Insights into the chloroplast genome diversity of the genus Isatis in China
Min Wei, Chengxiang Wang, Yong Su, Hongzhuan Shi, Liangju Ma, Tao Bao, Qiaosheng Guo

TL;DR
This study analyzes chloroplast genomes of seven Isatis species in China to resolve taxonomic confusion and identify useful genetic markers.
Contribution
The study provides new chloroplast genome data and identifies the rpl32–trnL spacer as a novel marker for species identification in Isatis.
Findings
Chloroplast genomes of seven Isatis species were sequenced and analyzed for structural and sequence variation.
The rpl32–trnL intergenic spacer was found to be the most polymorphic region and a promising molecular marker.
Phylogenomic analysis confirmed the monophyly of Isatis and revealed misidentifications in public databases.
Abstract
The genus Isatis contains medicinally important but taxonomically controversial species in China. Reliable genomic resources are urgently needed for accurate species identification and phylogenetic clarification. We assembled and characterized the complete chloroplast genomes of seven Isatis species. The genomes (153,260–153,872 bp) exhibited the typical quadripartite structure. Sequence variation was heterogeneous: the small single-copy (SSC) region was the most polymorphic (12.3 single-nucleotide polymorphisms (SNPs)/kb), followed by the large single-copy (LSC, 8.9 SNPs/kb) and inverted repeat (IR, 1.7 SNPs/kb) regions. Noncoding sequences showed 3.2-fold greater polymorphism than coding sequences. The rpl32–trnL intergenic spacer was identified as a promising molecular marker due to its high nucleotide diversity (π = 0.0582). While most protein-coding genes were under strong…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1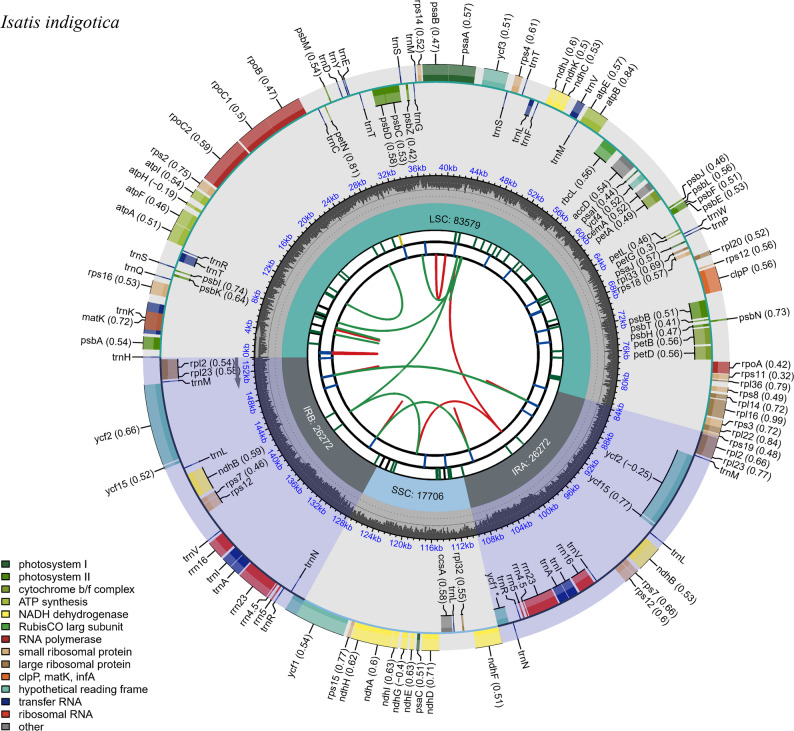 Figure 2
Figure 2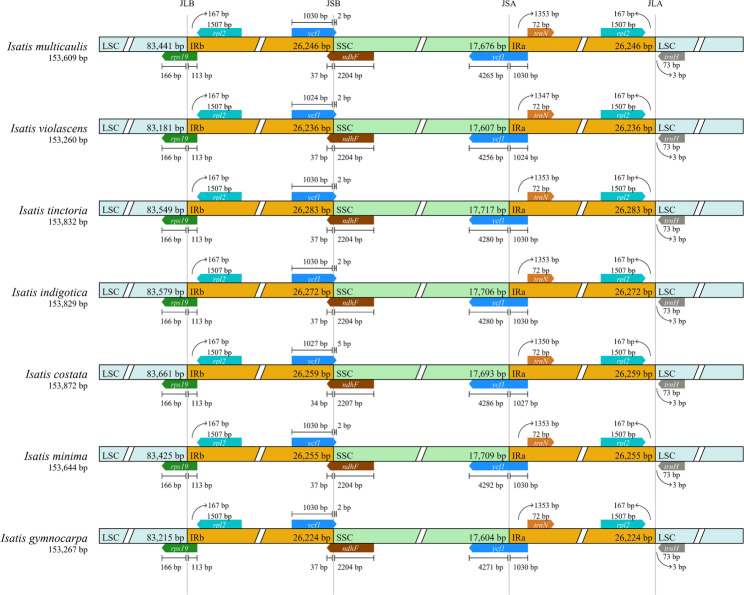 Figure 3
Figure 3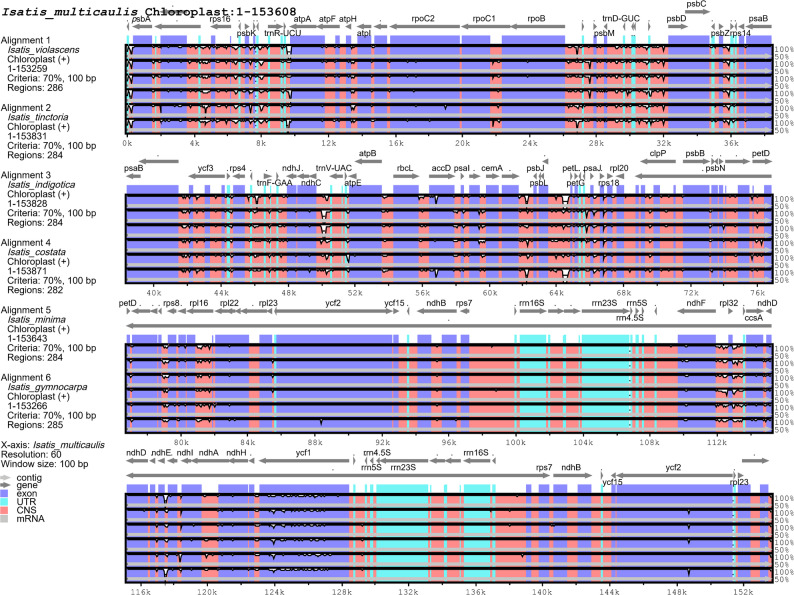 Figure 4
Figure 4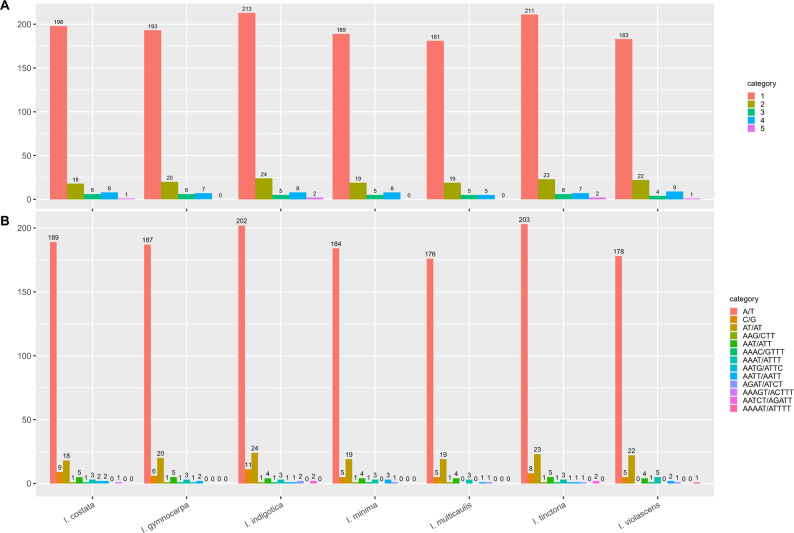 Figure 5
Figure 5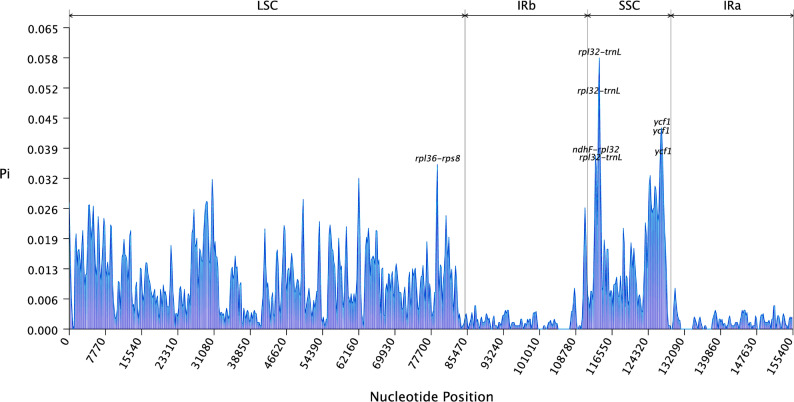 Figure 6
Figure 6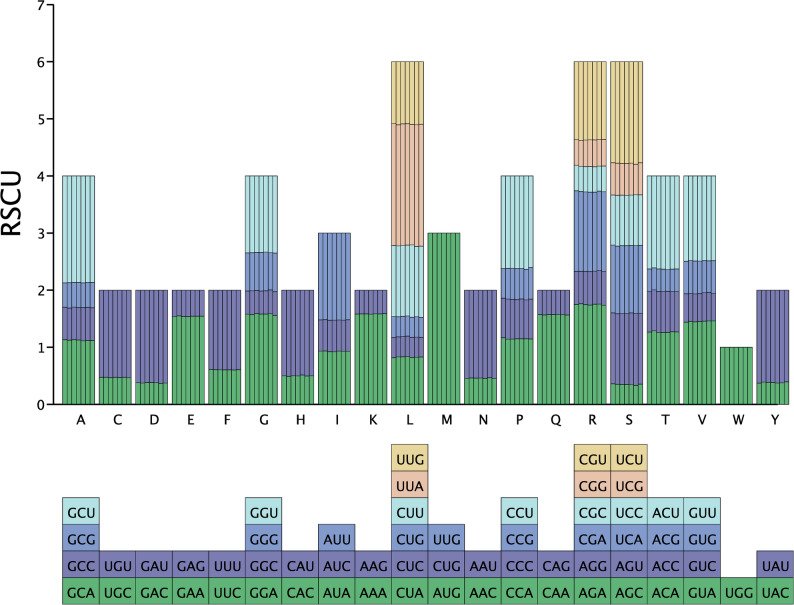 Figure 7
