Survey of bacteria associated with septic arthritis in beef feedlot cattle
Daniel Kos, Brian Warr, Danae M. Suchan, Danilo Wadt, Jennifer N. Russell, Mallory Norfield, Jenny Liang, Murray Jelinski, Andrew D. S. Cameron, Antonio Ruzzini

TL;DR
This study surveys bacteria in septic and healthy joints of beef cattle, finding that Mycoplasmopsis bovis is the most common cause of septic arthritis, along with other bacteria and antimicrobial resistance.
Contribution
The study introduces enhanced metagenomics via CapSeq to better detect pathogens and antimicrobial resistance in septic arthritis.
Findings
Mycoplasmopsis bovis was the most frequently detected pathogen in septic joints.
CapSeq revealed resistance determinants missed by conventional methods, including macrolide resistance in M. bovis and oxytetracycline resistance in T. pyogenes.
Polymicrobial infections and complex communities were observed in septic joints.
Abstract
Septic arthritis (SA) is a cause of lameness in cattle attributed to bacterial infections. Mycoplasmopsis bovis is the best known and characterized etiological agent of SA; however, cases caused by diverse bacteria have been reported. Accordingly, we surveyed bacteria associated with septic and healthy joints from animals in western Canadian feedlots. Microbial community profiling showed that M. bovis was the most frequently detected pathogen in septic joints, followed by Metamycoplasma alkalescens and Trueperella pyogenes. In most cases, disease was ostensibly caused by a single pathogen, though polymicrobial infections and complex communities were also observed in DNA isolated from septic joints. The application of enhanced metagenomics by target DNA hybridization capture sequencing (CapSeq) provided more robust pathogen detection and characterization. CapSeq revealed resistance…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
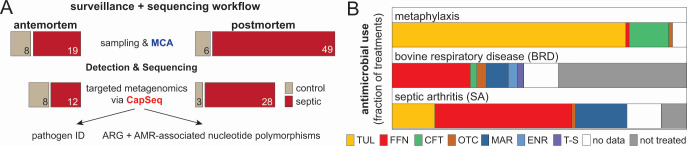 Fig 1
Fig 1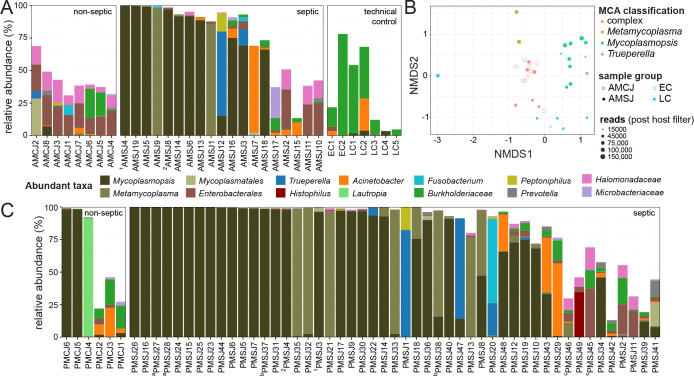 Fig 2
Fig 2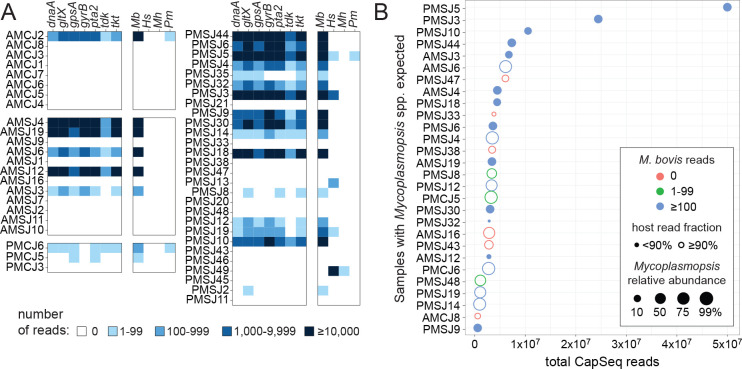 Fig 3
Fig 3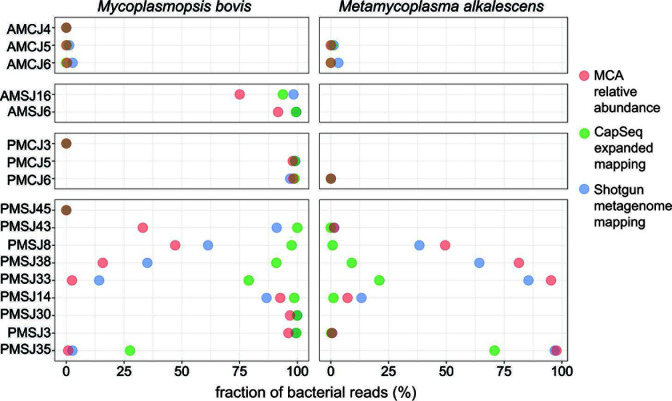 Fig 4
Fig 4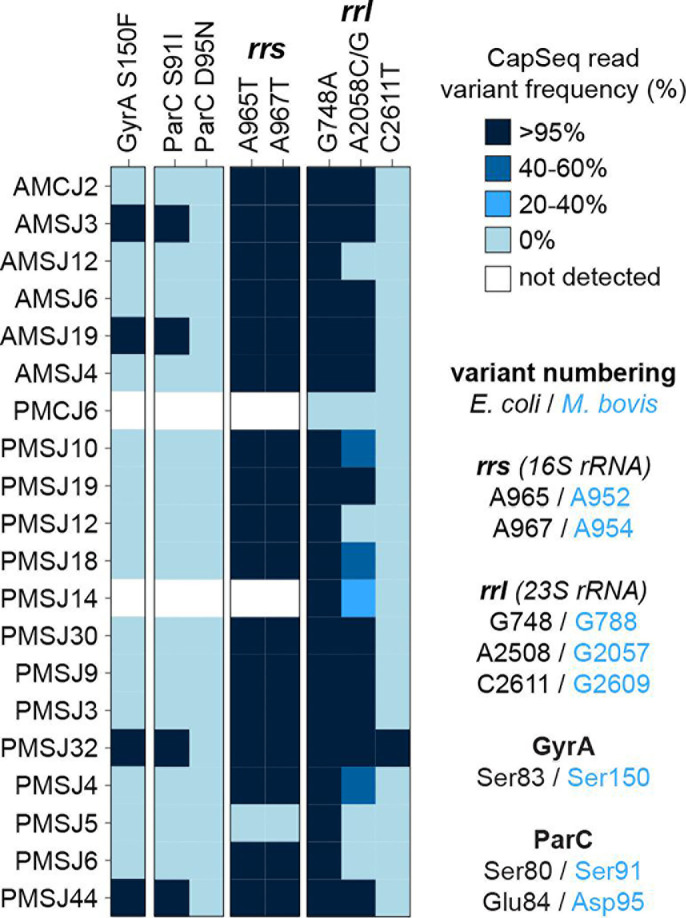 Fig 5
Fig 5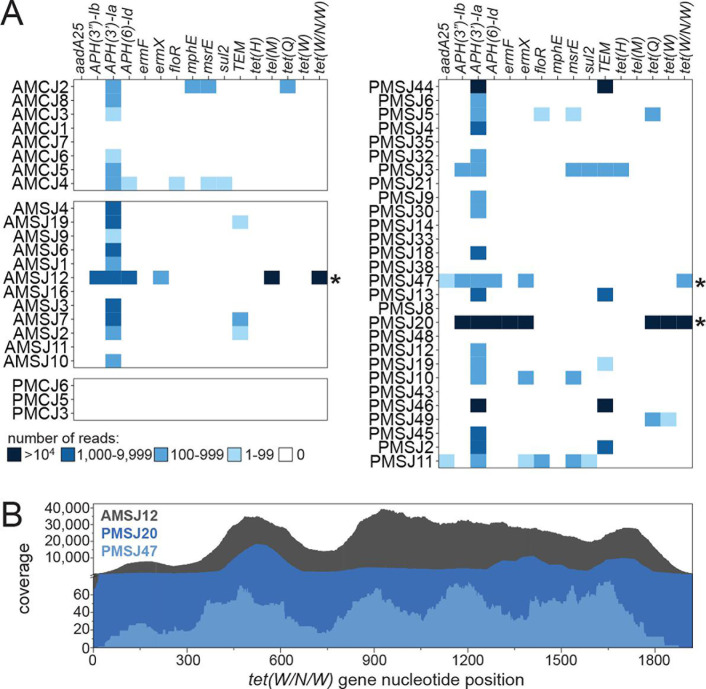 Fig 6
Fig 6 Fig 7
