The critical role of hcpR in regulating nitrosative stress defense in Clostridioides difficile
Sanjana Kalra, Toheeb O. Ayinde, Abiola O. Olaitan

TL;DR
The study shows that the hcpR gene helps Clostridioides difficile survive stress in the gut and increases toxin production.
Contribution
hcpR is identified as a novel regulator of nitrosative stress defense and virulence in C. difficile.
Findings
hcpR mutants show eightfold increased sensitivity to nitric oxide and elevated toxin production.
hcpR regulates hcp and frdX, which are involved in NO detoxification and iron-sulfur binding.
Metabolic changes in hcpR mutants include increased short-chain fatty acid production like butyrate.
Abstract
Clostridioides difficile is an anaerobic, toxin-producing pathogen that colonizes the host gastrointestinal tract. Within this hostile environment, the bacterium encounters stressors such as reactive nitrogen species (RNS), which impose nitrosative stress that must be mitigated for survival. This study aimed to elucidate the molecular mechanisms by which C. difficile defends against nitrosative stress. We screened an unordered transposon mutant library of the epidemic strain R20291 using a nitric oxide (NO) donor and identified a nitrosative stress-sensitive mutant with an inactivated hcpR, a transcriptional regulator of the Crp/Fnr family. Transcriptomic and metabolomic analyses were conducted, alongside the assessment of toxin production, a key virulence factor in C. difficile. Our results revealed that hcpR is critical for nitrosative stress adaptation, with the hcpR::Tn mutant…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
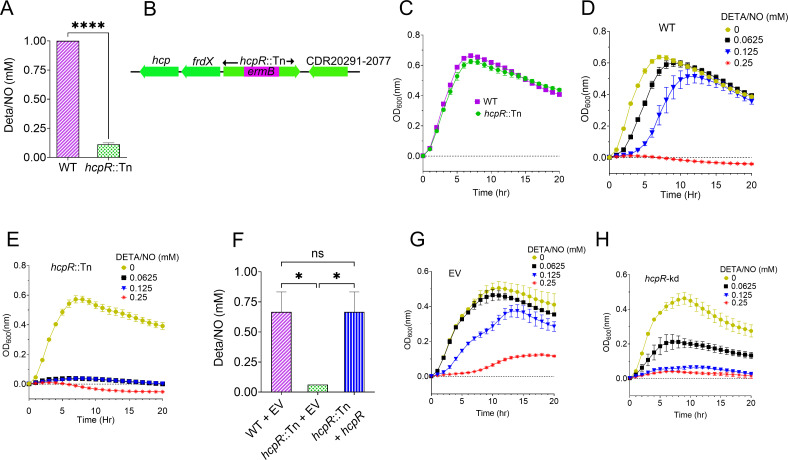 Fig 1
Fig 1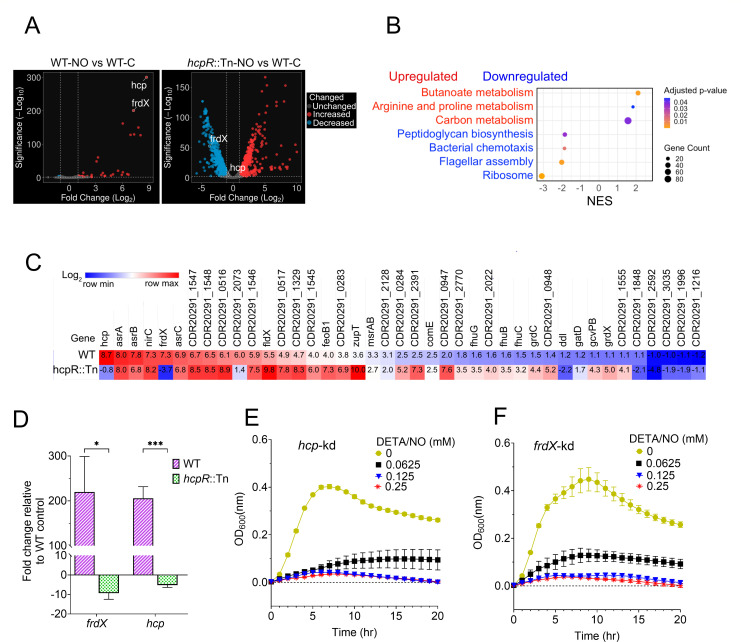 Fig 2
Fig 2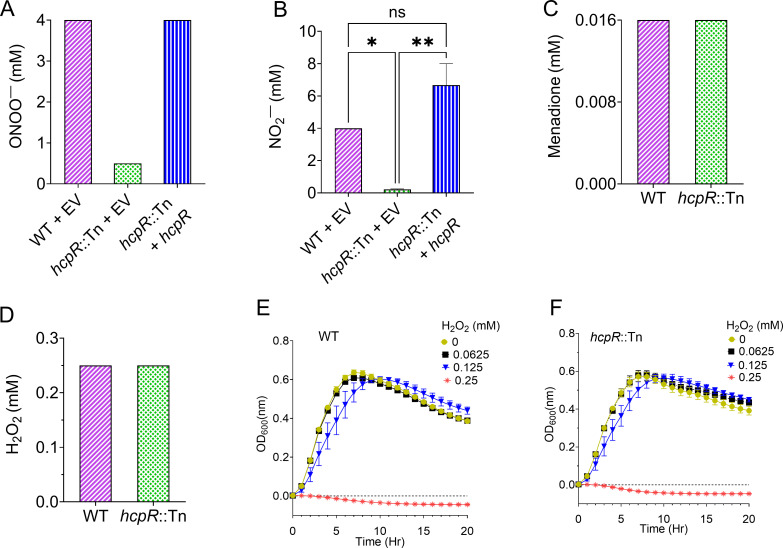 Fig 3
Fig 3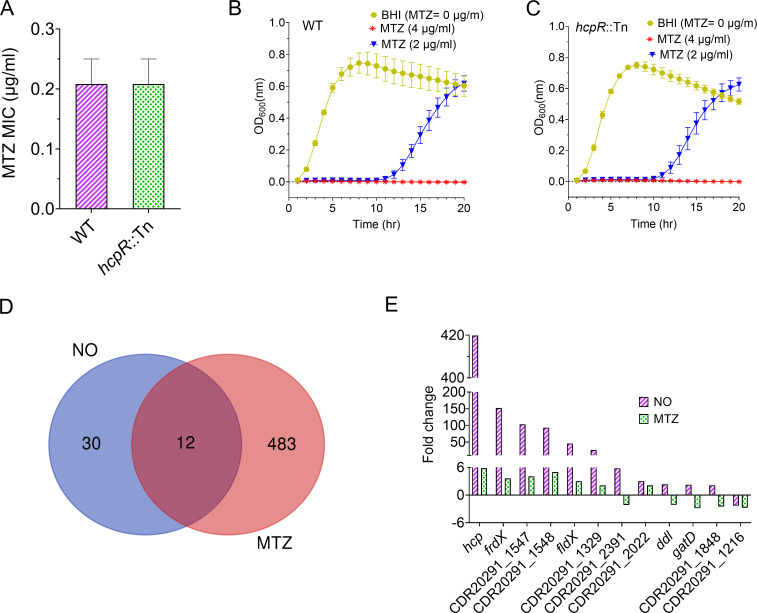 Fig 4
Fig 4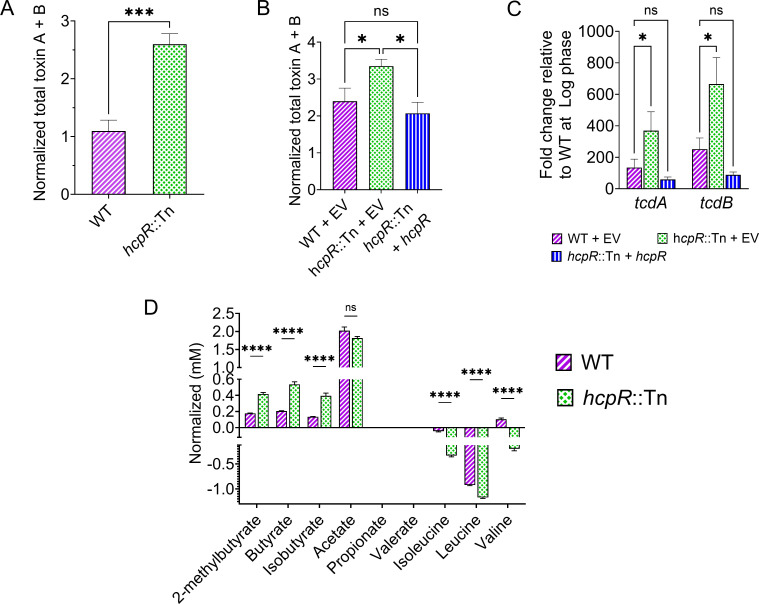 Fig 5
Fig 5| Gene ID | Log2 | Gene function | KEGG pathway |
|---|---|---|---|
|
| −4.59 | MBOAT family protein | Exopolysaccharide biosynthesis |
|
| −3.96 | AlgX/AlgJ SGNH hydrolase-like domain-containing protein | |
| −3.04 | Iron-sulfur binding protein | ||
|
| −2.62 | Hybrid-cluster protein | Nitrogen metabolism |
|
| −1.34 | Phosphotransferase system, IIc component | Phosphotransferase system/fructose and mannose metabolism |
|
| −1.29 | Phosphotransferase system, IId component | Phosphotransferase system |
|
| −1.27 | Phosphotransferase system, IIa component | |
|
| −1.17 | Phosphosugar isomerase | |
|
| 1.06 | ABC transporter, ATP-binding protein | |
|
| 1.18 | DUF917 domain-containing protein | |
|
| 1.18 | Hydantoinase | |
|
| 1.33 | LysR-family regulatory protein | |
| 1.42 | cNMP-binding regulatory protein | ||
|
| 1.64 | Histidinol-phosphate aminotransferase | |
|
| 1.69 | Probable permease | |
|
| 2.10 | HTH cro/C1-type domain-containing protein | |
|
