/4 | >8 | ≤0.5 | ≤0.12 | 16 | 16 | 32 | 0.5/4 | ≤2 | ≤0.5 |
| FUJA0986 | 323 |
| 8/4 | 4/4 | >8 | 1 | ≤0.12 | 8 | ≤1 | 2 | 0.5/4 | >8 | ≤0.5 |
| FUJA1146 | 37 |
| 16/8 | 4/4 | >8 | 2 | ≤0.12 | 32 | 2 | 32 | 1/4 | ≤2 | ≤0.5 |
| FUJA0024 | 9338 |
| ≤2/1 | 16/4 | ≤2 | 8 | ≤0.12 | ≤1 | ≤1 | ≤1 | 1/4 | ≤2 | ≤0.5 |
| FUJA1218 | 827 |
| ≤2/1 | 4/4 | 4 | 1 | ≤0.12 | ≤1 | ≤1 | ≤1 | 0.5/4 | ≤2 | ≤0.5 |
| FUJA1221 | 827 |
| ≤2/1 | 4/4 | ≤2 | ≤0.5 | ≤0.12 | ≤1 | ≤1 | ≤1 | 0.5/4 | ≤2 | ≤0.5 |
- —Japan Agency for Medical Research and Developmenthttp://dx.doi.org/10.13039/100009619
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntibiotic Resistance in Bacteria · Nosocomial Infections in ICU · Antibiotic Use and Resistance
INTRODUCTION
Klebsiella pneumoniae is a well-recognized opportunistic pathogen responsible for a variety of infections, including urinary tract infections, pneumonia, and bacteremia. In recent years, species belonging to the Klebsiella pneumoniae complex, primarily K. pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae, have received increasing attention due to their roles in antimicrobial resistance and hypervirulence. Among antimicrobial-resistant Klebsiella, carbapenem-resistant lineages harboring the blaKPC gene have spread globally, with sequence type (ST) 258 identified as the predominant clone in many countries (1). In addition, the prevalence of extended-spectrum β-lactamase (ESBL)-producing strains is increasing. In Japan, approximately 18% of Klebsiella clinical isolates are reported to produce ESBLs (1).
Another critical concern is the emergence of hypervirulent K. pneumoniae, which was initially identified in East Asia and has since become a significant global public health threat due to its propensity to cause severe community-associated infections, including liver abscesses, endophthalmitis, and meningitis (2). It possesses a distinctive ability to disseminate hematogenously to multiple organs, complicating both diagnosis and treatment, and is frequently associated with high mortality rates (3). Alarmingly, recent studies have identified the emergence of multidrug-resistant (MDR) hypervirulent K. pneumoniae strains (4). Given the combination of enhanced virulence and limited treatment options, continued surveillance of these strains is imperative.
Hypervirulent K. pneumoniae strains are typically characterized by the presence of multiple virulence-associated genes, most notably rmpA and rmpA2, which regulate the expression of the mucoid phenotype. These genes contribute to the hypermucoviscous phenotype often identified by a positive string test. However, this phenotypic test alone is insufficient for definitive identification of hypervirulent K. pneumoniae. A more reliable definition of hypervirulent K. pneumoniae involves the detection of both rmpA and rmpA2 genes (5). Additional key virulence factors include siderophore systems that facilitate iron acquisition, such as aerobactin (iucABCD-iutA) and salmochelin (iroBCDN). The peg-344 gene, which encodes a putative metabolite transporter, has also been reported as a robust marker of hypervirulence (6). These virulence genes are typically encoded on the prototypical virulence plasmid pLVPK (7). Moreover, capsular genotypes K1 and K2 identified through wzi gene sequencing are strongly associated with the hypervirulent phenotype.
Several clonal lineages are recognized as hypervirulent K. pneumoniae, most notably ST23 with capsular genotype K1. K1-ST23 strains frequently harbor genes encoding multiple siderophores (e.g., aerobactin, salmochelin, yersiniabactin, and colibactin) as well as rmpA and rmpA2. Other lineages, such as ST65, ST86, and ST375, typically associated with K2 capsular genotype also exhibit hypervirulence (8, 9). These lineages have been identified mostly in East Asia, including Japan (10, 11).