Fig 7| Isolate | MIC (μg/mL) of: | AMR genotype | ST | GenBank no. | |||
|---|---|---|---|---|---|---|---|
| ENR | FFN | OTC | Macrolides | ||||
|
| |||||||
| AMSJ16 | ≤0.25 | 2 | 2 | >256 | 239 |
| |
| AMSJ16-2 | ≤0.25 | 2 | 4 | >256 | |||
| AMSJ16-3 | ≤0.25 | 4 | 4 | >256 | |||
| AMSJ16-4 | ≤0.25 | 2 | 2 | >256 | |||
| AMSJ16-5 | ≤0.25 | 4 | 4 | >256 | |||
| PMSJ14 | ≤0.25 | 4 | 4 | >256 | 239 |
| |
| PMSJ14-2 | ≤0.25 | 4 | 4 | >256 | |||
| PMCJ5 | 4 | 8 | 4 | >256 | 293 |
| |
| PMCJ5-2 | 4 | 16 | 4 | >256 | 295 |
| |
| PMSJ48 | 8 | 4 | ≤2 | >256 | 60 |
| |
| PMSJ48-2 | 8 | 4 | ≤2 | >256 | |||
|
| |||||||
| PMSJ35 | ≤0.25 | 8 | ≤2 | >256 |
| ||
- —Saskatchewan Agriculture Development Fund
- —Saskatchewan Cattle Association
- —Natural Sciences and Engineering Research Council of Canadahttp://dx.doi.org/10.13039/501100000038
- —Beef Cattle Research Council of Canada
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial infections and disease research · Animal Behavior and Welfare Studies · Animal health and immunology
INTRODUCTION
Septic arthritis (SA) is broadly defined as an infectious disease of joints associated with inflammation. SA falls under the clinical umbrella of animal lameness and, in the beef production system, it occurs most frequently in calves that enter feedlots in fall (1). Although SA is not the principal lameness diagnosis, it has the highest lameness case fatality rate. In live animals, confirmation of a SA diagnosis relies on arthrocentesis. Synovial fluid collection and pathogen cultivation are considered the gold standard for SA diagnosis; however, positive culture rates for the disease are generally low (1, 2). More rapid tests of the synovial fluid can also lead to the diagnosis and differentiation of SA from non-infectious arthritis. Typically, SA diagnoses are based on the joint fluid having a change in viscosity, an increase in protein levels, and the presence of white blood cells (3, 4). Technological advancements to DNA-sequencing workflows, mass spectrometry, and antigen microarray have been proposed but not extensively evaluated for SA diagnoses (5). Thus, current treatment of SA in cattle relies on empirical medicine based on limited diagnostic and epidemiological data.