Figure 7 Figure 8
Figure 8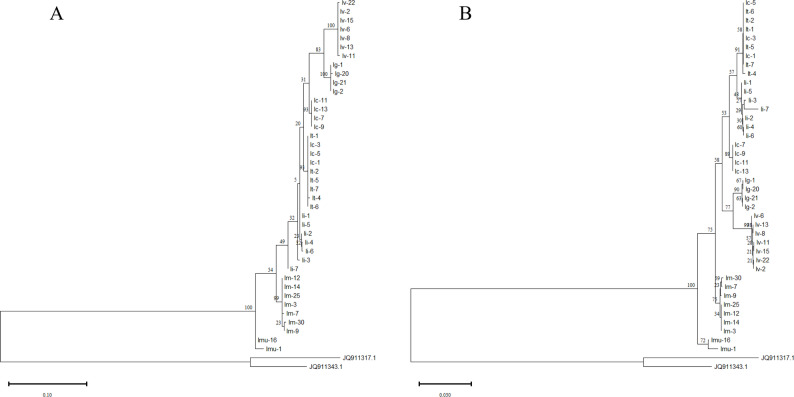 Figure 9
Figure 9- —the National Key Research and Development Program of China (Project Title: Spatiotemporal Analysis of the Quality Formation of Chinese Herbal Medicines and Demonstration of Pseudocultivation Research;
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Marine Sponges and Natural Products · Plant Diversity and Evolution
Introduction
The genus Isatis Tourn. ex L. comprises a group of plants within the family Brassicaceae Burnett that hold significant medicinal and economic value [1]. Widely distributed across Eurasia and the Mediterranean region, several species—such as Isatis cappadocica Desv. and Isatis tinctoria L.—serve as natural resources for the dye [2], cosmetics [3, 4], and pharmaceutical industries [5, 6]. However, long-standing controversies regarding the taxonomy and phylogenetic relationships within this genus have considerably hindered the accurate identification and quality control of medicinal materials. First, inconsistencies exist among authoritative taxonomic references: of the six species recorded in the Flora of China [7], two, Isatis multicaulis (Kar. & Kir.) Jafri and Isatis gymnocarpa (Fisch. ex DC.) Al-Shehbaz, Moazzeni & Mumm., were transferred from the neighbouring genera Pachypterygium Bunge and Tauscheria Fisch. ex DC., respectively. In contrast, the Chinese Pharmacopoeia recognizes only Isatis indigotica Fortune ex Lindl. as the official source of “Isatidis Radix” [8], whereas major taxonomic treatments such as the Flora of China regard I. indigotica as a synonym of I. tinctoria L [7]. This taxonomic controversy, combined with the high morphological similarity and overlapping distributions of many Isatis species, poses considerable challenges for traditional identification methods. Although attempts have been made to develop chloroplast mini-barcodes for distinguishing I. indigotica from I. tinctoria, their applicability and effectiveness across a broader range of Isatis species remain to be thoroughly evaluated [9, 10].
