Trio-sequencing Reveals High Germline Mutation Rates in the Colorado Potato Beetle (Leptinotarsa decemlineata)
Shuqing Xu, Somaia Al-Madhagy, Pablo Duchen, Alitha Edison

TL;DR
This study finds that the Colorado potato beetle has a high germline mutation rate, which may explain its rapid evolution of pesticide resistance.
Contribution
The paper provides the first quantification of germline mutation rates in beetles and links it to evolutionary dynamics.
Findings
The germline mutation rate in CPB is 5.8 × 10−9 per site per generation, twice the median for insects.
Mutation rate in insects is positively associated with genome-wide GC content.
One CPB female's brood can introduce nearly 141 new mutations into coding regions each generation.
Abstract
Germline mutation rates are fundamental to evolution, yet they remain unquantified for beetles (Coleoptera), the most speciose order including major pests. We sequenced genomes from 16 trios of the Colorado potato beetle (Leptinotarsa decemlineata, CPB)—a pest that has evolved resistance to many insecticides. We estimated a germline mutation rate of 5.8 × 10−9 (95% CI: 4.7 × 10−9, 7.2 × 10−9) per site per generation in CPB, a rate 2-fold higher than the median for other insects. Across 13 insect species, mutation rate was positively associated with genome-wide GC content (PGLS). The increased mutation rate in CPB is also consistent with drift-barrier expectations. Based on this mutation rate and the beetle’s fecundity, we estimate that the brood from just one CPB female can introduce nearly 141 new mutations into the coding regions each generation. These findings inform CPB’s rapid…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
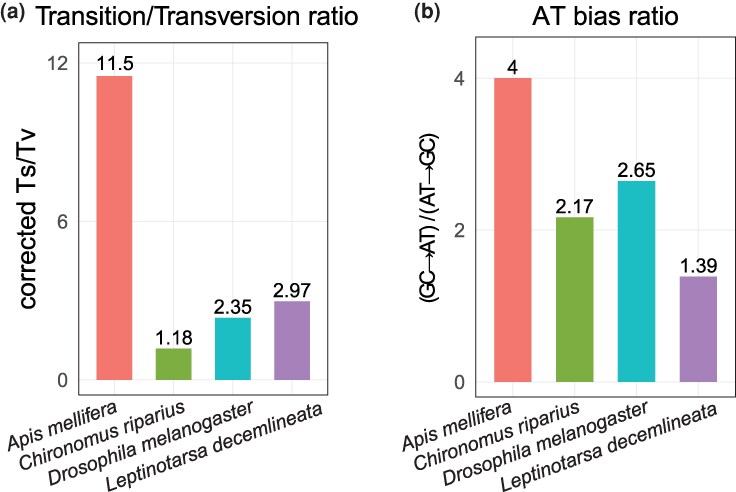 Fig. 1
Fig. 1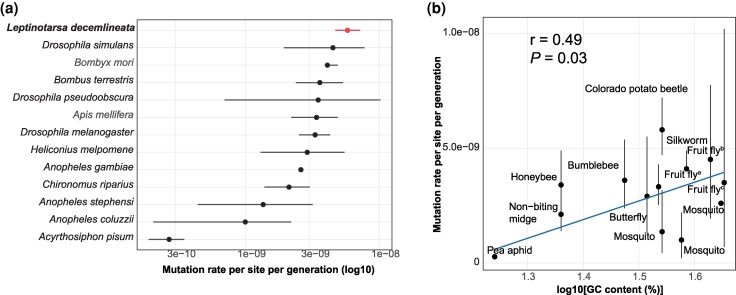 Fig. 2
Fig. 2 Fig. 3
Fig. 3- —German Research Foundation10.13039/501100001659
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInsect Resistance and Genetics · Evolution and Genetic Dynamics · Fungal Plant Pathogen Control
Introduction
Germline mutation rates are fundamental to evolutionary processes, shaping genetic diversity, adaptation, and speciation (Keightley and Eyre-Walker 2012). Pedigree-based estimates provide direct insights into per-generation changes, revealing rates around 10^−8^ per site per generation in vertebrates (Bergeron et al. 2023) but lower (∼2 to 4 × 10^−9^) in insects like Drosophila melanogaster (Keightley et al. 2014) and Anopheles (Rashid et al. 2022). These lower rates may stem from conserved DNA repair or fewer germline divisions in arthropods (Wang et al. 2020, 2022). Mutation rates also vary among insect lineages, and several nonexclusive factors have been proposed to explain this variation, including life history (e.g. generation time) (Wang and Obbard 2023; Lewin and Eyre-Walker 2025), population-genetic constraints on the evolution of replication fidelity (the drift-barrier hypothesis) (Lynch 2010; Sung et al. 2012; Lynch et al. 2016), and genomic features, such as GC content and genome size that can covary with mutational processes (Gu and Li 1994; Kiktev et al. 2018; Wang and Obbard 2023). However, data remain sparse for non-model insects, with none for beetles (Coleoptera), the most speciose order (∼400,000 species) that includes many pests (McKenna et al. 2019).