Mycoplasmopsis bovis (formerly Mycoplasma bovis) has been the most frequently reported cause of SA in cattle. Experimental infections via intraarticular or intravenous injections confirmed that M. bovis can cause SA (6, 7), and the organism has been identified in septic joints through direct isolation (6–10) and indirect assay (11–13). Additional causative agents and joint-associated bacteria have been identified through cultivation efforts from both beef and dairy animals. These include Histophilus somni (14), Mannheimia haemolytica, Trueperella pyogenes (2, 8), Streptococcus spp. (2), Helcococcus ovis (15), and Metamycoplasma alkalescens (16). Notably, M. bovis, M. haemolytica, H. somni, and T. pyogenes also participate in bovine respiratory disease (BRD), which is the most common infectious disease of cattle that is managed by antimicrobial use (AMU). Significantly, most cattle that develop SA do so subsequent to or concurrent with BRD (2, 17). In light of prior or overlapping needs to treat BRD and a relatively small number of reports on pathogens from different food production systems, antimicrobial treatment of SA cases varies between beef and dairy cattle. In feedlot cattle, the parenteral administration of antibiotics that generally demonstrate efficacy against M. bovis and H. somni alongside nonsteroidal anti-inflammatory drugs (NSAIDs) for pain management is recommended (18). In dairy animals, coagulase-negative streptococci are considered probable causative agents of disease when determining treatment strategies (2). An improved understanding of the presumptive etiological agents of SA and their expected antimicrobial susceptibilities could better inform AMU.
We undertook a survey of the causative agents of SA in western Canadian feedlots by sampling live and dead animals diagnosed with the disease. Microbial community profiles and two distinct metagenomic approaches were employed for pathogen surveillance. In particular, targeted metagenomic analyses by DNA capture sequencing (CapSeq) provided improved pathogen detection and broadened our understanding of clinically relevant antimicrobial resistance (AMR) therein. This combined microbial community and metagenomic analysis highlighted the prevalence of well- and less-appreciated bacterial pathogens that contribute to SA. Simple infections by M. bovis, M. alkalescens, T. pyogenes, and H. somni were observed. In the case of M. bovis and M. alkalescens, isolation, whole-genome sequencing, and phenotypic assays supported cultivation-independent observations of AMR genotypes. In addition to simple infections, mixed infections or complex bacterial communities were found in septic joints. The results call attention to underappreciated and potentially unreported causes of SA that require further study.
RESULTS AND DISCUSSION
Overview of sample collection
Ten feedlots in western Canada provided an opportunity to study cattle, resulting in a total of 82 joint samples from 68 SA and 14 control cases (Fig. 1A; Table S1). Antemortem septic and control joints (AMSJ, n = 19 and AMCJ, n = 8) as well as postmortem septic and control joints (PMSJ, n = 49 and PMCJ, n = 6) were sampled. In two cases, joint samples were obtained before and after death from the same animals and, in three cases, two infected joints from animals with polyarthritis were sampled. Sampling was conducted in the winter months (December–February) on calves that were placed in the fall. A veterinarian’s diagnosis guided sampling of presumptive septic and control joints from healthy animals or asymptomatic joints of culled individuals. SA can arise from a penetrating injury or as hematogenous infection: animals with grossly identifiable penetrating injuries were excluded from this study. Feedlot treatment histories were provided for most cases, indicating that at least 32 of the 63 animals suffering from SA that were sampled also had a history of BRD (Fig. 1C; Table S1). Tulathromycin (TUL) was the most frequently administered metaphylactic agent (≥80% of animals) to help manage BRD during the study, whereas florfenicol (FFN), oxytetracycline (OTC), ceftiofur, enrofloxacin, marbofloxacin (MAR), or trimethoprim/sulfadoxine were employed as individual treatments for respiratory disease. SA cases were reported to have been treated with FFN, MAR, OTC, or TUL. Data on the use of in-feed antimicrobials (e.g., OTC and tylosin) were not collected but is a common industry practice.
Overview of surveillance and characterization of septic arthritis cases in western Canada. (A) Samples and surveillance-based sequencing workflow highlighting the capacity to detect and characterize pathogens and AMR determinants via targeted metagenomics. (B) Stacked bar charts summarizing AMU data provided for animals that were part of the study. AMU included tulathromycin (TUL), florfenicol (FFN), ceftiofur (CFT), oxytetracycline (OTC), marbofloxacin (MAR), enrofloxacin (ENR), and trimethoprim-sulfadoxine (T-S).
Simple or complex bacterial community profiles are found in animals suffering from SA
To catalog contributors to SA, DNA isolated from septic and control joints was used to identify bacteria by partial 16S rRNA gene amplicon sequencing (Fig. 2). Microbial community analysis (MCA) revealed Mycoplasmopsis to be the most frequently SA-associated genus in both antemortem and postmortem septic joints based on relative bacterial abundance (Fig. 2A and C; Table S2). In 31 of 60 Mycoplasmopsis-containing samples, the taxon accounted for >90% of the observed bacterial community, including 24 instances in which ≥95% of the reads were assigned to the genus. In two cases, ante- and postmortem sampling was performed from the same joint. AMSJ4 and PMSJ3 were collected from one animal’s joint and AMSJ8 and PMSJ4 from another; the bacterial communities for all four samples were dominated by Mycoplasmopsis (Fig. 2). Of the 68 septic joint samples, Mycoplasmopsis co-occurred with other known causes of SA: Metamycoplasma (18 samples), Trueperella (5 samples), and Histophilus (1 sample). In other SA cases with limited to undetectable levels of Mycoplasmopsis, three genera, Metamycoplasma, Trueperella, and Acinetobacter, were observed as the most abundant taxon. Acinetobacter spp. were also detected in technical control samples, suggesting it may be an artifact; however, experimental samples (e.g., AMCJ and AMSJ) differed significantly from each other as well as from extraction and sequencing controls with respect to the compositions of their microbial communities (Fig. 2B). In contrast to diseased joints, the communities observed from healthy animals were more complex, with the exception of two postmortem controls dominated by Mycoplasmopsis. While no signs of SA were noted in these two animals, the presence of Mycoplasmopsis may reflect hematogenous spread from common sites of their residence, treatments with ceftiofur (a β-lactam to which Mycoplasmopsis are intrinsically resistant), and/or postmortem population expansion otherwise controlled in live healthy animal joints. M. bovis has also previously been isolated from cattle joints without evidence of arthritis (19). Generally, the survey demonstrated that Mycoplasmopsis, presumably M. bovis, is the most prevalent cause of SA in western Canadian feedlot cattle and that Metamycoplasmopsis, presumably M. alkalescens, is an often-overlooked contributor to this disease, appearing in around 20% of SA cases in this study and a recently completed UK-based survey (20). In fact, Metamycoplasmopsis accounted for >95% of the assigned taxa in five samples in which the detection of Mycoplasmopsis was limited (≤2.5%) or undetected.