Resolving these taxonomic controversies requires a well-characterized phylogenetic framework for Isatis. In recent years, a new classification system has divided the subfamily Brassicoideae into several supertribes, with the tribe Isatideae placed within the supertribe Brassicodae [11–13]. Nevertheless, the evolutionary position of Isatis within this system and the validity of previously proposed genus transfers based on morphology and nuclear genes (e.g., I. multicaulis and I. gymnocarpa) still require verification using independent data such as chloroplast genomes [14, 15]. Moreover, existing studies on interspecific relationships within the genus—particularly among morphologically and ecologically similar species such as Isatis minima Bunge and Isatis violascens Bunge—have relied largely on single-marker approaches [16, 17]. These studies provide limited phylogenetic information, making it difficult to discern whether these phenotypic similarities are due to close relationships or convergent evolution.
Given its compact structure, sequence conservation, and high information content, the chloroplast genome has emerged as a powerful tool for resolving phylogenetic relationships among closely related taxa and for developing highly discriminatory molecular markers [18–20]. To systematically address the aforementioned issues in the taxonomy and phylogeny of Isatis, this study employs Illumina high-throughput sequencing to sequence and compare the chloroplast genomes of seven representative species: I. tinctoria, Isatis costata C.A. Mey., I. minima, I. violascens, I. multicaulis, I. gymnocarpa, and I. indigotica. The aims are to assess and develop new chloroplast DNA barcodes suitable for species identification across the entire genus, to clarify the phylogenetic placement and monophyly of Isatis within the higher-level classification of Brassicaceae, and to evaluate the consistency of previous genus-level reclassifications based on nuclear data with chloroplast genome evidence. These findings provide a solid genomic foundation for the conservation of Isatis resources, accurate identification of medicinal materials, and further study of the evolutionary history of this genus.
Materials and methods
Plant materials
The plant materials utilized in this study are comprehensively summarized and illustrated in Fig. 1. A total of seven Isatis species were collected from their natural habitats. Specifically, I. indigotica was sampled from Huangshi City, Hubei Province, while the other six species (I. tinctoria, I. costata, I. minima, I. violascens, I. multicaulis, and I. gymnocarpa) were collected from various locations within the Xinjiang Uygur Autonomous Region, China. All the collected specimens were authenticated by Professor Qiaosheng Guo of Nanjing Agricultural University. Voucher specimens for each of the seven species are preserved in the research collection of the Institute of Chinese Medicinal Materials at Nanjing Agricultural University, Nanjing, China. For each sample, healthy young leaves were selected and rapidly dried in silica gel until DNA extraction. Detailed collection information, including geographical coordinates, is provided in Table S1.
Fig. 1. Morphological characteristics of seven Isatis species. The samples are labeled as follows: **A ** I. indigotica, **B ** I. costata, **C ** I. tinctoria, **D ** I. violascens, **E ** I. minima, **F ** I. gymnocarpa, **G ** I. multicaulis. Scale bar: 2 cm (applicable to A-G)
To construct a robust phylogenetic framework, we supplemented our data with all available complete chloroplast genome sequences of Isatis and related taxa retrieved from the NCBI Organelle Genome Database [4, 9, 21]. The final dataset encompasses sequences from congeneric species, representatives from closely related genera within the tribe Isatideae, and outgroup species from other tribes of Brassicaceae. The accession numbers, sources, and corresponding references for all the downloaded sequences are also listed in Table S1.
DNA extraction, sequencing, assembly, and annotation
Genomic DNA was extracted from leaf tissues using a Plant Genomic DNA Extraction Kit (Dp360; Tiangen Biotech, Beijing, China). High-throughput sequencing was performed on the BGISEQ-T7 platform (BGI-Shenzhen, China), generating approximately 7.1–9.0 Gb of raw data per sample. The raw reads were subsequently used for chloroplast genome assembly without prior quality filtering, as is standard practice for organelle genome assembly due to its uneven base composition [22]. The quality of the raw data was high, with Q20 and Q30 scores ranging from 95.7 to 98.5% and 89.3–95.9%, respectively.
The chloroplast genomes were assembled de novo using GetOrganelle (v1.7.7.1) [23] with the -F embplant_pt parameter. The assembly quality was rigorously assessed by (1) confirming the complete circularization of the genome; (2) verifying the presence of the typical quadripartite structure, consisting of a large single-copy (LSC), a small single-copy (SSC), and two inverted repeat (IR) regions; and (3) leveraging the inherent high coverage depth resulting from the high copy number of chloroplast DNA.
Annotation was performed using a dual-strategy approach: (1) structural annotation with GeSeq to identify IR regions and determine genome orientation [24] and (2) functional annotation with CPGAVAS2 for gene prediction [25]. Manual curation was subsequently applied to refine gene boundaries and intron/exon structures, and genome maps were generated using OGDRAW (v1.3.1) [26]. The complete chloroplast genome sequences were submitted to the NCBI database, and GenBank accession numbers were obtained.
Comparative genomic analysis and identification of divergent hotspots
The chloroplast genomes of seven Isatis species were compared using the mVISTA online tool (https://genome.lbl.gov/vista/mvista/) in Shuffle-LAGAN mode, with the I. multicaulis (GenBank: PQ059879) chloroplast genome serving as the reference. The expansion and contraction of the IR and SC region boundaries were visualized using IRscope (https://irscope.shinyapps.io/irapp/).
To identify mutation hotspots, the nucleotide diversity (π) across the aligned chloroplast genomes was calculated using DnaSP software (v6.12.03) [27]. The analysis was performed with a sliding window of 400 bp and a step size of 200 bp, using the I. multicaulis genome as the alignment reference. The region with the highest π value, the rpl32–trnL intergenic spacer, was identified as the most divergent hotspot.