The prevalence of hypervirulent K. pneumoniae clinical isolates varies geographically. Higher carriage rates have been reported in Asia, including 21.6–37.8% in China (12), 42% in Korea (13), and 29.6% in Taiwan (14). In contrast, lower rates are observed in other regions, such as 3.2% in Spain, 3.9% in the United States, and 4% in Iran. Several studies suggest that intestinal colonization by K. pneumoniae may precede subsequent systemic infection (15, 16), highlighting the need for surveillance of hypervirulent K. pneumoniae carriage.
In Japan, a unique system exists for monitoring intestinal carriage of pathogenic bacteria in the community: individuals working in food-related occupations are required to undergo routine stool testing, typically at least twice per year, to prevent the spread of etiologic agents of infections, such as dysentery and typhoid fever. In this study, we utilized this surveillance framework to assess the carriage rate of hypervirulent K. pneumoniae among community-dwelling individuals in Japan and identify potential risk of community transmission of hypervirulent and ESBL-producing K. pneumoniae.
RESULTS
Stool samples
A total of 646 stool samples were collected from community-dwelling individuals across multiple regions in Japan between August and September 2023. Of these, 250 samples were obtained from the Chubu region, primarily Aichi Prefecture (n = 217), and 242 were collected from the Kanto region. Bacteria suspected to belong to the order Enterobacterales were detected in 627 of the 646 samples (97.1%), yielding a total of 1,340 isolates. Nineteen samples that did not grow any Enterobacterales were considered inadequate and excluded. The remaining 627 samples were used as adequate specimens for subsequent analyses.
Species identification by ANI
Among the 1340 bacterial isolates, 407 strains detected from 368 samples (58.7%) were identified as Klebsiella species (Table S1). The species distribution was as follows: K. pneumoniae (n = 218, 53.6%), K. variicola (n = 137, 33.7%), K. quasipneumoniae (n = 51, 12.5%), and K. quasivariicola (n = 1, 0.25%). Of these, 39 samples contained two distinct Klebsiella species; 13 samples contained K. pneumoniae and K. quasipneumoniae; 25 samples contained K. pneumoniae and K. variicola; and one sample contained both K. quasipneumoniae and K. quasivariicola (Table 1). The estimated distribution of Klebsiella species based on random sampling was consistent with these findings. Among the 100 randomly selected specimens, K. pneumoniae complex species were detected in 60 samples, with the following species distribution: K. pneumoniae (n = 36, 60.0%), K. variicola (19, 31.7%), and K. quasipneumoniae (5, 8.3%) (Table 1; Table S2).
Sequence types of Klebsiella spp.
A total of 407 Klebsiella strains were classified into 285 distinct sequence types (STs). Among the 218 K. pneumoniae strains, ST37 was the most frequently identified (n = 15, 6.9%), followed by ST17 (n = 8, 3.7%) and ST36 (n = 6, 2.8%). Among the 137 K. variicola strains, ST616 was the most prevalent (n = 6, 4.4%), followed by ST697 (n = 5, 3.6%). In contrast, the 51 K. quasipneumoniae strains exhibited greater diversity. ST2355 was the most common ST within this species (n = 3, 5.9%), but 70.6% of strains belonged to distinct STs (Fig. 1). Overall, the majority of strains (54.8%) belonged to unique STs. Owing to the high degree of ST diversity and the low number of strains per ST across geographic regions, statistical comparison of ST distributions using the χ^2^ test lacked sufficient power and was, therefore, not considered appropriate for evaluating regional differences. Even so, no notable geographic clustering of STs was observed (Fig. 1). In the single nucleotide polymorphism (SNP)-based phylogenetic trees (Fig. 2), the frequencies of bloodstream infection (BSI)-derived strains differed significantly from those of stool-derived strains among clusters of K. pneumoniae (P = 0.00019). In contrast, no significant differences in the frequencies of BSI-derived strains among clusters were observed in the K. quasipneumoniae and K. variicola trees.
Geographical distribution of Klebsiella strains. Minimum-spanning trees generated using GrapeTree for each Klebsiella species based on MLST results. Circle colors represent the geographic regions where the subjects reside. Regional color coding in the trees corresponds to the map of Japan shown in the figure. Frequency of the sequence type of K. pneumoniae, K. quasipneumoniae, and K. variicola is also shown in the pie charts. The base map was obtained from https://www.kabipan.com/geography/whitemap/ distributed under a Creative Commons license, with source data derived from the Geospatial Information Authority of Japan.