Microbial community analysis from cattle with septic arthritis. (A) Bar plots showing abundant members of bacterial communities identified in antemortem control (left; AMCJ), septic joint (middle; AMSJ), and technical control samples (right). (B) Ordination plot showing non-metric multidimensional scaling of bacterial communities identified in AMCJ, AMSJ, and control samples. Dots are scaled to post-host filtered read count, colored by MCA classification, and either filled or unfilled to distinguish sample groups. (C) Bar plots showing abundant members of bacterial communities identified in postmortem control (left; PMCJ) and septic joints (right; PMSJ). All bar plots are arranged by increasing Shannon index values (Table S2) and show abundant taxonomic groups across all samples as well as known pathogenic groups, including Histophilus and Trueperella. Superscripts indicate samples taken from the same joint (numbers) or different joints (letters) from the same animal.
Septic joint samples were found to contain a range from simple to complex bacterial communities. Individual α-diversity values, measured using the Shannon diversity index (H′), ranged between 0 (e.g., all Mycoplasmopsis) and 4.0 for clinical and control samples (Table S2). Notably, the α-diversity values for a subset of five clinical samples (AMSJs; H′ = 2.72–3.30) were comparable to those of the eight live control animals (AMCJs; H′ = 2.49–4.00). A total of 619 bacterial taxa were identified in the 27 antemortem and 7 technical control samples with the aforementioned subset of complex clinical cases resembling healthy controls (Fig. 2B). Thus, whereas a primary agent or select few could explain disease in 14 of 19 live animals suffering from SA, about a quarter of cases were not simply correlated to known or presumptive disease-causing agents. Misdiagnoses may explain these cases; however, our analysis is blind to the potential contributions of non-bacterial infectious agents that have yet to be studied in the context of beef cattle SA. Finally, an improved or whole joint sampling strategy on postmortem animals might also help to explain these results as organisms associated with the synovial lining, for example, which might be underrepresented or more challenging to detect in the fluid samples. In the future, unbiased microbial isolation efforts may help to reveal and implicate additional causative agents that cannot be reliably predicted by DNA-based methods. To this end, postmortem sampling of two joints from the same afflicted animal revealed similar and distinct microbial communities. In two of three cases, similar communities were observed whether simple (PMSJ27/28) or complex (PMSJ45/46). In a third case, joints differed in that the communities were characterized as being dominated by either Mycoplasmopsis or a mix of Metamycoplasma and Mycoplasmopsis (PMSJ37/38).
CapSeq revealed the presence of antimicrobial-resistant M. bovis within joints
To gain more insight into the nature of the Mycoplasmopsis infections, we performed specific DNA target capture and sequencing experiments on a subset of joint samples (n = 51). The workflow, which was recently applied to feedlot water and clinical samples, includes seven-gene multilocus sequencing typing (MLST) schemes for M. bovis, H. somni, M. haemolytica, and Pasteurella multocida (21). Detection of the targeted Pasteurellaceae was sporadic, occurring in only 4 of 55 joints (AMCJ2, PMCJ6, PMSJ5, and PMSJ49; Fig. 3A, right panels). H. somni detection in PMSJ49 was expected based on the relative abundance of the genus observed by MCA (34%). In contrast to the dearth of Pasteurelleceae in the data set, we expected 28 samples to contain Mycoplasmopsis DNA. Presumptive detection of M. bovis was possible for only 22 of these 28 samples using CapSeq (Fig. 3A). In the other six (AMCJ8, AMSJ16, PMSJ33, PMSJ38, PMSJ43, and PMSJ47), discordance between the MCA and CapSeq data sets appeared to be multifactorial (Fig. 3B; Table S3). Total read count, relative abundance of Mycoplasmopsis, and the host read abundance appear to impact M. bovis detection by CapSeq. For example, all samples expected to contain but devoid of M. bovis reads by CapSeq were high in bovid DNA content (host read fraction ≥ 90%) and some also possessed relatively low read counts and/or relative abundance of the target genes based on the MCA. In contrast, we also noted the presence of M. bovis in an unexpected sample (AMCJ2), explained by misannotation to a close relative of Mycoplasmopsis during MCA (Table S2; taxid ID: Eperythrozoon). Overall, the CapSeq data sets not only supported the involvement of M. bovis in SA, they also provided a snapshot of prevalent sequence types (STs; Table S3). In general, a single M. bovis ST could be identified per joint, including ST2, ST16, ST60, ST65, and ST80. One animal (PMSJ10) appeared to have suffered from a mixed M. bovis infection (Table S3).
Detection of specific cattle-associated pathogens using CapSeq. (A) Heatmap showing the detection for M. bovis (Mb), H. somni (Hs), M. haemolytica (Mh), and P. multocida (Pm) based on seven-gene MLST schemes. The seven-gene scheme used for M. bovis is shown in the left panels with shades of blue coloring indicating numbers of reads associated with each target. The right panels show the combined counts for M. bovis as well as those for the other three pathogens’ MLST schemes. (B) Dot plot comparing Mycoplasmopsis detection via MCA and CapSeq analysis. Samples with Mycoplasmopsis spp. are plotted against the total number of CapSeq reads from each sample. The relative abundance (%; only samples ≥1% are shown) from MCA is scaled by dot size and the read fraction attributed to B. taurus is depicted using open or closed dots (90% cutoff). Coloring is used to highlight detection of M. bovis via CapSeq.