| 3.86 | Amino acid permease | |
|
| 4.04 | NAD-dependent 4-hydroxybutyrate dehydrogenase | Butanoate metabolism |
|
| 4.26 | 4-Hydroxybutyrate CoA transferase | Butanoate metabolism |
|
| 4.29 | Uncharacterized protein | |
|
| 4.30 | Gamma-aminobutyrate metabolism dehydratase/isomerase | Butanoate metabolism |
|
| 4.71 | Glutamine amidotransferase | |
|
| 5.25 | Succinyl-CoA:coenzyme A transferase | Butanoate metabolism |
|
| 5.28 | Succinate-semialdehyde dehydrogenase [NAD(P)+] | Butanoate metabolism |
|
| 5.45 | Membrane protein |
- —Natural Sciences and Engineering Research Council of Canadahttp://dx.doi.org/10.13039/501100000038
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsClostridium difficile and Clostridium perfringens research · Nosocomial Infections in ICU · Yersinia bacterium, plague, ectoparasites research
INTRODUCTION
Clostridioides difficile is an anaerobic bacterial pathogen that colonizes the gastrointestinal tract of humans and animals, particularly the colon. It is a leading cause of healthcare-associated diarrhea, which can progress to colitis—a severe inflammation of the colon (1). Within the gut environment of the host, C. difficile is exposed to a range of stressors, including reactive compounds generated by the host immune response, such as reactive nitrogen species (RNS) and reactive oxygen species (ROS), as well as fluctuations in pH, oxygen levels, and nutrient availability (2–6). Among these, elevated RNS levels impose nitrosative stress (7), a challenge C. difficile must overcome for survival.
Pathogenic and environmental bacteria must adapt to chemically hostile conditions. Nitrosative stress, in particular, arises from increased levels of RNS like nitric oxide (NO), produced by host inducible NO synthase, and peroxynitrite (ONOO^−^), formed by the reaction of superoxide with NO (8). These reactive species disrupt essential cellular functions, causing DNA damage, lipid peroxidation, and ultimately cell death (9, 10). To counteract these effects, many bacteria have evolved specialized defense mechanisms (11–14). For example, Escherichia coli and Salmonella sp. express the hmp gene, which encodes flavohemoglobin that detoxifies NO by converting it to nitrate under aerobic conditions or to nitrous oxide (N_2_O) anaerobically (15, 16). Other systems include regulatory proteins such as NorR, a NO reductase regulator in E. coli (17), and NsrR, a NO-responsive transcriptional repressor found in species like Streptomyces coelicolor (18).
Another major nitrosative stress regulatory defense system is the HcpR transcription factor, which has been identified in both environmental and pathogenic anaerobes, such as Desulfovibrio spp. and Porphyromonas gingivalis (19–22). In these organisms, hcpR not only promotes bacterial survival under nitrosative stress but is also associated with enhanced virulence (20).