The Colorado potato beetle (Leptinotarsa decemlineata, CPB) exemplifies rapid pest evolution, resisting >50 insecticides since invading North America in the 19th century (Schoville et al. 2018; Pélissié et al. 2022). As a model for agricultural genomics and resistance mechanisms, empirically estimating the CPB’s mutation rate will advance our understanding of the tempo and mode of its adaptive evolution.
Here, we sequenced the genomes of 16 trios that were derived from two CPB families to identify de novo mutations. We validated a subset via Sanger sequencing, and estimated the germline mutation rate.
Results and Discussion
We sequenced 20 individuals comprising two full-sib families (2 parents + 8 offspring each), yielding 16 parent–offspring trios, with ∼444.7 million clean reads per individual (Table S1). On average, 74.8% of reads mapped to the CPB reference genome, yielding a mean coverage of 32.8× per sample. Among all 16 trios (with ∼491.8 million bp callable sites per trio on average, Table S2), we detected 92 de novo mutations that are heterozygous in offspring and absent in parents (Table S3). To estimate the false-positive rate, we randomly selected a subset of SNVs for Sanger sequencing. Among the successfully sequenced SNVs (14), we confirmed all of them (Fig. S1), suggesting a low false-positive rate. Thus, the estimated mutation rate (μ) is 5.8 × 10^−9^ per site per generation (95% CI: 4.7 × 10^−9^, 7.2 × 10^−9^) (see Materials and Methods). No difference was observed between the two families (likelihood-ratio test, P > 0.1). As we only considered the de novo mutations that are unique to each offspring (see Materials and Methods), which omits the early germline mutations, this is likely a conservative estimate.
Among the identified SNVs (Table S3), the mutational spectrum (55 transitions, 37 transversions; opportunity corrected Ts/Tv = 2.97) is consistent with the spectrum observed in other insects but lower than bees (Fig. 1a) (Keightley et al. 2015; Liu et al. 2017; Oppold and Pfenninger 2017; Rashid et al. 2022; Han et al. 2023). Collapsing mutations onto a pyrimidine reference (A↔T, C↔G) showed that C > T/G > A transitions were the most common class (34/92; 37%), followed by T > C/A > G (21/92; 23%). Among transversions, T > A and A > T mutations were the most frequent category, contributing 12/37 (32%) of all transversions and 13% of all SNVs, followed by T > G or A > C (10/37; 27%), C > A/G > T (9/37; 24%), and C > G/G > C (6/37; 16%). The bias toward A/T mutation is similar to other insects, but the magnitude is much lower in CPB (Fig. 1b).
De novo single-nucleotide mutation spectra across four insect species. A, opportunity-corrected transition/transversion ratio, calculated as Ts/(Tv/2), where Ts = C > T + T > C and Tv = C > A + C > G + T > A + T > G (pyrimidine-oriented convention). B, mutational AT bias, expressed as (GC→AT)/(AT→GC), where GC→AT = C > T + C > A and AT→GC = T > C + T > G. Bars are colored by species; values above bars indicate the corresponding ratios. Species were included based on availability of published de novo mutation call sets. For Drosophila melanogaster, the estimate is a pooled (SNV-count–weighted) average across five studies (Keightley et al. 2009; Schrider et al. 2013; Huang et al. 2016; Sharp and Agrawal 2016; Wang et al. 2023).
To compare CPB mutations with other species, we compiled published mutation rate estimates and compared them to CPB. We then tested whether mutation rate covaries with genome-wide GC content, genome size, days to sexual maturity and life-span using phylogenetically informed models. Our findings suggest that the mutation rate in CPB is high (Fig. 2a), but below that of humans and other vertebrates (typically 1 to 1.5 × 10^−8^ per site per generation). The lower mutation rates in insects compared to vertebrates may be due to conserved DNA repair mechanisms or fewer germline cell divisions (Wang et al. 2020, 2022), or the overall lower GC content and smaller genome size.