Repeat sequence analysis
Simple sequence repeats (SSRs) were identified using the MISA tool (https://webblast.ipk-gatersleben.de/misa/), with minimum repeat thresholds set to 8, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, penta-, and hexanucleotides, respectively, and a minimum distance of 100 bp between adjacent SSRs. Dispersed repeats were detected using REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer/manual.html) [28], with the following parameters: maximum number of repeats = 50; minimum repeat length = 30; Hamming distance = 3; and four repeat types, including forward (f), reverse (r), complement (c), and palindromic (p).
Codon usage bias and selective pressure analysis
Relative synonymous codon usage (RSCU) was calculated for each sample using DAMBE software (v7.0.35), with redundant genes excluded and non-ATG start codons corrected during amino acid editing [29]. To investigate the patterns of natural selection, we determined the pairwise Ka/Ks ratios for all coding sequences (CDSs) using TBtools (v2.056). This analysis allowed for an assessment of selective pressure across the Isatis sample set [30].
Phylogenetic analysis
To ensure robust phylogenetic inference, we analysed 26 Isatis chloroplast genomes, incorporating all publicly available sequences from NCBI along with seven newly sequenced accessions. Sisymbrium altissimum L. (Sisymbrieae, Brassicodae) was selected as the outgroup for stable rooting. The ingroup consisted of 26 Isatis sequences to resolve infrageneric relationships, supplemented by three representatives from related Isatideae genera (Myagrum perfoliatum L., Schimpera arabica Hochst. & Steud., and Conringia planisiliqua Fisch. & C. A. Mey. (a synonym of Iljinskaea planisiliqua (Fisch. & C.A.Mey.) Al-Shehbaz, Özüdoğru & D.A. German)) to test monophyly. Goldbachia laevigata (M. Bieb.) DC. (Calepineae) was included as a contextual reference within supertribe Brassicodae [13].
Phylogenetic analyses were conducted using both maximum likelihood (ML) and Bayesian inference (BI) methods. The ML analysis was performed with IQ-TREE 2 (version 2.4.0; https://github.com/iqtree/iqtree2) [31], with the best-fit substitution model (TVM + F + I + R4) selected automatically according to the Bayesian information criterion and branch support assessed with 1000 bootstrap replicates [32]. The BI analysis was performed using MrBayes (version 3.2.7a; http://mrbayes.sourceforge.net/) under the GTR + Γ model of sequence evolution. Two independent runs of four Markov chain Monte Carlo (MCMC) simulations each were conducted for 200,000 generations, sampling every 500 generations. The first 25% of the trees were discarded as burn-in. The phylogenetic trees were visualized and annotated using the Interactive Tree of Life (iTOL) (accessed on [29 Sep. 2025]; https://itol.embl.de/) [33].
Analysis of sequence divergence in rpl32–trnL
To validate the utility of the identified rpl32–trnL hotspot for species discrimination, we employed a previously published primer pair (Forward: 5’-ACCTTGATGCAATAATAAACAAAGA-3’; Reverse: 5’-AAAATGAAAACTTCTCCAAAATGC-3’) [9]. These primers were used to amplify and sequence the region from 40 samples (Table S2). PCR amplification was performed in a 50 µL reaction mixture containing 47 µL of Tsingke Golden Mix, 1 µL each of 10 µM forward and reverse primers, and 1 µL of genomic DNA. The thermal cycling protocol consisted of an initial denaturation at 98 °C for 2 min; 35 cycles of denaturation at 98 °C for 10 s, annealing at 54 °C for 10 s, and extension at 72 °C for 10 s; followed by a final extension at 72 °C for 5 min. The PCR amplicons were sequenced bidirectionally on an AB 3730Xl DNA Sequencer (Applied Biosystems, U.S.A.) by Tsingke Biological Co., Ltd.
To root the phylogenetic trees, the rpl32–trnL intergenic spacer sequences of two closely related species, Myagrum perfoliatum L. (GenBank: JQ911317.1) from the tribe Isatideae and Sisymbrium orientale L. (GenBank: JQ911343.1) from the tribe Sisymbrieae, were retrieved from NCBI and included in the alignment as outgroups. The raw bidirectional sequencing reads were assembled and base-called using CExpress to generate consensus sequences for each sample. All 40 consensus sequences of the rpl32–trnL intergenic spacer and 2 outgroups were aligned using the ClustalW algorithm implemented in MEGA version 11.0.13 with default parameters. The alignment was manually checked and trimmed.
Phylogenetic trees were reconstructed from this alignment using both ML and Neighbor-Joining (NJ) methods in MEGA 11.0.13. For the ML analysis, the Kimura 2-parameter model was employed, with rate heterogeneity among sites modeled using a discrete Gamma distribution (+ G, 5 categories). Branch support was assessed from 1,000 bootstrap replicates. For the NJ tree, pairwise distances were computed using the Maximum Composite Likelihood method under a uniform rates model, with gaps/missing data handled via pairwise deletion. Node support was also evaluated by 1,000 bootstrap replicates.
Results
General characteristics of the Isatis chloroplast genomes
The complete chloroplast genomes of seven Isatis species were successfully assembled, and their circular structures are illustrated in Fig. 2. The genome lengths ranged from 153,260 bp (I. violascens) to 153,872 bp (I. costata), with highly conserved GC contents ranging from 36.46 to 36.52%. Further comparison revealed that the maximum difference in length among the genomes was 612 bp, whereas the minimum difference was only 7 bp.
Fig. 2. Circular chloroplast genome map of Isatis species. The diagram comprises six concentric rings, each depicting distinct genomic features. From the innermost to outermost layers, these include spatially distributed repeats (forward orientation in red, reverse in green), tandem repeats (blue bars), short tandem repeats (green bars), the quadripartite organization (LSC, SSC, and paired IR regions labeled IRA/IRB) with annotated lengths, GC content gradients, and color-coded gene annotations. Numeric values in parentheses adjacent to gene labels reflect codon usage bias
Gene annotation revealed four functional categories: self-replication, photosynthesis, uncharacterized genes, and others (Table 1). Intron analysis revealed 17 genes with splicing elements: 15 single-intron genes (rpl2,* rpl16*,* rps16*,* rpoC1*,* trnA-UGC*,* trnE-UCC*,* trnK-UUU*,* trnL-UAA*,* trnV-UAC*,* trnT-UGU*,* ndhA*,* ndhB*,* petB*,* petD*, and atpF) and two double-intron genes (clpP and ycf3).