Discordance observed between the MCA and MLST-based CapSeq detection pipeline prompted investigation beyond the MLST targets. An alternative read-mapping strategy to a custom database that included the B. taurus genome and genomes of known cattle pathogens was employed. We rationalized that the CapSeq probes, which were designed to enrich for additional M. bovis genes (21), would provide greater insight (Table S4). In parallel, we performed conventional shotgun metagenomic sequencing on a subset of samples (n = 16) and used the same custom database for M. bovis detection. In contrast to the MLST strategy, read mapping to B. taurus and a library of full bacterial genome sequences revealed concordance between the data sets and showed that CapSeq outperformed the shotgun approach for M. bovis detection (Fig. 4). With the expanded genome mapping approach, M. bovis was readily detected in samples characterized by low or no detection using the seven-gene MLST approach (e.g., PMSJ8, PMSJ33, PMSJ35, and AMSJ16). Bacterial reads assigned to rrs (16S rRNA) and rrl (23S rRNA) loci typically outnumbered assignment to all seven MLST targets combined (Table S3) as well as other specific M. bovis target (Table S4). These additional loci also afforded detection of M. alkalescens by CapSeq (Fig. 4). Thus, although M. alkalescens targets were not included in the experimental design, relatively high sequence identity with M. bovis (e.g., 85% for rrs and rrl genes) was sufficient for capture and provided support for the presence of M. alkalescens in septic joints. Finally, it should be noted that the dominance of ribosomal targets in captured sequence space provides a generalizable example of the utility of including multicopy universal target sequences in more specific analyses, but this can come with the negative consequence of diluting capture and sequencing of other informative loci.
A comparison of M. bovis and M. alkalescens detection via three different sequencing approaches. Dot plot showing the fraction (%) of M. bovis (left) and M. alkalescens (right)-assigned reads from microbial community analysis (pink), CapSeq (green), and shotgun metagenomic data sets (blue).
To exploit sequencing beyond identification and typing, the M. bovis CapSeq probes were designed to investigate nucleotide polymorphisms implicated in AMR (21). M. bovis typically evades the action of antibiotics through adaptive mutation to their targets. This includes mutations to the rrs and rrl genes as well as mutations in quinolone resistance-determining regions (QRDRs) of gyrase (e.g., gyrA and gyrB) and topoisomerase (e.g., parC and parE) genes (22, 23). Indeed, CapSeq afforded the identification of a series of nucleotide polymorphisms in M. bovis rrs, rrl, gyrA, and parC genes (Fig. 5). Specifically, the majority of rrs sequences contained A965T and A967T mutations (Escherichia coli numbering convention), which is correlated to reduced susceptibility to tetracyclines in M. bovis (24). All M. bovis rrl sequences contained the G748A polymorphism, which confers resistance to macrolides, and the majority of sequences also contained at least one other nucleotide polymorphism, A2057G/C or C2611T, associated with macrolide resistance (25–27). In four instances, co-occurrent mutations to the QRDR encoded within gyrA and parC (S83F and S80I; E. coli numbering) were observed. Accordingly, all SA-associated M. bovis are predicted to be macrolide resistant, less susceptible to the action of tetracyclines, and a subset with GyrA and ParC variants is predicted to be resistant to fluoroquinolones. The observations of these genotypes are in line with known AMU: macrolides and tetracyclines have long served as conventional treatments and cattle feed additives, whereas fluoroquinolones (e.g., enrofloxacin and marbofloxacin) are employed to explicitly treat disease (Table S1).
Detection of M. bovis determinants of AMR via CapSeq. Heatmap showing genes with polymorphisms associated with AMR in M. bovis. Protein variants of GyrA and ParC are shown using M. bovis numbering, whereas nucleotide variants in the rrs and rrl genes are listed following the E. coli convention. For clarity, equivalent sites of mutations between M. bovis and E. coli are listed at the right.
CapSeq suggests the presence of AMR T. pyogenes in septic joints
The CapSeq panel included probes for additional ARGs known to be encoded by other cattle pathogens. Overall, detection of these ARGs was relatively limited (Fig. 6A), consistent with the prevalence and abundance of organisms known to lack them (M. bovis and M. alkalescens). For example, the floR gene, which encodes for a FFN efflux pump, was detected in only three joint samples despite FFN use by at least 8 of the 10 feedlots during the study (Table S1). Nevertheless, CapSeq detection of ARGs from this clinical material greatly outperformed a conventional shotgun metagenomic approach, which failed at reliable detection of resistance determinants (Table S5). Accordingly, this approach could be implemented to better address pathogen and AMR detection in future investigations of agricultural systems, matching similar efforts in human medicine (28, 29).
Detection of ARGs by CapSeq. (A) A focused summary of the results of ARG detection in joint DNA samples highlighting clinically relevant and commonly observed genes in the feedlot environment. Asterisks denote the detection of a T. pyogenes-specific ARG. (B) Mapping of CapSeq reads across the tet(W/N/W) gene observed in samples AMSJ12, PMSJ20, and PMSJ47.
The utility of the CapSeq approach was highlighted by specific ARG detection suggesting the presence of OTC-resistant T. pyogenes in septic joints. Coincidental detection of tet(W/N/W) by CapSeq and T. pyogenes by MCA was remarkable. The tet(W/N/W) gene is nearly confined to T. pyogenes and often co-occurs with ermX (30–32) as observed in the SA joint samples (Fig. 6A), and was detected in three samples expected to possess the organism via MCA: AMSJ12, PMSJ20, and PMSJ47 (Fig. 6B). Few other bacterial species encode for the tet(W/N/W) gene (based on nucleotide sequence identity >98%), though many related ribosomal protection protein-encoding genes exist (e.g., tetW). Consensus nucleotide sequences for reads mapped to tet(W/N/W) from AMSJ12 and PMSJ47 data sets were identical to the reference sequence (GenBank: AY049983.2, 8,309…10,228) whereas that constructed for PMSJ20 was 98.9% identical. Thus, the sequence identity of the tet(W/N/W) reads observed in the joint samples suggested that they originated from T. pyogenes. The identification of tetracycline-resistance gene encoding sequences is consistent with a recent report that ~60% T. pyogenes isolated from cattle carry the tet(W/N/W) gene (30) as well as the in-feed delivery of OTC and its specific use for SA treatments.