Germline mutation rates in insects. A, germline mutation rate estimates for 13 insect species. Mean and 95% confidence interval are shown. The x axis is log-scaled. The CPB (in red) is at the upper end of published direct estimates for insects: silkworm (Bombyx mori) (Han et al. 2023), honey bee (Apis mellifera), bumblebee (Bombus terrestris) (Liu et al. 2017), fruit fly (three species) (Keightley et al. 2009, 2014; Schrider et al. 2013; Huang et al. 2016; Sharp and Agrawal 2016; Krasovec 2021; Wang et al. 2023), butterfly (Heliconius melpomene) (Keightley et al. 2015), nonbiting midge (Chironomus riparius) (Oppold and Pfenninger 2017), mosquito (Anopheles, three species) (Rashid et al. 2022; Wang and Obbard 2023), and pea aphid (Acyrthosiphon pisum) (Fazalova and Nevado 2020). B, correlation between GC content and mutation rate. The P-value is estimated from phylogenetic generalized least squares (PGLS) (F1,10 = 6.3). Adjusted r2 = 0.24. aDrosophila pseudoobscura; bDrosophila simulans; cDrosophila melanogaster.
Across 13 insect species, mutation rate was positively associated with genome-wide GC content in a phylogenetic generalized least squares analysis (P = 0.03, Fig. 2b), indicating that variations in GC content in the genome might have contributed to the mutation variations in insects. This is consistent with the theoretical model that was built based on replication/mutagenesis kinetics and nucleotide precursor pool composition (Gu and Li 1994), and observations in yeast (Kiktev et al. 2018) and human (Schaibley et al. 2013). The correlations between mutation rate and genome size, life span and days to sexual maturity are not statistically significant (P > 0.1).
Interestingly, although the GC content in CPB genome is less than silkworm and fruit flies, its mutation rates are higher, suggesting that additional factors, such as effective population size, might have contributed to its elevated mutation rate. The drift-barrier hypothesis predicts that mutation rates decline until further refinement is prevented by the power of random genetic drift; higher rates can persist where effective population size (Ne) is constrained (Lynch et al. 2016). This hypothesis is consistent with the CPB’s recent invasion history; the European populations from which our strains derive experienced a population bottleneck, resulting in a smaller effective population size (Ne) (Schoville et al. 2018). With an estimated genetic diversity in CPB (π = 0.005), the average ancestral effective population size (Ne) of CPB would be 2.4 × 10^5^ under a neutral evolutionary model. A pairwise sequentially Markovian coalescent (PSMC) analysis (Cohen et al. 2022), which assumed a mutation rate of, 2.8 × 10^−9^, places the present-day Ne from US population around 10^4^. Recalculating with our higher mutation rate (μ), the present-day Ne would be approximately 2-fold smaller, as Ne is inversely proportional to μ [Ne = θ/(4μ)].
With a mutation rate of 5.8 × 10^−9^ and 1Gb haploid genome, CPB can generate 11.6 (95% CI: 9.4 to 14.4) new single-nucleotide mutations per diploid zygote each generation. In coding sequence (∼21.4 Mb total), the expectation is ∼0.25 (95% CI: 0.20 to 0.31) new point mutations per diploid genome. A female CPB can produce an average of 569.4 (± 80.0) larvae over a 16-week period when fed on plants (Thorpe and Bennett 2003). Therefore, the brood from just one female can introduce, on average, 141 (95% CI: 114.6 to 175.6) new mutations into the coding sequences of the next generation. This continual influx of potentially functional variants provides a vast reservoir of genetic diversity upon which selection can act, helping to explain the beetle’s remarkable capacity for rapid adaptation.
Materials and Methods
Establishment and Sequencing of Parent-offspring Trios
The parental lines of CPBs were obtained from Dr. Ralf Nauen's lab, Bayer AG (Monheim, Germany). The insect rearing, plant growing, and experiments were carried out in conditions similar to those in our previous work (Edison et al. 2024, 2025). In brief, all insects were reared on 5-week-old potato (Solanum tuberosum) plants (Annabelle variety, seed potatoes purchased from Ellenberg's Kartoffelvielfalt GmbH & Co. KG, Barum) in 85 cm × 45 cm × 55 cm-sized insect cages. The climate chamber with a long day photoperiod (16:8 L:D) at a temperature of 24 °C was used.