Table 1. Gene composition of the Chloroplast genomes in the genus IsatisCategoryGene groupGene namePhotosynthesisSubunits of photosystem IpsaA, psaB, psaC, psaI, psaJSubunits of photosystem IIpsbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZSubunits of NADH dehydrogenasendhA*, ndhB*(2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhKSubunits of cytochrome b/f complexpetA, petB*, petD*, petG, petL, petNSubunits of ATP synthaseatpA, atpB, atpE, atpF*, atpH, atpILarge subunit of rubiscorbcLSubunits photochlorophyllide reductase-Self-replicationProteins of large ribosomal subunitrpl14, rpl16*, rpl2*(2), rpl20, rpl22, rpl23(2), rpl32, rpl33, rpl36Proteins of small ribosomal subunitrps11, rps12**(2), rps14, rps15, rps16*, rps18, rps19, rps2, rps3, rps4, rps7(2), rps8Subunits of RNA polymeraserpoA, rpoB, rpoC1*, rpoC2Ribosomal RNAsrrn16S(2), rrn23S(2), rrn4.5(2), rrn5S(2)Transfer RNAstrnA-UGC*(2), trnC-GCA, trnD-GUC, trnE, trnF-GAA, trnG-GCC, trnH-GUG, trnI*(2), trnK-UUU*, trnL-CAA(2), trnL-UAA*, trnL-UAG, trnM-CAU(4), trnN-GUU(2), trnP-UGG, trnQ-UUG, trnR-ACG(2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-CGU*, trnT-GGU, trnT-UGU, trnV-GAC(2), trnV-UAC*, trnW-CCA, trnY-GUAOther genesMaturasematKProteaseclpPEnvelope membrane proteincemAAcetyl-CoA carboxylaseaccDc-type cytochrome synthesis geneccsATranslation initiation factor-Other-Genes of unknown functionConserved hypothetical chloroplast ORF#ycf1, ycf1, ycf15(2), ycf2(2), ycf3, ycf4Gene*: Gene with one intron; Gene**: Gene with two introns; #Gene: Pseudogene; Gene (2): Number of copies of multicopy genes
Comparative analysis of chloroplast genomes in Isatis
The size of the LSC region varies from 83,181 bp (I. violascens) to 83,661 bp (I. costata), with a maximum difference of 480 bp and a minimum difference of 16 bp. Additionally, the size of the SSC region ranged from 17,604 bp (I. gymnocarpa) to 17,717 bp (I. tinctoria), with a maximum difference of 113 bp and a minimum difference of 3 bp. The IR regions varied in size from 26,224 bp (I. gymnocarpa) to 26,283 bp (I. tinctoria), with a maximum difference of 59 bp and a minimum difference of 4 bp. A total of 132 genes were identified, including 37 tRNAs, 8 rRNAs, and 87 protein-coding genes (PCGs). Among these, 14 tRNAs and 8 rRNAs are located within the IR regions, as detailed in Table 2.
Analysis of the inverted repeat sequences revealed that the rps19 gene is located at the junction of the LSC and IRb regions. The ndhF and #ycf1 genes are located at the boundary of IRa and SSC, with the majority of the ndhF gene extending into the SSC region. The ycf1 gene is positioned at the junction of SSC and IRb, predominantly extending into the SSC region. The trnH gene is located at the junction of IRa and LSC, with a distance of 3 bp, whereas the rpl2 gene slightly contracts towards the IRa region. As shown in Fig. 3, the IR/SC junctions are nearly identical in length, with only minor expansions and contractions, indicating a high degree of similarity among the chloroplast genomes of the seven Isatis species.
Fig. 3. Comparison of the junction sites between the single-copy and IR regions in Isatis chloroplast genomes. The expansion and contraction of the IR boundaries are shown
The overall sequences of the chloroplast genomes of the seven Isatis species were mapped using mVISTA, with the annotation of I. gymnocarpa serving as the reference. These seven species demonstrate a high level of sequence similarity, with the IR regions being more conserved than the single-copy regions. Although coding regions generally exhibited higher conservation than noncoding regions did, we identified four genes with relatively high levels of variation as hypervariable regions: ycf1 (located at the SSC-IRA boundary), ycf2, ndhF, and rpoC2. This phenomenon may be related to the functional specificity of these genes and relaxed selective constraints. Furthermore, significant divergence was observed in noncoding intergenic spacers (IGSs), including highly variable regions near the trnF-GAA, trnV-UAC, petD, and rpl16 loci, as depicted in Fig. 4.
Fig. 4. Whole-genome alignment and sequence divergence analysis of the seven Isatis chloroplast genomes. The chloroplast genome of I. multicaulis was used as the reference. The x-axis represents the genome coordinates, and the y-axis shows the percent identity (50–100%) of the aligned regions to the reference
Table 2. Summary of the Chloroplast genome characteristics of 7 species of the Isatis genusSpeciesSize (bp)GenBankAccessionNo.GCContent(%)LSCLength(bp)SSCLength(bp)IRLength(bp)GeneNumberProteinCodingGeneNumberrRNA GeneNumbertRNA GeneNumber I. indigotica 153,829PQ15847436.4783,57917,70626,27213287837 I. costata 153,872PQ15847536.5183,66117,69326,25913287837 I. minima 153,644PQ09398536.4683,42517,70926,25513287837 I. violascens 153,260PQ15847236.5083,18117,60726,23613287837 I. gymnocarpa 153,267PQ09398636.5283,21517,60426,22413287837I. multicaulis153,609PQ05987936.4983,44117,67626,24613287837 I. tinctoria 153,832PQ15847336.4783,54917,71726,28313287837
SSR analysis of Chloroplast genomes in Isatis
In this study, MISA software was used to detect SSRs within the chloroplast genomes of Isatis species, and the results are detailed in Fig. 5. Analysis of SSRs in the chloroplast genomes of seven Isatis species revealed that the total number of SSRs ranged from 210 (I. multicaulis) to 252 (I. indigotica), as shown in Table 3. Mononucleotide repeats were the most abundant type across all species (ranging from 181 to 213), significantly outnumbering other repeat types. The numbers of dinucleotide, trinucleotide, and tetranucleotide repeats were relatively low and comparable, whereas pentanucleotide repeats were the rarest and were detected only in I. costata, I. indigotica, I. violascens, and I. tinctoria. Furthermore, the predominant A/T repeats were highly abundant (176 to 203), further highlighting a strong base composition bias in the SSR motifs. Notably, the A/T-only pentanucleotide repeat AAAAT/ATTTT was specifically found only in I. violascens, as detailed in the complete SSR dataset (Supplementary Material 3).