Studies have shown that C. difficile possesses stress defense response mechanisms that enable it to withstand a range of environmental challenges. One such mechanism involves the alternative sigma factor σ^B^ (sigB), a general stress regulator in Gram-positive bacteria. sigB modulates defenses against various stressors, including oxygen tension, RNS, ROS, acidification, cationic antimicrobial peptides, and antibiotic exposure (2, 23, 24). Despite these insights, the specific systems that C. difficile employs to combat RNS remain largely underexplored.
In this study, we identified hcpR, a transcriptional regulator in C. difficile, through genetic screening. Its inactivation increased sensitivity to nitrosative stress and caused significant transcriptional perturbations upon NO exposure. Functional analyses demonstrated that hcpR specifically mediates resistance to RNS, but not ROS. Furthermore, inactivation of hcpR led to elevated toxin production, accompanied by a metabolic shift characterized by increased levels of short-chain fatty acids (SCFAs), including butyrate. These findings indicate that hcpR plays a central role in nitrosative stress defense and metabolic remodeling, with potential impact on the modulations of C. difficile virulence factors.
RESULTS
hcpR protects C. difficile from nitrosative stress
We first assessed C. difficile tolerance to nitrosative stress by determining the minimum inhibitory concentration (MIC) of the NO donor diethylenetriamine/NONOate (DETA/NO) for the hypervirulent epidemic strain R20291 (25); the MIC was found to be 1 mM. Subsequently, in a screen of ~2,800 transposon (Tn) mutants from an unarrayed library derived from strain R20291 (26) to identify mutants sensitive or resistant to various stresses, including nitrosative stress, we found that most mutants exhibited only a modest twofold increase in sensitivity to DETA/NO. However, one mutant showed a striking eightfold increase in sensitivity, with an MIC of 0.125 mM compared to the R20291 wild-type (WT) MIC of 1 mM (Fig. 1A). Due to its significantly heightened sensitivity to the NO donor, we focused on this mutant. Whole-genome sequencing identified a single insertion of the ermB cassette in gene CDR20291_2076 at position 2,431,817 (corresponding to nucleotide 641 of 729 within the coding sequence) of the R20291 genome (FN545816.1), resulting in gene inactivation (Fig. 1B). CDR20291_2076 is annotated as a putative cNMP-binding regulatory protein, and domain analysis in NCBI shows that it belongs to the Crp/Fnr transcriptional regulator family. According to PaperBLAST and confirmed through BLAST searches in NCBI (27), the protein shows homology to HcpR from P. gingivalis (27% identity) and Desulfovibrio vulgaris (26% identity), as well as to HcpR2 from Desulfovibrio desulfuricans (34% identity). We found that CDR20291_2076 was indeed annotated as hcpR in RegPrecise (https://regprecise.lbl.gov/regulon.jsp?regulon_id=62803) along with a previous in silico analysis (28). We therefore reannotated CDR20291_2076 as hcpR and its Tn mutant as hcpR::Tn.
*hcpR mediates protection against nitrosative stress in C. difficile. (A) MICs of DETA/NO, a NO donor, were determined for WT and hcpR::Tn strains in BHI after 24 hours. The hcpR::Tn mutant showed increased sensitivity (MIC = 0.125 mM) compared to the WT (MIC = 1 mM). ****: P < 0.0001 (two-tailed unpaired t-test). (B) Schematic representation of insertional inactivation of hcpR (hcpR::Tn). The mutant contains an ermB insertion that inactivates the hcpR coding sequence. Flanking genes are depicted, with arrows indicating their transcriptional orientation. (C) Growth curves of WT and hcpR::Tn strains in BHI without NO. Both strains showed comparable growth under non-nitrosative stress condition. (D and E) Growth of WT (D) and hcpR::Tn (E) strains in BHI with varying DETA/NO concentrations. WT tolerated up to 0.125 mM, whereas hcpR::Tn growth was inhibited at 0.0625 mM. Controls (0 mM) contained DMSO equivalent to that in 0.25 mM DETA/NO. (F) Complementation of hcpR restores nitrosative stress tolerance in C. difficile. MICs of DETA/NO were determined for the complemented strain (hcpR::Tn + hcpR), the hcpR::Tn mutant, and WT with EV (pRPF185). The mutant was highly sensitive to DETA/NO (MIC = 0.0625 mM), whereas complementation restored tolerance to WT levels (0.5–1 mM). ns: not significant; : P < 0.05 (one-way analysis of variance with Tukey’s multiple comparison test). (G and H) Growth of WT EV (G) and hcpR CRISPRi knockdown; hcpR-kd (H) strains under nitrosative stress. WT EV tolerated 0.0625 mM DETA/NO, whereas hcpR-kd showed sensitivity or growth inhibition at the same concentration. All data are shown as mean ± SEM from three biological replicates.
Growth curve assays showed that the WT and hcpR::Tn strains grew similarly under normal conditions without the NO donor (Fig. 1C). Under nitrosative stress, however, the WT strain tolerated DETA/NO concentrations up to 0.125 mM (Fig. 1D). In contrast, hcpR::Tn failed to grow even at 0.0625 mM (Fig. 1E), indicating that hcpR inactivation impairs NO tolerance. To confirm that the increased sensitivity to nitrosative stress was specifically due to hcpR inactivation and to rule out polar effect, we complemented the hcpR::Tn mutant with an intact hcpR gene under its native promoter. This complementation restored hcpR::Tn tolerance to NO donor, with the MIC returning to 1 mM, comparable to the WT empty vector (EV) strain (Fig. 1F). We further performed hcpR knockdown using CRISPR interference (CRISPRi). At 0.0625 mM DETA/NO, the WT EV control showed comparable growth to the dimethyl sulfoxide (DMSO) control (Fig. 1G). In contrast, a hcpR knockdown strain (hcpR-kd) exhibited impaired growth at the same concentration and failed to grow at 0.125 mM, which was tolerated by the WT EV (Fig. 1G and H). These results demonstrate that hcpR is required for C. difficile adaptation to nitrosative stress.
Transcriptional regulation by hcpR during nitrosative stress
To investigate the regulatory role of hcpR in C. difficile under both unstressed and nitrosative stress conditions, we conducted transcriptome analysis comparing WT and hcpR::Tn strains. Under unstressed conditions, there were 24 significantly differentially expressed genes (DEGs) in the hcpR::Tn strain relative to WT, excluding the inactivated hcpR gene (Table 1). Several of the most upregulated genes were associated with butanoate (also called butyrate) metabolism according to Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. The downregulated genes included hcp, CDR20291_2075 (reannotated as frdX, a ferredoxin-like iron-sulfur binding protein based on RegPrecise), and genes associated with fructose and mannose metabolism (Table 1). To identify potential direct targets of HcpR among these DEGs, we analyzed their promoter regions with the HcpR binding motif from RegPrecise using FIMO (MEME Suite). A putative binding site was identified only upstream of the frdX-hcp gene pair, which co-localizes with hcpR, suggesting that hcpR directly regulates these genes through a shared upstream promoter (29).