SNP-based phylogenetic trees incorporating strains isolated from bloodstream infections (BSIs). (A) Klebsiella pneumoniae, (B) Klebsiella variicola, and (C) Klebsiella quasipneumoniae. Clusters containing five or more strains are indicated by circles or squares. Same colors indicate clusters defined by TreeCluster. Strains identified in this study are shown with open symbols, while strains collected from BSI patients are indicated by filled symbols.
Virulence genes and capsular genotypes
The capsular polysaccharide regulatory genes rmpA and rmpA2 genes were detected in three of 218 K. pneumoniae strains (1.4%). Two of these strains (FUJA0307 and FUJA0714) belonged to ST23 and the capsular genotype K1, and another (FUJA1256) belonged to ST412 and K57. All three strains also harbored the multiple iron acquisition genes, including iroBCDN (salmochelin siderophore biosynthesis), iucABCD (aerobactin siderophore biosynthesis), iutA (aerobactin transporter), and peg-344 (metabolite transporter). Comparative analysis of hybrid genome assemblies from these three strains against the reference virulence plasmid pLVPK (GenBank Accession number AY378100) revealed the presence of plasmids closely related to pLVPK (Fig. S1). The plasmids from the two ST23 strains (FUJA0307 and FUJA0714) covered approximately 96% of pLVPK, while the plasmid from the ST412 strain (FUJA1256) covered about 84%. All virulence genes mentioned above were located on these plasmids in each strain. None of the strains of K. variicola and K. quasipneumoniae carried these virulence-associated genes.
Resistance genes
Among the 407 strains, three strains (0.74%; FUJA1146, FUJA0986, and FUJA0800) harboring CTX-M type ESBL genes (blaCTX-M-14, n = 2 [ST37 and ST323] and blaCTX-M-15, n = 1 [ST9372]) were identified from isolates grown on MacConkey agar supplemented with cefotaxime. Antimicrobial susceptibility testing revealed that all three strains were resistant to cefotaxime and cefazolin, and the strain harboring the blaCTX-M-15 gene was also resistant to ceftazidime (Table 2). Furthermore, AMRFinderPlus detected blaSHV-38 as a potential ESBL gene in three K. pneumoniae strains (FUJA0024, FUJA1218, and FUJA1221). However, the strain did not exhibit an ESBL phenotype.
DISCUSSION
In this study, we investigated the distribution of K. pneumoniae complex species among individuals who resided in the community and were presumed to be healthy. Members of the K. pneumoniae complex were detected in approximately 60% of stool samples. This colonization rate is comparable to those reported in pregnant women in Cambodia (66.4%), where detection was based on rectal swabs or stool samples enriched in amoxicillin-supplemented medium, and in healthy individuals without recent healthcare exposure in several Asian countries (41.3% to 87.7%) (17, 18). In contrast, considerably lower colonization rates have been reported in Europe and North America. For example, a study from Norway documented a prevalence of 16.3% based on stool samples stored at −80°C from randomly selected citizens (19), and a U.S. study reported a rate of 23% among hospitalized patients in intensive care or hematology/oncology units (16). Although differences in culture media, colony selection strategies, and identification methods may influence detection rates, the findings of our study support the observation that colonization with K. pneumoniae complex species is generally more common in Asian populations. Lin et al. reported a colonization rate of 18.8% in Japan; however, that study included only 32 specimens collected from Chinese residents in Japan, which may not accurately reflect the general Japanese population. Our results align with previous reports suggesting that K. pneumoniae complex members may constitute part of the normal intestinal microbiota in these regions.
In this study, approximately half of the strains were identified as K. pneumoniae and one-third as K. variicola. Compared to previous studies analyzing clinical isolates, detection rates of K. variicola in our cohort were notably higher, as prior reports have documented rates ranging from 17.1 to 24.4% (10, 20, 21). Our findings suggest that K. variicola may constitute a component of the normal gut microbiota in the community. Certain K. variicola strains have been implicated in severe infections associated with high mortality rates (21, 22). However, no virulence genes were detected in any of the K. variicola strains in our study. Moreover, the genetic characteristics of the K. variicola strains isolated from BSIs in a previous study from Japan (10) were indistinguishable from those identified here. This indicates that the virulence potential of the K. variicola strains in our study is comparable to that of clinical isolates. A recent study suggested that K. variicola may generally exhibit lower virulence (23). Together, these findings support the notion that most K. variicola lineages detected in our study likely represent commensal strains within the intestinal microbiota, and that high-risk lineages appear to be rare in the community setting.