Cultivation from SA joints corroborates detection of antimicrobial-resistant bacteria
To complement the DNA-based detection of AMR, M. bovis were isolated from a subset of control and septic joints. A total of 11 M. bovis isolates were subjected to antimicrobial susceptibility tests (ASTs; Table 1), 5 of which were characterized using whole-genome sequencing (Table 1; Table S6). We focused on M. bovis since it was the major pathogen found in SA cases and can be readily isolated in the laboratory through the use of selective media. A panel of feedlot relevant antibiotics was used to determine minimum inhibitory concentrations (MICs) in pleuropneumonia-like organism (PPLO) broth. All joint isolates were determined to be resistant to both 15- and 16-atom-containing macrolides (tulathromycin, gamithromycin, tylosin, and tildipirosin; MICs > 256 μg/mL) whereas only a subset demonstrated elevated MICs for other classes of antibiotics. Overall, the ranges of measured MICs were similar to previous reports. MIC_50_ values for ENR, FFN, and tetracyclines, for example, were reported as 0.25, 2, and 2 μg/mL, respectively, from isolates originating from Germany, Canada, and Australia (33–35). In particular, M. bovis from PMSJ14, PMSJ48, and PMCJ5 was capable of growing in the presence of 4–8 μg/mL ENR. This phenotype was explained by GyrA S150F and ParC S91I or D95N variants in the QRDR (M. bovis numbering). MICs ≥4 μg/mL were also observed for FFN and OTC; whereas reduced susceptibility to FFN is not well understood at a molecular level in M. bovis, OTC resistance can be explained by the rrs A952T and A954T mutations (E. coli numbering). In most cases, the AMR genotypes were also observed in CapSeq data; however, we noted that isolates from a control joint, M. bovis PMCJ5 and PMCJ5-2, belonged to previously unreported STs (ST293 and ST295; Table S6) and carried mutations that were not dominant in CapSeq data sets (e.g., rrl C2611G and ParC D95N). The differences in M. bovis abilities to be cultivated in the laboratory may or may not reflect their true abundance in septic joints. Furthermore, many clinical joint samples were heterogeneous in nature, showing signs of inflammation and requiring physical disruption (e.g., mincing with sterile scissors thereby dividing the material) during sample preparation for DNA and cultivation, which might explain differences in cultivation-dependent and independent methods.
In addition to M. bovis, M. alkalescens has been reported to cause SA. Isolation of M. alkalescens from symptomatic dairy cattle and intraarticular, but not intravenous, inoculations of healthy animals demonstrated it as the cause of SA nearly five decades ago (16). In this first report, which documented ~30 SA cases in a herd of 215 animals, the disease was suspected to be linked to umbilical exposure with M. alkalescens-induced omphaloarteritis preceding SA. Additional reports have implicated M. alkalescens as a cause of SA (20, 36), respiratory disease (37), mastitis (38), and abortion (39) in cattle. Here, Metamycoplasma was the second most abundant SA-associated genus, and we isolated M. alkalencens from a sample expected to be enriched in the genus (PMSJ35; Fig. 1B; 98% relative abundance). MICs for the PMSJ35 isolate were within the reported ranges for other cattle-associated M. alkalescens (40, 41). Similar to M. bovis, M. alkalescens PMSJ35 was resistant to macrolides, and whole-genome sequencing revealed that it contained the rrl G748A polymorphism correlated to macrolide resistance. The isolate was also characterized by phenotypic sensitivities to OTC and ENR while showing reduced susceptibility to FFN. Notably, there are only two other reported M. alkalescens genomes currently available: a closed genome for the type strain NCTC 10135 (ATCC 2910), which was isolated from a nasal cavity (42) and a draft genome of M. alkalescens 14918, which was isolated from lung tissue from a calf that suffered from pneumonia (43). The M. alkalescens PMSJ35 closed circular 767,707 bp genome is ~57 kb smaller than the NCTC 10135 sequence with which it shares 98.5% average nucleotide identity (ANI; Fig. 7). Future studies should focus on M. alkalescens isolation and characterization considering the relatively limited information that is available for this species, which is frequently present in SA cases.
Comparison of M. alkalescens genomes. Linearized M. alkalescens genome maps showing 233 orthologous matching segments used to determine ANI between the PMSJ35 isolate and type strain.
Improving and implementing surveys and characterizations of SA in cattle
This DNA-focused survey of causative agents of SA in feedlot cattle confirmed the prominent role of M. bovis and revealed the involvement of underappreciated taxa in this seriously debilitating disease. Importantly, the identification of M. alkalescens in samples taken from seven different feedlots implicates it as a common contributor to SA in western Canada (Table S1). Moreover, T. pyogenes, including AMR genotypes, were identified more frequently and at higher relative abundance than H. somni, which is anecdotally thought of as a prominent cause of SA in the region. Finally, Acinetobacter spp. was routinely detected in SA samples, including as the dominant taxon in AMSJ7, co-infecting with M. bovis in PMSJ48, and as a member of more complex communities like PMSJ15 (Fig. 2; Table S2). Whether or not Acinetobacter spp. contribute directly to disease remains to be determined. More broadly, how complex communities that lack known causative agents of SA generate inflammation remains a significant epidemiological and molecular question. Ultimately, a better understanding of SA cases is likely to help guide the empirical medicine that is used to treat animals suffering from this disease.
MATERIALS AND METHODS
Animals, case selection, and sampling
All participating feedlots were typical of the western Canadian industry, receiving diets formulated to meet or exceed National Research Council standards (44), and included monensin, an ionophore, at 22–33 ppm based on dry matter. AMU to manage disease, including BRD and SA, occurred in treatment facilities (hospital pens) where veterinarians confirmed the case definitions and enrolled animals for sampling. The treatment histories of animals, collection year, and additional metadata (e.g., anonymized feedlot identifiers) were recorded when possible (Table S1). Samples collected in 2021 were immediately mixed 1:1 with RNAlater, whereas those collected in 2022 were not. All samples were frozen at −20°C at a veterinary clinic before transportation in the frozen state to a research laboratory, where they were stored at −80°C until further processing.