Under nitrosative stress, we assessed the role of hcpR by comparing the transcriptomes of NO-treated WT and NO-treated hcpR::Tn strains, each relative to untreated WT. The WT showed a modest response, with 42 DEGs (~1.2% of the genome; Fig. 2A; Table S1), whereas hcpR::Tn exhibited extensive transcriptional disruption, with 1,481 DEGs (~42% of the genome; Fig. 2A; Table S2). Comparison of NO-treated hcpR::Tn and NO-treated WT strains revealed that 1,350 genes were uniquely differentially expressed due to hcpR inactivation under nitrosative stress (Table S3). Gene set enrichment analysis (GSEA) of KEGG pathways using a stringent cutoff (p.adjust < 0.05) identified significant enrichment of multiple metabolic pathways. Upregulated genes were enriched in arginine/proline and carbon metabolism (excluding butanoate metabolism impacted mainly by hcpR inactivation). In contrast, downregulated genes were strongly enriched for ribosome function, flagellar assembly, bacterial chemotaxis, and peptidoglycan biosynthesis (Fig. 2B). Using a relaxed threshold (q ≤ 0.25) revealed additional enriched pathways, including glycerophospholipid and nicotinate/nicotinamide metabolism among upregulated genes, and amino acid and fatty acid biosynthesis pathways among downregulated genes (Table S4). These results indicate that hcpR inactivation broadly impacts both metabolic and structural processes during nitrosative stress.
*hcpR-mediated regulation of gene expression during nitrosative stress. (A) Volcano plots of DEGs in WT (left) and hcpR::Tn (right), both under nitrosative stress induced by exposure to DETA/NO at 0.0625 mM for 30 minutes. Expression changes are relative to WT untreated condition. Dashed lines indicate significance thresholds (log2 fold change ≥ 1 and FDR ≤ 0.01). NO exposure caused minimal transcriptional changes in WT, whereas hcpR inactivation led to extensive transcriptional perturbation. (B) GSEA with KEGG pathway enrichment in NO-treated hcpR::Tn relative to NO-treated WT. GSEA revealed pathways significantly enriched (adjusted P < 0.05) exclusively in the hcpR::Tn mutant under nitrosative stress. (C) Transcriptome profiles of DEGs shared between NO-treated WT and NO-treated hcpR::Tn strains, each relative to the untreated WT. The heatmap highlights differences in gene induction by nitrosative stress between strains with intact versus inactivated hcpR. Among these genes, hcp and frdX, known targets of hcpR, were induced in WT but not in hcpR::Tn. hcp was modestly downregulated in the hcpR::Tn strain (log2 fold change = –0.8; below the cutoff of ≥1) but was included for comparison with its expression in the WT. (D) Transcriptional analysis of hcpR targets (hcp and frdX) in NO-treated WT and NO-treated hcpR::Tn strains, each relative to the untreated control via qPCR. Both targets were strongly induced by NO in WT but downregulated under the same condition in hcpR::Tn. Expression was normalized to the 16S rRNA housekeeping gene. All data are presented as means ± SEM from four biological replicates. *: P < 0.05; **: P ≤ 0.001 (two-tailed multiple unpaired t-test with Holm-Šídák multiple comparison test). (E and F) Growth of CRISPRi knockdown strains targeting hcp (E) and frdX (F) under varying concentrations of the NO donor DETA/NO. Both knockdowns were sensitive to nitrosative stress relative to the same strains grown in the DMSO control lacking the NO donor. All data are presented as means ± SEM from three biological replicates.
Among the genes shared between NO-treated hcpR::Tn and NO-treated WT strains (Table S5), only hcp and frdX were identified as direct targets of hcpR. Both genes were strongly induced under nitrosative stress in the WT strain but remained uninduced in the hcpR::Tn mutant (Fig. 2C). Quantitative PCR (qPCR) confirmed this trend; in the WT, hcp and frdX were upregulated by ~206 ± 26-fold and 220 ± 79-fold, respectively. In contrast, their expression in the mutant was reduced by about ~5 ± 1fold and 9 ± 3-fold, respectively (Fig. 2D). To investigate the role of the frdX-hcp gene cluster in nitrosative stress defense, we individually knocked down hcp and frdX. Under nitrosative stress, both knockdown strains showed impaired growth compared to unstressed controls (Fig. 2E and F). The negative control, knockdown nimB, a gene not known to contribute to NO detoxification under the NO levels used in this study, was not impacted by NO exposure (Fig. S1). Overall, these findings demonstrate that hcpR inactivation leads to extensive transcriptional dysregulation under nitrosative stress and that it activates the frdX-hcp cluster in response to NO.
hcpR protects against nitrosative stress but not oxidative stress
Building on the observed sensitivity of the hcpR::Tn mutant to NO, we examined its response to other sources of nitrosative stress. Specifically, hcpR::Tn exhibited an eightfold increase in sensitivity to peroxynitrite (ONOO^−^), with an MIC of 0.5 mM compared to 4 mM for the WT strain (Fig. 3A). Complementation of hcpR restored the mutant’s tolerance, increasing the MIC to 2 mM. The mutant also exhibited a 16- to 32-fold increase in sensitivity to nitrite (NO_2_^−^), with MIC values ranging from 0.125 to 0.25 mM, while the WT strain maintained an MIC of 4 mM (Fig. 3B). Complementation with intact hcpR similarly reversed this sensitivity, restoring MIC values to 4–8 mM, similar to the WT. To determine whether hcpR contributes to oxidative stress defense, WT and hcpR::Tn strains were exposed to ROS, including menadione (a superoxide generator) and hydrogen peroxide (H_2_O_2_), which produces hydroxyl radicals (30). Both strains showed similar sensitivity to menadione and H_2_O_2_, with MICs of ~0.0156 and 0.25 mM, respectively (Fig. 3C and D). Growth assays under H_2_O_2_-induced stress confirmed that both strains experienced identical growth inhibition at 0.25 mM, with growth resuming below this concentration (Fig. 3E and F). Taken together, these results reveal that hcpR specifically mediates in nitrosative stress defense in C. difficile but does not modulate defense against oxidative stress.