The most common STs of K. pneumoniae identified in this study were ST37 (6.9%) and ST17 (3.7%), consistent with patterns previously reported in clinical isolates (24–27). Several additional STs detected in this study have also been documented in clinical settings, suggesting that the overall ST distribution among individuals in the community may reflect that observed in clinical populations (25, 28–31). Although Klebsiella populations are generally characterized by high ST diversity without clearly dominant clones, ST37 is notable because it has been frequently reported in BSIs and is often associated with ESBL production (10, 32, 33). Thus, ST37 may represent an important clinical lineage with respect to both antimicrobial resistance and virulence. In contrast, some clusters in the SNP-based phylogenetic tree exhibited a lower frequency of BSI-derived strains. This observation suggests that commensal Klebsiella strains may include lineages with lower virulence potential compared to those commonly found in clinical isolates.
Notably, three hypervirulent K. pneumoniae strains (0.74%) were identified in this study, belonging to ST23-K1 and ST412-K57. These strains harbored virulence plasmids closely related to pLVPK, a well-known marker of hypervirulence. Despite their identification, the overall prevalence of hypervirulent lineages among community-dwelling individuals was substantially lower than that observed in clinical K. pneumoniae isolates based on MALDI-TOF MS identification, where approximately 20% were reported to carry rmpA (34). In contrast, a study from Korea reported a hypervirulent K. pneumoniae colonization rate of 4.6% among healthy individuals (35). Our findings suggest that colonization with hypervirulent K. pneumoniae strains is relatively uncommon in the community in Japan. Nevertheless, individuals colonized with hypervirulent K. pneumoniae strains may still be at increased risk of developing invasive infections compared to those harboring less virulent Klebsiella strains.
In recent years, the prevalence of ESBL-producing K. pneumoniae has been increasing among clinical isolates (36, 37). In contrast, ESBL-producing K. pneumoniae strains were rarely detected among individuals in the general community in this study, suggesting that ESBL-producing K. pneumoniae has not spread in the community in Japan. This is in contrast to Escherichia coli, for which significant carriage rates of ESBL-producing strains have been reported among healthy individuals in many countries including Japan (38, 39).
We acknowledge limitations of our study. Stool samples used in this study were obtained from a commercial clinical laboratory that routinely tests specimens submitted by clients, most of whom are food industry workers. However, no detailed information was available regarding the individuals who provided the samples, aside from the locations to which the test results were returned; therefore, duplicate sampling cannot be fully excluded. Although most samples were likely collected from healthy individuals, some may have originated from persons with underlying medical conditions, including those who had been recently hospitalized or were receiving antimicrobial therapy. In addition, the geographic distribution of the samples was skewed toward central Japan, which may have introduced a location-based sampling bias. Finally, only one representative isolate was further worked up from each stool sample unless different morphologies were observed, potentially underestimating the genetic diversity of K. pneumoniae complex in these stool samples.
In conclusion, K. pneumoniae complex strains colonized approximately 60% of the community-dwelling individuals in Japan. The overall ST distribution was similar to that of clinical isolates, but hypervirulent and ESBL-producing strains were rare and each comprised less than 1%. The findings suggest that, although carriage of K. pneumoniae complex is common, transmission of high-risk K. pneumoniae strains is predominantly occurring in healthcare than community settings in Japan.
MATERIALS AND METHODS
Stool samples
Stool samples were collected from community-dwelling individuals across multiple regions in Japan. These samples were submitted to a commercial diagnostic laboratory mostly by workers in the food industry on a routine basis to rule out Shigella spp., Salmonella Typhi, Salmonella Paratyphi A, and enterohemorrhagic E. coli. Residual stool samples received over a 6-week period between August and September 2023 were transported to the research laboratory once a week for processing. The study was approved by the institutional review board of Fujita Health University on an opt-out consent basis (HM23-065).