Live animals were sampled by arthrocentesis after a 15 cm^2^ area was clipped and scrubbed with 4% chlorhexidine gluconate and 70% rubbing alcohol solutions. Animals were also provided with a local anesthetic, lidocaine, before a veterinarian used a 3.8 cm needle to sample the joint capsule. Synovial fluid was aspirated into a syringe before animals were administered an oral dose of meloxicam, an NSAID, at 1 mg/kg BW. While a 14- or 16-gage needle was required for animals suffering from SA, fluid samples from healthy joints were retrieved using 18- and 20-gage needles. For the presumptive SA cases, animals had at least one swollen joint, no signs of lesions, and no previous history of lameness. Control animals were selected from the same pens as infected animals.
Dead animal samples, cases, and controls were identified by evening postmortem exams conducted by a feedlot veterinarian. SA cases were grossly identified by joint swelling, whereas postmortem control cases did not show joint swelling but succumbed to another illness. A clean knife was used to reflect the skin from the joint with manual traction applied to reduce contact and potential contamination of the subcutaneous tissue over the joint. A 14- or 16-gage needle was then inserted into the joint capsule and synovial fluid aspirated into a syringe. All cases were samples <24 h from time of death. All animals, live or dead, were fall-placed calves sampled in early winter.
Extraction of DNA from joint samples
DNA was extracted from joint fluid samples using two protocols: a phenol:chloroform extraction of sedimented materials or a Qiagen Blood & Tissue kit. Samples, in particular those from SA cases, were cut up using sterile surgical scissors in an aseptic environment. These minced samples were weighed to ~25 mg to comply with kit specification before extraction and pooling as necessary to meet yield requirements for DNA sequencing experiments. In nearly all instances, DNA obtained by phenol:chloroform extraction was re-purified using an AMPure XP magnetic bead kit (Beckman Coulter Inc, CA, USA). Technical controls were performed alongside DNA extractions and sequencing library preparations (see below) using common kits and reagents.
Microbial community profiling
PCRs (30 cycles) were performed with Q5 High-Fidelity 2X Master Mix (New England Biolabs, MA, USA) containing 2 ng DNA and 1 μM 515F/806R 16S V4 rRNA primers as previously described (45). Amplicon library clean-up was completed using AMPureXP magnetic beads. An index PCR (10 cycles) was performed with Q5 High-Fidelity 2X Master Mix and i5 and i7 indices following the Nextera XT Index Kit v2 (Illumina, San Diego, CA, USA) specification. Indexed libraries, purified using AMPureXP magnetic beads, were pooled and size-selected for a ~440 bp band using an E-Gel Size Select 2% gel (Invitrogen, CA, USA). The size-selected library was quantified with the NEBNext Library Quant Kit for Illumina (New England Biolabs) and diluted to 1.6 pM for sequencing. Paired-end (2 × 150 bp) sequences were generated on either an Illumina NovaSeq 6000 or Miniseq platform (300 cycles).
Sequencing reads that mapped to a Bos taurus reference genome (ARS-UCD2.0; GCF_002263795.3) were removed from data sets using a local alignment with Bowtie2 (v2.5.1; --no-mixed --no-discordant [46]). TrimGalore (v0.6.10) was utilized with the Nextera and polyA tags to remove adaptors (47) from the reads before they were imported into Qiime2 (Amplicon, v2024.5) and additional filtering of reads with homopolymeric sequences >8 bp was removed using rescript cull-seqs (48, 49). Amplicon sequence variants were generated with DADA2 with minimum overlap set to 7 bp (50), and classified using the Greengenes2 (v2024.09) database (51). Qiime2 results were imported into R using qiime2R (52). After calculating proportional abundance through dividing abundance of taxa by the sum abundance within each sample, Shannon diversity index (H) was calculated with the formula H = −∑(proportion × ln(proportion)). Genera were considered detected when they made up ≥1% of the total abundance classified.
Metagenomic sequencing
CapSeq sequencing libraries with a DNA probe set targeting BRD pathogens were prepared as previously described (21), following technical specifications provided by the DNA probe manufacturer (Twist Biosciences, San Francisco, CA, USA). Briefly, following library preparations and quantification, the libraries were pooled for hybridization with a Twist kit (Twist Biosciences, 104180) with the following thermal cycling conditions: 95°C for 5 min and 60°C for 30 min, and post-hybridization PCR amplification was performed after which they were cleaned. CapSeq libraries underwent pair-ended (2 × 150 bp) sequencing on an Illumina NovaSeq6000 platform. DNA libraries for shotgun sequencing were prepared using TruSeq DNA PCR-Free Kit (Illumina) followed by pair-ended (2 × 150 bp) sequencing on one lane of a NovaSeqX Series 10B flow cell.
Gene detection from metagenomic data sets
Initial read processing for both the CapSeq data (n = 51) and the shotgun metagenomic (n = 17) data sets started with using fastp to (v0.23.4) (53) trim illumina adaptors with a minimum output length of 50 bp. Bowtie2 was used to remove host reads as described above for the 16S rDNA amplified data set. Reads were classified with Kraken2 (2.1.3) (54) with a NCBI RefSeq database constructed out of available complete genomes for B. taurus, M. bovis, M. alkalescens, M. haemolytica, P. multocida, H. somni, B. trehalosi, T. pyogenes, M. wenyonii, and U. diversum (data fetched May 2025). MLST scheme genes (55) and genes with AMR associated polymorphisms for M. bovis, M. haemolytica, P. multocida, and H. somni were used as references for mapping with processed sequencing data using BBMap (BBTools v39.08; local alignment, 95% identity, kiflter = 25, subflter = 20, maxindel = 3, insfilter = 0, and delfilter = 0) (56). Consensus MLST gene sequences (cutoffs: >95% conservation across all positions, ≥100 mapped reads) were queried using the PubMLST sequence typing tool (pubmlst.org) to identify M. bovis STs.