*hcpR specifically defends against nitrosative stress and does not provide protection against oxidative stress. (A and B) MICs of ONOO− (A) and NO2− (B) were determined for WT EV, hcpR::Tn EV, and the complemented strain (hcpR::Tn + hcpR). The hcpR::Tn mutant was sensitive to both RNS sources, whereas complementation restored tolerance to the same level as WT EV. ns: not significant; *: P < 0.05; *: P ≤ 0.01 (one-way analysis of variance with Tukey’s multiple comparison test). (C and D) MICs of menadione (C) and H2O2 (D) were determined for WT and hcpR::Tn. Both strains exhibited similar tolerance to these ROS. (E and F) Growth curves of WT (E) and hcpR::Tn (F) under oxidative stress with varying concentrations of H2O2 (0.0625–0.25 mM). Strains were grown in BHI supplemented with H2O2 and untreated control. Both strains displayed similar growth patterns, tolerating 0.125 mM H2O2 or being inhibited at 0.25 mM. All data are presented as means ± SEM from three biological replicates.
hcpR does not mediate defense against MTZ-induced stress
Metronidazole (MTZ), a nitroheterocyclic antibiotic effective against C. difficile, is believed to exert its bactericidal effect by generating cytotoxic nitro radical intermediates that mimic nitrosative stress (31, 32). To investigate whether hcpR influences MTZ sensitivity, we compared the susceptibility of WT and hcpR::Tn strains to MTZ. Both strains showed similar MICs of 0.125–0.25 µg/mL (Fig. 4A). Growth assays showed similar response, with both strains inhibited at 4 µg/mL MTZ and reduced growth at lower concentration (Fig. 4B and C). To further explore the relationship between MTZ and NO-mediated nitrosative stress defenses, we analyzed previously published RNA-seq data (26) comparing transcriptomes of MTZ-treated and NO-treated WT C. difficile R20291. MTZ exposure resulted in 495 DEGs, whereas NO exposure affected only 42 DEGs. Twelve DEGs were shared between the two treatments (Fig. 4D). Furthermore, hcp and frdX, known hcpR targets, were among the most strongly upregulated genes by NO (~420- and 152-fold, respectively) but were only modestly induced by MTZ (~6- and 4-fold, respectively; Fig. 4E). These findings suggest that hcpR is not essential for protection against MTZ-induced toxicity or its associated nitro radical intermediates in C. difficile.
hcpR does not provide protection against MTZ. (A) The MIC of MTZ, a nitroheterocycle-based antibiotic, was determined for WT and hcpR::Tn strains. Both strains exhibited the same level of sensitivity to MTZ, with an MIC of 0.25 µg/mL. (B and C) Growth curves of WT (B) and hcpR::Tn (C) in response to MTZ. Strains at exponential phase were grown in BHI supplemented with varying concentrations of MTZ and BHI containing DMSO as the control. Both strains exhibited similar growth response to MTZ. All data are presented as means ± SEM from three biological replicates. (D) Venn diagram showing the transcriptomes of significantly DEGs in C. difficile R20291 exposed to NO or MTZ for 30 minutes, each relative to the DMSO control. The intersection shows that only 12 genes were shared between the two stressors. (E) Comparison of the 12 DEGs shared between the transcriptomes of NO- and MTZ-treated C. difficile R20291. NO induced stronger upregulation of certain genes, including the hcpR targets (hcp and frdX), compared to MTZ.
hcpR modulates C. difficile toxin production
Nitrosative stress defense mechanisms have been linked to bacterial virulence (33). To assess the impact of hcpR inactivation on toxin virulence factor, we quantified toxin production. The hcpR::Tn mutant produced ~2.4-fold higher levels of toxins (TcdA/TcdB) compared to the WT strain (Fig. 5A). To further validate this observation, we examined toxin levels in the hcpR::Tn strain carrying either an EV or a complementation construct. The hcpR::Tn with an EV maintained elevated toxin production (Fig. 5B), whereas introduction of an intact hcpR gene restored toxin levels to those observed in the WT EV strain. We further quantified the expression levels of tcdA and tcdB toxin genes using qPCR. In the hcpR::Tn EV strain, expression of both tcdA and tcdB was significantly elevated compared to the WT EV strain. In contrast, these elevated expression levels were restored to WT EV levels in the hcpR::Tn + hcpR-complemented strain (Fig. 5C).
*Impact of hcpR on toxin production. (A) Quantification of toxins (TcdA/TcdB) in WT and hcpR::Tn strains revealed that the mutant produced higher levels of toxin compared to WT. Data are presented as means ± SEM from six biological replicates in technical duplicates. ***: P ≤ 0.001 (two-tailed unpaired t-test). (B) Quantification of toxins (TcdA/TcdB) in WT EV, hcpR::Tn EV, and complemented hcpR::Tn (hcpR::Tn + hcpR) strains. The hcpR::Tn EV produced higher toxin levels than WT EV, whereas complementation of the mutant with intact hcpR restored toxin production to WT levels. Data are presented as means ± SEM from eight biological replicates in technical duplicates. ns: not significant; *: P < 0.05 (one-way analysis of variance with Tukey’s multiple comparison test). (C) qPCR quantification of tcdA and tcdB expression levels in WT EV, hcpR::Tn EV, and hcpR::Tn + hcpR strains. Overnight cultures were grown to exponential phase (OD600 = 0.3) and to stationary phase (24 hours) for gene expression. The hcpR::Tn EV strain exhibited higher expression of toxin genes compared to WT EV, whereas complementation of the mutant with intact hcpR restored expression to WT levels. Gene expression levels are shown relative to WT EV at exponential/log phase. Data are presented as means ± SEM from five biological replicates. ns: not significant; *: P < 0.05 (two-way analysis of variance with Tukey’s multiple comparison test). (D) Targeted metabolomics of WT and hcpR::Tn strains using 1H-NMR. The hcpR::Tn mutant exhibited increased levels of SCFAs and reduced BCAAs compared to WT. Acetate and propionate were not detected by 1H-NMR, which may indicate their absence or concentrations below the detection limit. Data are presented as means ± SEM from six biological replicates. ns: not significant; ***: P ≤ 0.0001 (two-tailed multiple unpaired t-test with Holm-Šídák multiple comparison test).
Given that genes involved in butanoate metabolism were upregulated in the hcpR::Tn transcriptome (Table 1) and considering that increased butyrate levels are known to enhance toxin production (34, 35), we investigated whether SCFA metabolites were elevated in the mutant strain. Targeted metabolomics confirmed increased levels of SCFAs, including butyrate, isobutyrate, and 2-methylbutyrate, in the hcpR::Tn mutant. Concurrently, levels of branched-chain amino acids (BCAAs), such as valine, leucine, and isoleucine were reduced (Fig. 5D). Overall, these findings indicate that hcpR impacts C. difficile toxin production and contributes to metabolic changes affecting SCFA levels.