Fig. 5. Analysis of SSRs in Isatis chloroplast genomes. A Number of different SSR types. B Distribution of SSRs across LSC, SSC, and IR regions
Table 3. Distribution of SSRs in the Chloroplast genomes of seven Isatis speciesSpeciesTotalMononucleotideDinucleotideTrinucleotideTetranucleotidePentanucleotideA/T Repeats I. costata 23119818681189 I. gymnocarpa 22619320670187 I. indigotica 25221324582202 I. minima 22118919580184 I. multicaulis 21018119550176 I. violascens 21918322491178 I. tinctoria 24921123672203
Nucleotide diversity analysis of chloroplast genomes in Isatis
Nucleotide diversity analysis was conducted on 132 genes within the chloroplast genomes of Isatis species. As shown in Fig. 6, the average nucleotide variability (π) for the genus was 0.0083, ranging from 0.0000 to 0.0650. The rpl32–trnL region presented a high π value of 0.0582, suggesting its potential as a candidate marker for distinguishing Isatis species.
Fig. 6. Sliding window analysis of nucleotide diversity (π) in Isatis chloroplast genomes. The analysis was conducted with a window size of 400 bp and a step size of 200 bp. The x-axis represents the position in the chloroplast genome, and the y-axis shows the nucleotide diversity (π) value per window. Peak regions indicate highly variable loci. Key regions with high π values are labeled
Codon usage characteristics of Isatis chloroplast genomes
Codon distribution analysis revealed amino acid frequencies ranging from 1.63% (rare) to 9.38% (abundant). Three residues dominated the codon composition: leucine (Leu, 9.38%) preferentially used UUA, arginine (Arg) favoured AGA, and serine (Ser) predominantly employed UCU. Among the 20 amino acids, methionine (Met) and tryptophan (Trp) exclusively relied on the AUG and UGG codons, whereas the other amino acids presented 2–5 synonymous variants. Four-codon amino acids (Ala, Gly, Pro, Thr, and Val) showed distinct preferences for GCU, GGA, CCU, ACU, and GUA, respectively. Among its three synonymous options, isoleucine (Ile) demonstrated AUU dominance. Analysis of 61 codons revealed 35 with RSCU > 1 and 26 with RSCU < 1, indicating codon-ending preferences: 22.95% A/T, 50.82% C/G, and 26.23% U (Fig. 7).
Fig. 7. Codon usage bias in Isatis chloroplast protein-coding genes. The RSCU values for all 64 codons are shown. Codon families are color-coded by the encoded amino acid
Evolutionary constraints on chloroplast genome evolution
The Ka/Ks ratio (nonsynonymous/synonymous substitution rates) serves as an evolutionary pressure indicator, where ratios > 1 and < 1 indicate positive and purifying selection, respectively. All the examined Isatis species presented Ka/Ks values less than 0.4 across chloroplast genes (Table 4), revealing two predominant evolutionary patterns: (1) the universal presence of purifying selection and (2) strong functional conservation during angiosperm evolution. Notably, a few exceptions were observed; for instance, the rpoA gene exhibited the highest Ka/Ks value (2.36) in the comparison between I. costata and I. minima. This conservation pattern suggests the stringent elimination of deleterious mutations through natural selection.
Table 4. Genes under positive selection and genome-wide selection pressure in pairwise comparisons of Isatis speciesSpecies pairGene under positive selectionKa/Ks (gene)Genome-wide KaGenome-wide KsGenome-wide Ka/KsI. costata vs. I. minima rpoA 2.35500.11540.66490.1736I. multicaulis vs. I. gymnocarpa cemA 1.65440.25391.09770.2313I. indigotica vs. I. minima ccsA 1.40440.10460.44340.2360I. tinctoria vs. I. costata accD 1.40250.11130.55720.1998I. violascens vs. I. costata rps16 1.38020.18630.80580.2312I. violascens vs. I. gymnocarpa ndhG 1.29520.09050.34750.2605I. indigotica vs. I. costata ccsA 1.17630.10240.47550.2154I. multicaulis vs. I. violascens cemA 1.17440.21560.93410.2308I. multicaulis vs. I. tinctoriaNANA0.17910.82190.2179I. multicaulis vs. I. indigoticaNANA0.16860.77650.2172I. multicaulis vs. I. costataNANA0.18150.85170.2132I. multicaulis vs. I. minimaNANA0.15950.78760.2025I. violascens vs. I. tinctoriaNANA0.15510.64350.2410I. violascens vs. I. indigoticaNANA0.14620.61640.2371I. violascens vs. I. minimaNANA0.18630.80580.2312I. tinctoria vs. I. indigoticaNANA0.15510.64350.2410I. tinctoria vs. I. minimaNANA0.11390.52720.2161I. tinctoria vs. I. gymnocarpaNANA0.18090.92580.1954I. indigotica vs. I. gymnocarpaNANA0.17050.84820.2010I. costata vs. I. gymnocarpaNANA0.22050.97500.2262I. minima vs. I. gymnocarpaNANA0.19970.84850.2353The genome-wide values are averages across all protein-coding genes. NA indicates that no gene with a Ka/Ks > 1 was detected
Phylogenetic relationships based on chloroplast genome sequences
To elucidate the phylogenetic position of the genus Isatis within the Brassicodae supertribe and the relationships among its species, we constructed phylogenetic trees from chloroplast genome sequences using both ML and BI methods (Fig. 8A, B). The topologies of the ML and BI trees were highly congruent, with only minor differences in the placement of a few conspecific accessions. The phylogenetic analysis clearly resolved several major clades among the studied Brassicodae species. First, the tribe Isatideae was robustly supported as a monophyletic group with 100% bootstrap support. This tribe comprised a core Isatis clade, along with Myagrum perfoliatum, Schimpera arabica, and Iljinskaea planisiliqua, which together formed a highly supported (100% bootstrap support (BS)) monophyletic clade, thereby clarifying their systematic positions within Isatideae. The genus Isatis itself was also strongly supported as monophyletic (100% BS), indicating that the species studied here share a most recent common ancestor. Notably, I. gymnocarpa and I. multicaulis, which were previously transferred into Isatis, were robustly nested within the Isatis clade, confirming their current taxonomic placement within this genus. As expected, Sisymbrium altissimum from the tribe Sisymbrieae, used as the outgroup, was positioned outside the Isatideae clade. Furthermore, Goldbachia laevigata, representing another tribe within the Brassicodae supertribe, was clearly separated from the core clade (comprising Isatideae and Sisymbrieae). This further validates the reliability of our phylogenetic framework and provides a clear context for the circumscription of the Isatideae.