For ARG detection from CapSeq data sets, the resistance gene identifier (RGI, bwt, v6.0.3, bowtie2 aligner) from the Comprehensive Antibiotic Resistance Database (CARD, v4.0.0) was employed (57). Positive ARG detection was considered if ≥100 mapped reads/gene was observed along with ≥80% coverage. Shotgun metagenomic data were analyzed using the RGI tool and a more specific search for selected genes (see Table S5) was performed using BBSplit (BBTools v39.08) (56) as described above. To detect and quantify nucleotide polymorphisms in targeted ARGs, only data sets that included ≥100 reads at the genes of interest were considered.
Isolation of M. bovis and M. alkalescens
The isolations of M. bovis and M. alkalescens were guided by results of the 16S rRNA community profiling. A 20 μL volume of a joint fluid from samples showing “simple” infection types was used to inoculate 2 mL of PPLO broth enriched with 10% yeast extract (wt/vol), 10% horse serum (vol/vol), penicillin G (10,000 IU), and thallium acetate (0.0005% wt/vol; abbreviated hereafter as PPLO broth/agar). The inoculated broth was incubated at 37°C in a humidified 5% CO_2_ environment for 7 days. Then, a 50 μL aliquot was plated on PPLO agar and incubated at 37°C in a humidified 5% CO_2_ environment for 3–5 days before individual colonies were selected for further propagation and cryopreservation.
DNA isolation, sequencing, and analysis of M. bovis and M. alkalescens genomes
A PureLink Genomic DNA Mini Kit (Invitrogen) was employed to isolate DNA from 5-day-old cultures. DNA sequencing was performed using Oxford Nanopore Technologies by Plasmidsaurus Inc. (South San Francisco, CA, USA). Amplification-free long-read sequencing libraries were constructed using v14 library chemistry and then sequenced using an R10.4.1 flow cell. High-quality 45–125× coverage genomes were assembled by downsampling data sets. Briefly, read removal was performed by Filtlong v0.2.1 (58) and preliminary assemblies were constructed using Miniasm v0.3 (59) before further downsampling, weighting against low-quality reads. Flye v2.9.1 (60) and Medaka v1.8.0 (61) were then used to generate the final, polished assemblies. Nucleotide polymorphisms in AMR determining genes were identified by sequence alignment and manual inspection. Pairwise genome comparisons of M. alkalescens were performed using FastANI (62) within the Proksee platform (63).
Determination of antimicrobial susceptibilities
Antimicrobial susceptibility testing was performed using PPLO broth amended with alamarBlue (ThermoFisher) at 37°C in a humidified 5% CO_2_ environment. For M. alkalescens, the PPLO broth was supplemented with 21.25 mg/L pyruvate. Bacteria were prepared at 10^3^ CFU/mL from single-use glycerol stocks of known viable M. bovis or M. alkalescens counts and arrayed into 200 μL volumes in 96-well microtiter plates containing twofold dilution series of ENR, FFN, OTC, GAM, TUL, TYL, TIL, and TIP. MICs were assigned to conditions in which no bacterial growth was detected using alamarBlue (ThermoFisher) fluorescence as a proxy. Three independent measurements were performed for each strain. Positive (no antibiotic) and negative (no bacteria) controls were performed in parallel.
Data visualization
Plots were routinely generated using R, Microsoft Excel, Origin v2024b, and then re-organized and re-colored using Adobe Illustrator to generate multi-panel figures.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Desrochers A, Francoz D. 2014. Clinical management of septic arthritis in cattle. Vet Clin North Am Food Anim Pract 30:177–203, doi:10.1016/j.cvfa.2013.11.00624534665 · doi ↗ · pubmed ↗
- 2Constant C, Nichols S, Desrochers A, Babkine M, Fecteau G, Lardé H, Fairbrother J-H, Francoz D. 2018. Clinical findings and diagnostic test results for calves with septic arthritis: 64 cases (2009–2014). J Am Vet Med Assoc 252:995–1005. doi:10.2460/javma.252.8.99529595396 · doi ↗ · pubmed ↗
- 3Rohde C, Anderson DE, Desrochers A, St-Jean G, Hull BL, Rings DM. 2000. Synovial fluid analysis in cattle: a review of 130 cases. Vet Surg 29:341–346. doi:10.1053/jvet.2000.560510917284 · doi ↗ · pubmed ↗
- 4Weaver AD. 1972. Disease of the bovine stifle joint. Bov pract 7:41–45. doi:10.21423/bovine-vol 1972 no 7p 41-45 · doi ↗
- 5Pearson GB, Ysebaert MP, Papa B, Reesink HL. 2023. Synovial sepsis diagnostics and antimicrobial resistance: a one-health perspective. J Am Vet Med Assoc 261:1115–1120. doi:10.2460/javma.23.05.022737380157 · doi ↗ · pubmed ↗
- 6Stalheim OH, Stone SS. 1975. Isolation and identification of Mycoplasma agalactiae subsp. bovis from arthritic cattle in Iowa and Nebraska. J Clin Microbiol 2:169–172. doi:10.1128/jcm.2.3.169-172.19751176624 PMC 274165 · doi ↗ · pubmed ↗
- 7Stalheim OH, Page LA. 1975. Naturally occurring and experimentally induced mycoplasmal arthritis of cattle. J Clin Microbiol 2:165–168. doi:10.1128/jcm.2.3.165-168.19751176623 PMC 274164 · doi ↗ · pubmed ↗
- 8Anholt RM, Klima C, Allan N, Matheson-Bird H, Schatz C, Ajitkumar P, Otto SJ, Peters D, Schmid K, Olson M, Mc Allister T, Ralston B. 2017. Antimicrobial susceptibility of bacteria that cause bovine respiratory disease complex in Alberta, Canada. Front Vet Sci 4:207. doi:10.3389/fvets.2017.0020729255716 PMC 5723070 · doi ↗ · pubmed ↗