DISCUSSION
In the host gastrointestinal tract, C. difficile is exposed to nitrosative stress caused by elevated RNS, which are harmful and must be counteracted for bacterial survival (7). In this study, we demonstrated that the inactivation of hcpR (hcpR::Tn) increased C. difficile susceptibility to multiple RNS, including NO, ONOO^−^, and NO_2_^−^, a related nitrogen metabolite known to contribute to RNS generation. These findings align with previous reports of HcpR-mediated nitrosative stress defense in other bacteria, such as Desulfovibrio spp. and P. gingivalis, which inhabit environmental and oral niches, respectively (19, 20). Inactivation of hcpR rendered these bacteria sensitive to nitrosative stress in the presence of elevated nitrite or S-nitrosoglutathione. Together, these results paint a picture that hcpR-mediated defense against nitrosative stress is a conserved survival strategy across both diverse bacterial species and ecological niches. Our data showed that hcpR specifically confers protection against RNS, but not ROS, consistent with observations in P. gingivalis (20). This functional specificity might be explained by the fact that C. difficile encodes dedicated ROS defense systems, including the peroxide stress regulator PerR and various oxidative stress detoxification enzymes (36, 37), which likely operate independently of RNS-responsive pathways. This indicates that C. difficile uses distinct regulatory systems to manage different stresses in the host environment.
Our findings show that inactivation of hcpR markedly reshapes the global transcriptional landscape of C. difficile under nitrosative stress and demonstrated that hcpR is essential for maintaining transcriptional homeostasis during nitrosative stress, protecting both metabolic and structural processes from widespread dysregulation. Under nitrosative stress, the hcpR::Tn mutant exhibited increased amino acid catabolism and central carbon metabolism, reflecting an attempt to increase energy production under stress. Concurrent downregulation of energy-intensive processes, including ribosome biogenesis and cell wall synthesis, indicates a shift toward energy conservation. Together, these changes provide insight into the stress-adaptive metabolic reprogramming driven by hcpR inactivation.
We found that hcpR regulates the expression of hcp and frdX in response to nitrosative stress. These genes are located directly upstream of hcpR (Fig. 1B), a genomic arrangement similar to that seen in Desulfovibrio spp., such as D. vulgaris Hildenborough and Desulfovibrio alaskensis G20, although with some variation in gene content (29, 38). The distinct but coordinated expression patterns of hcp and frdX in WT and hcpR::Tn strains, together with a shared HcpR binding site and similar nitrosative stress–responsive phenotypes, support their co-regulation by hcpR in C. difficile. This pattern is consistent with findings in D. vulgaris Hildenborough (39).
The hcp gene is a well-established HcpR target that plays a key role in NO detoxification (40). In P. gingivalis, hcp is the primary HcpR-regulated gene and is essential for nitrosative stress adaptation and intracellular survival (41). In E. coli, deletion of hcp increases NO sensitivity and impairs its reductive detoxification to nitrous oxide (N_2_O) (42). Similarly, in D. vulgaris, N_2_O binds Hcp, reinforcing its role in NO detoxification (43). Consistent with these findings, we showed that knockdown of hcp in C. difficile increased sensitivity to NO. Although our data suggest that frdX also contributes to nitrosative stress adaptation, its exact function remains unclear. frdX encodes a ferredoxin-like iron-sulfur binding protein typically involved in electron transfer via Fe-S clusters (28, 44). It is plausible that frdX participates in redox regulation or electron transfer during hcpR-mediated stress defense responses (29).
Our observation that the hcpR mutant displayed similar susceptibility to MTZ suggests that hcpR is unlikely to play a major role in the bacterial defense against MTZ, which is thought to induce nitrosative stress. Transcriptomic analysis showed that MTZ exposure elicited a broader and more extensive transcriptional response than NO exposure in the WT, with distinct expression profiles. Additionally, the hcpR target genes hcp and frdX were strongly induced by NO but only modestly upregulated by MTZ. These findings suggest that MTZ and NO elicit separate or unique regulatory pathways to deal with these stressors. The cellular response to MTZ detoxification likely involves mechanisms that are largely independent of hcpR. This is intriguing given that MTZ is believed to generate reactive nitro radicals capable of inducing nitrosative stress (32).
Beyond its role in stress defense, our findings indicate that hcpR impacts a key virulence factor in C. difficile. The hcpR::Tn mutant showed increased production of toxins (TcdA/TcdB), including at the transcriptional level. This virulence factor is central to C. difficile pathogenesis and contributes to inflammation of the colon (45). The increased expression of butanoate (butyrate) metabolism genes in hcpR::Tn correlates with elevated butyrate levels. Additionally, we observed increased concentrations of other SCFAs and decreased levels of BCAAs, suggesting enhanced BCAA catabolism. Given that BCAAs such as valine and isoleucine are precursors for SCFAs like isobutyrate and 2-methylbutyrate, respectively (46), this pattern indicates that hcpR inactivation triggered a metabolic shift toward increased amino acid fermentation. Although our analysis focused on targeted metabolites, we observed additional metabolic changes beyond SCFAs between WT and hcpR::Tn strains, including differences in 3-phenylpropionate, isocaproate, and trehalose (Table S6). These findings point to broader metabolic remodeling resulting from the loss of a functional hcpR. The overall metabolic shift may serve as a compensatory adaptive mechanism to support energy production and maintain redox balance in the stress-sensitive mutant. Butyrate and other SCFAs are known to enhance toxin production in C. difficile (34, 35, 47), which may explain the elevated toxin levels observed in the hcpR::Tn mutant. The increased toxin production is likely a secondary consequence of the metabolic reprogramming advantages conferred by hcpR inactivation, rather than a direct effect of hcpR loss. Indeed, among the DEGs in the hcpR::Tn mutant, only the frdX-hcp cluster contains a putative HcpR binding site, suggesting that hcpR does not directly regulate butyrate or other SCFA biosynthesis. Hence, the upregulation of butanoate metabolism likely reflects broader metabolic changes triggered by increased nitrosative stress sensitivity.
In summary, our study demonstrates that hcpR plays a key role in nitrosative stress defense in C. difficile. Inactivation of hcpR compromises tolerance to RNS and leads to widespread transcriptional and metabolic changes, including increased SCFAs and elevated toxins. These findings highlight hcpR as a crucial transcriptional regulator that contributes to stress adaptation and also modulates virulence-related processes.