Fig. 8. Phylogenomic reconstruction of Isatis and the tribe Isatideae using whole chloroplast genomes. Numbers at branches indicate ML bootstrap values (left) and BI posterior probabilities (right). Sections are color-coded, and Sisymbrium altissimum was used as the outgroup. GenBank accession numbers are shown. For full taxonomic details (genus and tribe), see Table S1
Within Isatis, the BI tree confirmed the same complex interspecific relationships and phylogenetic structures as revealed by the ML analysis. The seven core species self-determined in this study and verified by morphology (I. indigotica, I. tinctoria, I. costata, I. minima, I. multicaulis, I. gymnocarpa, and I. violascens) were confirmed in their phylogenetic positions with high support, showing clear differentiation. Specifically, the self-determined sequences of I. violascens and I. gymnocarpa each formed independent, fully supported (100% BS) clades, affirming their status as distinct species entities. The self-determined I. multicaulis formed a distinct branch, indicating its unique genetic background. However, upon the inclusion of congeneric sequences from public databases (NCBI), clustering patterns that were inconsistent with their species labels were observed. For instance, the self-determined I. minima clustered in a clade with some NCBI sequences labelled as I. minima, as well as NCBI sequences labelled as I. violascens and I. oblongata. The self-determined I. costata clustered with one NCBI sequence labelled as I. tinctoria, whereas the self-determined I. tinctoria clustered with two NCBI sequences labelled as I. costata. The self-determined I. indigotica clustered into a large, highly supported clade with all NCBI-derived I. indigotica sequences, a subset of NCBI-derived I. tinctoria sequences, and I. cappadocica. These clustering results indicate that the genetic boundaries within Isatis for certain species (e.g., I. costata, I. tinctoria, and I. indigotica) may be more complex than those defined by traditional morphological taxonomy. Furthermore, an interesting phylogenetic structure was observed: I. minima and I. violascens, which are highly similar in morphology and distribution, did not form a direct sister clade. Instead, their respective clades were separated by those of I. gymnocarpa and I. multicaulis. This may be attributed to their similar habitats, which requires further investigation.
The rpl32–trnL intergenic spacer effectively discriminates most Isatis species
To evaluate the discriminatory power of the highly variable “rpl32–trnL” intergenic spacer—identified through chloroplast genome screening—at the species level, we sequenced 40 samples representing seven core species. Two outgroups (Myagrum perfoliatum and Sisymbrium orientale) were included, and phylogenetic trees were reconstructed using both ML and NJ methods.
The ML and NJ trees revealed largely congruent topologies regarding species-level relationships (Fig. 9A, B). The marker successfully delineated several species into highly supported monophyletic clades, including I. violascens (ML-BS = 100%), I. gymnocarpa (ML-BS = 100%), I. minima (ML-BS = 99%), I. multicaulis (ML-BS = 100%), and two distinct, highly supported lineages within the I. costata complex (both ML-BS = 93%). These results confirm that the “rpl32–trnL” spacer is highly polymorphic and possesses strong discriminatory power among most Isatis species.
Fig. 9. Phylogenetic analysis based on the rpl32–trnL intergenic spacer. The analysis included 40 Isatis accessions and two outgroup species (Myagrum perfoliatum and Sisymbrium orientale). Trees were reconstructed using (A) ML and (B) NJ methods. Numbers at branches represent bootstrap support values from ML (left) and NJ (right) analyses
The analysis also revealed complex relationships. Although all samples of I. indigotica formed a monophyletic cluster in both trees, internal branches received low support (BS < 60%). Furthermore, one lineage of the I. costata complex (comprising Ic-1, Ic-3, and Ic-5) was phylogenetically indistinguishable from all samples of I. tinctoria. In all analyses, the two outgroups were positioned at the outermost branches.
Discussion
Comparative analysis of chloroplast genomes in Isatis
Chloroplasts, the organelles responsible for photosynthesis and biosynthesis in photosynthetic organisms, provide organic compounds and energy essential for their life processes [34]. These semiautonomous organelles possess a closed circular double-stranded DNA structure and a chloroplast genome independent of the nuclear genome [35]. Characterized by structural stability, sequence conservation, abundant variable sites in intergenic regions, and a relatively slow molecular evolutionary rate, chloroplast genomes are widely utilized in studies of phylogenetic relationships, systematic evolution, and genetic diversity [36]. The chloroplast genomes of more than a thousand plant species have been sequenced to date. Among these, traditional Chinese medicinal (TCM) plants are being increasingly characterized, constituting a valuable resource for phylogenetic and evolutionary studies. The genomic data from TCM species such as Cistanche deserticola Y.C. Ma [37], Platycodon grandiflorus (Jacq.) A. DC [38]. , Polygala tenuifolia Willd [39]. , and Clematis henryi Oliv [40]. provide critical references, thereby facilitating future research on a broader range of species. These sequencing results have played a significant role in elucidating plant phylogenetic positions, species identification, and evolutionary analyses [41]. Investigating codon usage patterns in plant chloroplast genomes provides valuable data for enhancing the efficiency of gene expression vector construction, exploring species evolutionary relationships, understanding the molecular mechanisms of biological adaptation to the environment, and improving crop germplasm [9]. By constructing the chloroplast genomes of Isatis, sequence variations among different germplasms can be identified, enabling more precise species discrimination within the genus and resolving taxonomic controversies. Furthermore, this approach provides molecular markers and genetic resources for the genetic breeding of Isatis species, accelerating the process of varietal improvement.
Comparative analysis of the Isatis chloroplast genomes revealed a conserved quadripartite architecture with limited IR variation (< 2% length difference), in contrast to the relatively high divergence in single-copy regions (LSC/SSC: 5.8–7.4% boundary shifts). Sequence variation exhibited spatial gradients: SSC (12.3 single-nucleotide polymorphisms (SNPs)/kb) > LSC (8.9) > IRs (1.7) and functional partitioning, with noncoding regions showing 3.2× greater polymorphism than coding sequences (P < 0.001). Molecular signatures, including codon usage bias (RSCU = 1.15–1.82) and SSR distribution (42–47 loci/genome), followed core eudicot patterns. These findings collectively support a dual evolutionary model, namely, strong functional constraints maintaining IR/coding region stability versus neutral evolution driving diversification in single-copy/noncoding regions, which is consistent with genus-wide evolutionary trajectories.
Analysis of codon usage in the chloroplast genomes of Isatis
Codon usage in chloroplast genomes refers to the triplet nucleotide sequences encoding proteins within chloroplast DNA [42]. Although these codons are largely similar to those in the nuclear genome, some differences exist [43]. Codon usage can be analysed for preferences, selection pressures, and polymorphic sites [44]. In-depth studies on codon usage patterns and their influencing factors in plant chloroplast genomes provide a theoretical basis for vector selection in genetic engineering and gene expression [45]. Such analyses also aid in predicting the expression of unknown genes or identifying potential functional genes, which is highly important for studies on species evolution and genetics [46].