Screening using selective isolation media
Stool samples were plated onto MacConkey agar supplemented with 1 µg/mL cefotaxime, as well as deoxycholate hydrogen sulfide lactose (DHL) and Salmonella-Shigella (SS) agar plates. The plates were incubated at 37°C for 24 h. Up to five colonies displaying morphological characteristics consistent with E. coli or Klebsiella spp. were selected for further analysis. Selected colonies were subcultured onto CHROMagar Orientation plates and incubated under the same conditions. Colonies exhibiting metallic blue coloration were considered presumptive members of the K. pneumoniae complex; however, this phenotype may also include other Enterobacterales, such as Enterobacter spp. Stool samples from which any Enterobacterales were detected were considered valid for inclusion in the study.
Identification of Klebsiella species by PCR
Presumptive Klebsiella isolates were subjected to species-level identification using polymerase chain reaction (PCR) with Klebsiella-specific marker primers: (K. pneumoniae, F: 5′-TGACTGCGTTGTAAAAAGCG-3′, R: 5′-AATTTAGGTTTACCGTCTGCG-3′, K. variicola, F: 5′- ATGCAGGCCAATTTCGAC-3′, R: 5′-CCATGGCCAAATCGACTT-3′, and K. quasipneumoniae, F: 5′-ACGGAACATTCTCTCTGAAGCC- 3′, R: 5′-ACAGATTTAAAGGCGCTGGA-3′) (40). Template DNA was prepared by suspending bacterial cells in Tris-EDTA buffer at McFarland 0.5, followed by heat lysis at 95°C for 10 min and centrifugation. The resulting supernatant was used as the PCR template. PCR reactions were performed using GoTaq Hot Start Polymerase (Promega) following the manufacturer’s protocol. Thermal cycling conditions were 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min, repeated for 30 cycles. Amplicons were visualized via electrophoresis on 2.5% (w/v) agarose gels stained with ethidium bromide and examined under UV illumination.
Whole genome analysis
Whole-genome sequencing was performed on PCR-confirmed Klebsiella strains. Duplicate strains of the same species from the same individual were excluded. Genomic DNA was extracted using the Gentra Puregene Yeast/Bact Kit (Qiagen). DNA libraries were prepared with the QIAseq FX DNA Library Kit (Qiagen) and sequenced on the Illumina NextSeq 2000 platform (Illumina, San Diego, CA, USA). Reads were assembled using SPAdes version 3.13.1. Strains carrying the rmpA gene additionally underwent long-read sequencing using Oxford Nanopore MinION (O. Hybrid assemblies were generated with Unicycler v0.5.1 using both Illumina and Nanopore reads, with the depth_filter parameter adjusted to 0.1 to prevent the loss of low-coverage sequences.
Species identification
Species-level identification was carried out using FastANI (https://github.com/ParBLiSS/FastANI) (41). Strains showing average nucleotide identity (ANI) value of 95% or higher against a reference genome (K. pneumoniae strain HS11286 [CP003200], K. quasipneumoniae strain KqPF26 [CP065838], K. variicola strain LEMB11 [CP045783], and Klebsiella quasivariicola strain KPN1705 [CP022823]) were classified as the same species (42).
Rate of K. pneumoniae complex species using randomized samples
The number of colonies selected from each sample varied depending on the culture conditions. To estimate the prevalence of K. pneumoniae complex species, 100 specimens were randomly selected, and the number of Klebsiella isolates recovered from each sample was recorded. For samples containing multiple Klebsiella species, a single strain was randomly chosen for subsequent analyses.
Genotypic characterization
Multilocus sequence typing (MLST) was performed in accordance with protocols from the Institut Pasteur MLST and Whole-Genome MLST Database (https://bigsdb.pasteur.fr/klebsiella/). Geographical distribution was visualized using minimum-spanning trees based on MLST profiles generated with GrapeTree (https://github.com/achtman-lab/GrapeTree). To assess regional differences in ST distribution, a χ^2^ test was conducted comparing the Chubu and Kanto areas using STs represented by three or more isolates. Capsular genotypes were determined based on wzi sequences using the kaptive command-line interface (https://github.com/klebgenomics/Kaptive). Antimicrobial resistance genes, including those conferring cephalosporin resistance, as well as virulence genes, were detected using AMRFinderPlus with the National Center for Biotechnology Information Bacterial Antimicrobial Resistance Reference Gene Database (43). Kleborate 3.2.4 (https://github.com/klebgenomics/Kleborate) was also used to determine additional genetic features. Core genome single nucleotide polymorphism (SNP) analysis was performed using SNIPPY version 4.6.0, incorporating strains isolated from BSIs in our previous study (10). Phylogenetic trees for each species were constructed using FastTree version 2.2 and visualized with MEGA version 7.0.26. Clusters within the phylogenetic trees were defined using TreeCluster version 1.0.4 (https://github.com/niemasd/TreeCluster). The frequency of BSI-derived strains within clusters consisting of five or more strains was evaluated using the χ^2^ test.