MATERIALS AND METHODS
MIC assay and Tn mutant screening
C. difficile strains were cultured in pre-reduced Brain Heart Infusion (BHI) medium, supplemented with 1.5% agar when required, and incubated overnight at 37°C in a Whitley A35 anaerobic workstation (Don Whitley Scientific) under a gas mixture of 85% N_2_, 5% CO_2_, and 10% hydrogen. An unordered Tn-mutant library of C. difficile R20291, previously generated using plasmid pRPF215 (26, 48), was used for nitrosative stress screening. Briefly, an overnight culture of R20291 carrying pRPF215, a Himar1 mariner delivery vector, was subcultured and grown to an OD_600_ of 0.2 in the absence of thiamphenicol. Himar1 transposition was induced with 100 ng/mL anhydrotetracycline and supplemented with 20 µg/mL lincomycin, followed by overnight incubation. The following day, several serial dilutions of the culture containing Tn mutants were plated on pre-reduced BHI agar supplemented with 20 µg/mL lincomycin. After incubation, individual Tn colonies were picked into 96-deep-well plates containing BHI with 20 µg/mL lincomycin and incubated. The unordered Tn-mutant library was subsequently stored for screening. Tn mutants were maintained on BHI agar containing 20 µg/mL lincomycin, and spore stocks were revived on BHI supplemented with 0.1% taurocholate, 250 µg/mL cycloserine, and 8 µg/mL cefoxitin. Screening of the Tn-mutant library was performed using overnight cultures grown in BHI with lincomycin and exposed to 1/4× and 1× MIC concentrations of test compounds in liquid BHI supplemented with 0.003% neutral red. The NO-sensitive mutant was whole-genome sequenced to identify the gene disrupted by ermB. Sequencing was performed at SeqCenter, LLC, in Pittsburgh, USA. MICs of test compounds were determined using broth dilution in BHI with 0.003% neutral red. Doubling dilutions were prepared in 196 µL volumes in microplate wells, pre-reduced for 3 hours in an anaerobic chamber, and inoculated with 4 µL of overnight cultures. MICs were recorded after 24 hours of incubation. Compounds tested included RNS sources, such as DETA/NO (Cayman Chemical, catalog no. 82120), peroxynitrite (Calbiochem, catalog no. 516620), and sodium nitrite (Sigma Aldrich, catalog no. S2252), and ROS sources including menadione (Sigma Aldrich, catalog no. M5625) and H_2_O_2_ (ThermoFisher Scientific, catalog no. H325500). MTZ (Sigma Aldrich, catalog no. M3761) MIC was determined using the agar dilution method as previously described (26). Stock solutions of all compounds were prepared in DMSO (Bioshop, Canada, catalog no. DMS555). All strains and plasmids used in this study are listed in Table S7.
Growth curve assay
Doubling dilutions of test compounds (DETA/NO, H_2_O_2_, and MTZ) were prepared in 96-well microplates containing 100 µL of BHI broth, followed by a 3-hour pre-reduction in an anaerobic chamber. Overnight cultures of WT and hcpR::Tn strains were grown to early exponential phase (OD_600_ = 0.2–0.3, or back-diluted to 0.1 for MTZ testing). Each well was inoculated with 100 µL of culture, bringing the final volume to 200 µL. Plates were sealed with ELISA Plate Sealers (R&D Systems, Cat. No. DY992), and growth was monitored by measuring absorbance at 600 nm every 30 minutes over 24 hours using an Alto portable plate reader (Cerillo, USA) inside the anaerobic chamber at 37°C. OD_600_ readings were normalized to zero at the start of the growth curve. All experiments were performed in three technical and three biological replicates.
Gene complementation
To complement hcpR::Tn, a 500 bp upstream region containing the native hcpR promoter along with the hcpR coding sequence from strain R20291 was PCR-amplified and cloned between the KpnI and BamHI sites of the pRPF185 plasmid (49). The plasmid was first introduced into electrocompetent E. coli SD46 (50) and then transferred to hcpR::Tn via conjugation. Briefly, 1 mL of overnight E. coli SD46 culture carrying the plasmid was pelleted and washed twice with phosphate-buffered saline. A 200 µL overnight culture of hcpR::Tn was added to the pellet, and the mixture was spotted onto BHI agar and incubated overnight. The next day, cells were harvested into 500 µL BHI broth and mixed, and 100 µL was spread on BHI agar containing 8 µg/mL cefoxitin, 250 µg/mL cycloserine, 15 µg/mL thiamphenicol, and 20 µg/mL lincomycin to select for transconjugants. After 2–3 days, colonies were transferred to fresh selective BHI agar for purification. An EV control (pRPF185) was also conjugated into hcpR::Tn and WT strains (no lincomycin added). Sensitivity assays for RNS were performed as already described. All primers used are listed in Table S8.
Gene knockdown
Gene knockdown was carried out using CRISPRi. Two guide RNAs targeting hcpR, frdX, and hcp were synthesized by Bio Basic Inc. (Ontario, Canada) and cloned into the PmeI site of the pXWxyl-dcas9 vector (51). The resulting plasmids were conjugated into C. difficile strain R20291. Gene silencing was induced by culturing the bacteria in 2% xylose (BioShop Canada Inc., Cat. No. XYL001.500) and 15 µg/mL thiamphenicol, both during overnight growth and during subsequent growth to exponential phase prior to performing growth curve assays.
Toxin quantification
WT and hcpR::Tn strains were cultured overnight on BHI agar and then grown in BHI broth for 24 hours. Cultures were centrifuged at 5,000 × g for 5 minutes at 4°C, and supernatants were stored at −80°C until toxin analysis. Toxins A and B were simultaneously quantified using an ELISA kit (tgcBIOMICS, Cat. No. TGC-E001-1) following the manufacturer’s instructions. Toxin levels were normalized to total protein content, measured using the Pierce BCA Protein Assay Kit (Thermo Scientific, Cat. No. LSG23227). For the hcpR-complemented strain and EV controls, a similar method was followed, except that cultures were grown in BHI supplemented with 2 µg/mL thiamphenicol to minimize the impact of the antibiotic on toxin production.