The chloroplast genome of Isatis exhibits adaptive codon usage bias characterized by A/U-ending preference (RSCU: 1.15–1.82) and low GC content at the third codon position (< 42%) [47], which aligns with conserved translational optimization strategies in higher plants such as Trigonella [48], Glehnia [49], and Sorghum [50]. This A/T-rich codon bias is correlated with elevated gene expression levels [10], reflecting genomic adaptation to environmental and evolutionary pressures [44]. Structural analysis revealed a conserved quadripartite architecture, with IRs showing < 2% length variation, in contrast with 5.8–7.4% boundary shifts in single-copy regions (LSC/SSC). These findings are consistent with the dual evolutionary model of functional constraint and neutral divergence.
Ka/Ks analysis across seven Isatis species revealed stringent purifying selection (mean < 0.3) in core photosynthesis-related genes, including photosystem components (psbD–Z), the cytochrome b/f complex (petB–N), and ATP synthase subunits (atpB–I), with near-zero substitution rates (P < 0.001). Conversely, uncharacterized genes (ycf1–2: 0.68–0.89) and protease-related loci (clpP, matK: 0.51–0.63) presented moderate positive selection signals. This functional dichotomy underscores stronger codon bias in highly expressed metabolic genes than adaptive flexibility in accessory genes, mirroring eudicot chloroplast evolutionary trajectories [44, 51].
Phylogenetic implications and taxonomic challenges
By constructing the first comprehensive phylogeny of Isatis based on complete chloroplast genomes, this study provides an independent and robust assessment of the evolutionary history of the genus, addressing several longstanding taxonomic questions. Our analysis not only confirms the monophyly of the tribe Isatideae and the genus Isatis with maximum support (100% BS) but also corroborates the phylogenetic placement of the tribe within the Brassicodae supertribe, aligning with and reinforcing previous findings from multilocus nuclear data [13]. This establishes a reliable evolutionary context for resolving relationships within this complex genus.
A key finding concerns the recent reclassification of I. gymnocarpa and I. multicaulis into Isatis. Their stable nesting within the core Isatis clade in our chloroplast genome phylogeny provides compelling independent evidence that validates these genus transfers, which were initially proposed based on morphological and limited nuclear evidence [14, 15, 52]. However, the infrageneric relationships revealed significant complexity. Although the seven morphologically verified core species formed well-supported, distinct clades, the inclusion of publicly available sequences revealed substantial discrepancies. We observed pronounced incongruence between molecular clustering and species labels for critical taxa such as I. costata, I. tinctoria, and I. indigotica. This pattern most parsimoniously points to widespread misidentification in public databases, an issue exacerbated by the morphological similarity within the genus. Although incomplete lineage sorting could contribute, the scale of the inconsistency strongly suggests that the current morphology-based species boundaries for these taxa are untenable and require critical re-evaluation.
The resolution offered by the complete chloroplast genome also enables the identification of highly variable regions suitable for DNA barcoding. Our discovery of the hypervariable rpl32–trnL region (π = 0.0582) aligns with and extends prior work, which identified the homologous region as an effective mini-barcode for distinguishing I. indigotica and I. tinctoria [9]. To empirically validate its discriminatory power, we conducted a phylogenetic analysis of 40 samples across the seven core species using this specific region. This analysis confirmed its effectiveness as a mini-barcode, robustly resolving most species into highly supported monophyletic clades. However, it also revealed a more complex pattern within I. costata, which was split into two highly supported lineages, one of which was inseparable from I. tinctoria. This incongruence with the species boundaries suggested by the chloroplast genome phylogeny points to a complex evolutionary history, potentially involving hybridization or chloroplast capture, that merits future investigation. This convergence of evidence underscores the dual utility of the rpl32–trnL region as both a practical identification tool and a probe for uncovering deeper evolutionary dynamics within the genus. Furthermore, our phylogeny offers insights into evolutionary processes beyond simple relationships. The observation that the morphologically similar I. minima and I. violascens do not form sister clades is particularly instructive. Given their shared dune habitat, this phylogenetic separation strongly implies that their phenotypic similarity is a result of convergent evolution driven by adaptation to analogous ecological pressures rather than a shared recent ancestry.
Conclusion
In this study, a multifaceted analysis of the chloroplast genomes in the genus Isatis was conducted through comparative genomics, codon usage bias, and phylogenetic analyses. The results demonstrate that the Isatis chloroplast genome adheres to the typical evolutionary patterns of core eudicots, which exhibit spatial heterogeneity in structural variation (highly conserved IR regions vs. significantly divergent single-copy regions) and sequence polymorphism (noncoding > coding regions), driven collectively by functional constraints and neutral evolution. Codon usage preference analysis further revealed an adaptive A/U-ending bias and strong purifying selection on core photosynthesis-related genes, collectively underscoring the evolutionary conservation of the genome. Phylogenomic analyses strongly supported the monophyly of the tribe Isatideae and the genus Isatis and confirmed the taxonomic placement of I. gymnocarpa and I. multicaulis within the genus. However, significant species misidentification in public databases was detected, particularly for critical taxa such as I. costata, I. tinctoria, and I. indigotica, indicating that current morphology-based species boundaries may not reflect their genetic delimitations. The rpl32–trnL marker further revealed the complex nature of the I. costata group, where some individuals were inseparable from I. tinctoria, suggesting potential hybridization or chloroplast capture. Furthermore, the morphologically similar I. minima and I. violascens were not sister species, implying that their similarity likely resulted from convergent evolution in analogous habitats rather than recent shared ancestry. This study provides crucial genomic resources and a theoretical foundation for species identification, systematic evolution, and genetic breeding in Isatis.
Supplementary Information
Supplementary Material 1.
Supplementary Material 2.
Supplementary Material 3.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1German DA, Hendriks KP, Koch MA, Lens F, Lysak MA, Bailey CD, Mummenhoff K. Al-Shehbaz IA.An updated classification of the brassicaceae (Cruciferae). Phyto Keys. 2023;220:12-144.1.10.3897/phytokeys.220.97724 PMC 1020961637251613 · doi ↗ · pubmed ↗
- 2Al-Shehbaz IA. The brassicaceae then and now: advancements in the past three decades, a review. Ann Bot-London, mcaf 055. Advance online publication. 2025. 10.1093/aob/mcaf 055.10.1093/aob/mcaf 05540138325 · doi ↗ · pubmed ↗
- 3Carillo P, Ferrante A. Decoding the intricate metabolic and biochemical changes in plant senescence: a focus on chloroplasts and mitochondria. Ann Bot. 2025:mcaf 003. 10.1093/aob/mcaf 003. 10.1093/aob/mcaf 00339883076 · doi ↗ · pubmed ↗