Antimicrobial susceptibility testing
Antimicrobial susceptibility testing was conducted using the broth microdilution method in accordance with the Clinical and Laboratory Standards Institute (CLSI) guidelines. Minimum inhibitory concentrations (MICs) were determined using K. pneumoniae ATCC 700603 as the quality control strain. The antimicrobial agents tested included: amoxicillin-clavulanate, piperacillin-tazobactam, cefazolin, cefmetazole, flomoxef, cefotaxime, ceftazidime, cefepime, ceftolozane-tazobactam, gentamicin, and levofloxacin.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Arcari G, Carattoli A. 2023. Global spread and evolutionary convergence of multidrug-resistant and hypervirulent Klebsiella pneumoniae high-risk clones. Pathog Glob Health 117:328–341. doi:10.1080/20477724.2022.212136236089853 PMC 10177687 · doi ↗ · pubmed ↗
- 2Russo TA, Marr CM. 2019. Hypervirulent Klebsiella pneumoniae. Clin Microbiol Rev 32:e 00001-19. doi:10.1128/CMR.00001-1931092506 PMC 6589860 · doi ↗ · pubmed ↗
- 3Namikawa H, Yamada K, Sakiyama A, Imoto W, Yamairi K, Shibata W, Yoshii N, Niki M, Nakaie K, Oinuma KI, Tsubouchi T, Niki M, Tochino Y, Takemoto Y, Kaneko Y, Shuto T, Kakeya H. 2019. Clinical characteristics of bacteremia caused by hypermucoviscous Klebsiella pneumoniae at a tertiary hospital. Diagn Microbiol Infect Dis 95:84–88. doi:10.1016/j.diagmicrobio.2019.04.00831256940 · doi ↗ · pubmed ↗
- 4Tang M, Kong X, Hao J, Liu J. 2020. Epidemiological characteristics and formation mechanisms of multidrug-resistant hypervirulent Klebsiella pneumoniae Front Microbiol 11:581543. doi:10.3389/fmicb.2020.58154333329444 PMC 7714786 · doi ↗ · pubmed ↗
- 5Alcántar-Curiel MD, Girón JA. 2015. Klebsiella pneumoniae and the pyogenic liver abscess: implications and association of the presence of rpm A genes and expression of hypermucoviscosity. Virulence 6:407–409. doi:10.1080/21505594.2015.103010125951089 PMC 4601161 · doi ↗ · pubmed ↗
- 6Bulger J, Mac Donald U, Olson R, Beanan J, Russo TA. 2017. Metabolite transporter PEG 344 Is required for full virulence of hypervirulent Klebsiella pneumoniae strain hv KP 1 after pulmonary but not subcutaneous challenge. Infect Immun 85:e 00093-17. doi:10.1128/IAI.00093-1728717029 PMC 5607406 · doi ↗ · pubmed ↗
- 7Compain F, Babosan A, Brisse S, Genel N, Audo J, Ailloud F, Kassis-Chikhani N, Arlet G, Decré D. 2014. Multiplex PCR for detection of seven virulence factors and K 1/K 2 capsular serotypes of Klebsiella pneumoniae. J Clin Microbiol 52:4377–4380. doi:10.1128/JCM.02316-1425275000 PMC 4313302 · doi ↗ · pubmed ↗
- 8Bialek-Davenet S, Criscuolo A, Ailloud F, Passet V, Jones L, Delannoy-Vieillard A-S, Garin B, Le Hello S, Arlet G, Nicolas-Chanoine M-H, Decré D, Brisse S. 2014. Genomic definition of hypervirulent and multidrug-resistant Klebsiella pneumoniae clonal groups. Emerg Infect Dis 20:1812–1820. doi:10.3201/eid 2011.14020625341126 PMC 4214299 · doi ↗ · pubmed ↗