1H-NMR -based metabolomics
A portion of the supernatant used for toxin quantification was filtered through a 0.2 µm membrane filter. ^1^H nuclear magnetic resonance (NMR) experiments were conducted at the University of Guelph NMR Center. The 4,4-dimethyl-4-silapentane-1-sulfonate (DSS) was used as the internal standard. Briefly, sterile-filtered extracts (540 µL) were mixed with 60 µL Chenomx IS-2 stock solution (4.94 mM DSS-d_6_; Chenomx, AB, Canada) to achieve a final DSS concentration of 0.494 mM. A 560 µL aliquot was transferred to a 5 mm NMR tube for analysis. Spectra were acquired at 298 ± 1 K on a Bruker AVANCE III spectrometer equipped with a 5 mm TCI cryoprobe, using a noesypr1d pulse sequence with 128 scans, a 2 s relaxation delay with water presaturation, and a 3 s acquisition period. Data were processed in Chenomx NMR Suite v10.0 with 0.2 Hz line broadening, manual phase and baseline correction, automated shim correction, and automated CSI calibration. Because 2-methylbutyrate is not included in the Chenomx library, it was manually added using a reference spectrum from a pure standard, as per the Chenomx user manual. Metabolite levels were normalized to total protein content, measured using the Pierce BCA Protein Assay Kit (Thermo Scientific, Cat. No. LSG23227).
Reverse transcription quantitative real-time PCR
Overnight cultures of hcpR::Tn and WT strains were grown in BHI broth to OD_600_ = 0.2 and then treated with 0.0625 mM NO donor or DMSO control for 30 minutes. Cells were harvested by centrifugation at 4,000 rpm for 5 minutes, and pellets were resuspended in 1 mL of RNAprotect Bacterial Reagent (Qiagen, Cat. No. 76506) to preserve RNA integrity. Pellets were stored for RNA extraction following a final centrifugation. Total RNA was extracted using the HiPure Total RNA Mini Kit (GeneBio Systems, Canada, Cat. No. R401102) and treated with TURBO DNase (Thermo Fisher, Cat. No. AM1907) to eliminate genomic DNA. cDNA synthesis was performed using qScript cDNA SuperMix (Quantabio, Cat. No. 95048-100), and qPCR was conducted with PerfeCTa SYBR Green SuperMix (Quantabio, Cat. No. 95074-012) on a QuantStudio 6 Flex Real-Time PCR System. Gene expression was normalized to 16S rRNA gene and fold change calculated using the 2^−ΔΔCt^ method (52). Primers used are listed in Table S8 (26, 53, 54).
RNA sequencing
WT and hcpR::Tn strains were treated with the NO donor or DMSO control (unstressed condition) for 30 minutes as described above. RNA extraction and purification were done as already described for qPCR. Samples were sent to The Center for Applied Genomics (Toronto) for sequencing. Libraries were prepared using the Illumina Stranded Total RNA Prep kit with rRNA depletion using the Ribo-Zero Plus Microbiome kit. Paired-end sequencing was performed on the NovaSeq X system using a 10B flow cell to generate 150 bp reads. RNA-seq analysis was performed using the previously described methods (26). Sequencing data were uploaded to the Galaxy platform (https://usegalaxy.org/), quality-checked with FastQC, and trimmed using Trim Galore. Reads were mapped to the C. difficile R20291 genome (accession no. FN545816.1) using BWA-MEM2. Read counts were obtained with htseq-count, and differential expression analysis was performed using edgeR in Degust (http://degust.erc.monash.edu/) with cutoffs of log_2_ fold change ≥ 1 and false discovery rate (FDR) ≤ 0.01. Because hcpR is inactivated in the hcpR::Tn strain, residual reads mapping to its sequence were detected by RNA-seq. Although hcpR appeared in the DEG output, it was excluded from downstream analyses. A volcano plot was generated using VolcaNoseR (https://huygens.science.uva.nl/VolcaNoseR/), and a heatmap was created using Morpheus (https://software.broadinstitute.org/morpheus/). GSEA with KEGG pathway was performed using ClusterProfiler, with significance defined as adjusted P < 0.05 and gene sets classified as upregulated (NES > 0) or downregulated (NES < 0) (55). A relaxed threshold of q ≤ 0.25 was also used. KEGG metabolic pathway analysis of the DEGs was performed using KEGG Mapper via KofamKOALA.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Di Bella S, Sanson G, Monticelli J, Zerbato V, Principe L, Giuffrè M, Pipitone G, Luzzati R. 2024. Clostridioides difficile infection: history, epidemiology, risk factors, prevention, clinical manifestations, treatment, and future options. Clin Microbiol Rev 37:e 0013523. doi:10.1128/cmr.00135-2338421181 PMC 11324037 · doi ↗ · pubmed ↗
- 2Kint N, Alves Feliciano C, Martins MC, Morvan C, Fernandes SF, Folgosa F, Dupuy B, Texeira M, Martin-Verstraete I. 2020. How the anaerobic enteropathogen Clostridioides difficile tolerates low O 2 tensions. m Bio 11:e 01559–20. doi:10.1128/m Bio.02678-2032900801 PMC 7482061 · doi ↗ · pubmed ↗
- 3Wetzel D, Mc Bride SM. 2020. The Impact of p H on Clostridioides difficile sporulation and physiology. Appl Environ Microbiol 86:e 02706–19. doi:10.1128/AEM.02706-1931811041 PMC 6997743 · doi ↗ · pubmed ↗
- 4Marshall A, Mc Grath JW, Graham R, Mc Mullan G. 2023. Food for thought-The link between Clostridioides difficile metabolism and pathogenesis. P Lo S Pathog 19:e 1011034. doi:10.1371/journal.ppat.101103436602960 PMC 9815643 · doi ↗ · pubmed ↗
- 5Hastie JL, Hanna PC, Carlson PE. 2018. Transcriptional response of Clostridium difficile to low iron conditions. Pathog Dis 76:fty 009. doi:10.1093/femspd/fty 00929390127 PMC 6251574 · doi ↗ · pubmed ↗
- 6Nibbering B, Gerding DN, Kuijper EJ, Zwittink RD, Smits WK. 2021. Host Immune responses to Clostridioides difficile: toxins and beyond. Front Microbiol 12:804949. doi:10.3389/fmicb.2021.80494934992590 PMC 8724541 · doi ↗ · pubmed ↗
- 7Abt MC, Mc Kenney PT, Pamer EG. 2016. Clostridium difficile colitis: pathogenesis and host defence. Nat Rev Microbiol 14:609–620. doi:10.1038/nrmicro.2016.10827573580 PMC 5109054 · doi ↗ · pubmed ↗
- 8Bogdan C, Röllinghoff M, Diefenbach A. 2000. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr Opin Immunol 12:64–76. doi:10.1016/s 0952-7915(99)00052-710679404 · doi ↗ · pubmed ↗