For the establishment of parent−offspring trios, two different parental lines were chosen as starting generations to create two families (Fig. 3). The genome-wide per base divergence between the parents is 0.019 in both families (estimated from genome-wide SNP data and callable regions). In each family, DNA from eight offspring (four males and four females, at adult stage) and their parents (at adult stage) were isolated using the Monarch Genomic DNA purification kit #T3010 (New England BioLabs) following the manufacturer's protocol. The concentration of DNA was assessed using NanoDrop 1000 (Thermo Scientific, Germany). Whole genome sequencing (150 bp PE) was performed using Illumina Novaseq sequencer at Biomarker (Beijing, China).
Study organism and experimental design. A, A CPB pair; B, the crossing design of the two trio families.
Read Trimming, Filtering and Mapping
Raw Illumina reads were adapter- and quality-trimmed with Skewer v0.2.2 (Jiang et al. 2014) using the -m pe setting, which enforces synchronized clipping of read pairs and removes trailing bases with Phred < 20. Parameter values (insert-size estimation window = 400 bp; minimum post-trim length = 35 bp) followed the developer's recommendations. Trimmed FASTQs were retained in gzip format and their integrity confirmed with FastQC (Brown et al. 2017).
Trimmed reads were aligned to an improved L. decemlineata reference genome (Wilhelm et al. 2025) with BWA-MEM v0.7.17 (16 threads, default seed length). As we noticed that some mitochondrial sequences were miss-assembled to the original reference genome, we downloaded the raw PacBio long reads (SRR20519124) (Yan et al. 2023) and reassembled the genome using hifiasm (v0.19.3-r572). Prior assembly, we removed all reads that mapped more than 90% to mitochondrial genome of L. decemlineata (MZ189364) (Dai et al. 2022) using minimap2 (Li 2018). The hifi reads were extracted using ccs (v6.4.0) function (−min-passes 3 −min-rq 0.99 −min-length 50 −max-length 200,000 −top-passes 60) and used for genome assembly with hifiasm (v0.19.3-r572) (Cheng et al. 2021). We then used Hi-C reads anchored the contigs to chromosomes using YaHS (v1.2.2) (Zhou et al. 2023). The final assembly achieved more than 98% BUSCO score, with 884 Mb sequences anchored to 18 chromosomes.
Primary alignments were converted to BAM, restricted to mappings with MAPQ ≥ 30 to suppress ambiguous placements, and coordinate-sorted with SAMtools v1.17 (Li et al. 2009). Duplicate reads were marked and removed using samtools markdup.
Joint Variant Calling and Variants Filtering
A major challenge in identifying de novo mutations is to minimize false positives. To this end, we adopted several strategies. First, we performed joint variant calling for all samples using both bcftools mpileup and GATK pipelines. For the bcftools mpileup pipeline, variants were called with bcftools (v1.14), producing a compressed VCF file. Hard filters were applied with bcftools view, retaining sites with QUAL > 30, total trio depth (INFO/DP) > 60, mapping-quality MQ > 30, and missing-data fraction F_MISSING < 0.10. For the GATK pipeline, we followed the best practice using GATK v4.6.0.0. We first generated gvcf files for each sample with options “−linked-de-bruijn-graph true −native-pair-hmm-threads 12 −standard-min-confidence-threshold-for-calling 40 -ERC BP_RESOLUTION -G AS_StandardAnnotation”. Then the variants were called using joint calling functions. Variants were then filtered using VariantFiltration with following options: -filter “QD < 2.0” -filter “QUAL < 30.0” -filter “SOR > 3.0” -filter “FS > 60.0” -filter “MQ < 40.0” -filter “MQRankSum < −12.5” -filter “ReadPosRankSum < −8.0”. Then for the variants identified from both pipelines, we only kept the variants that have exactly one alternative allele using bcftools view function with “−min-ac = 1 −max-ac = 1″ option. This filtering step specifically isolates putative de novo mutations: variants that are heterozygous in a single offspring but absent (i.e. homozygous for the reference allele) in both parents. Then, the consensus variants identified by both pipelines were selected using the bcftools isec function and kept for the downstream analysis. For all analyses, we specifically focused on single-nucleotide variant, as the sequencing depth is not sufficiently high to accurately call insertions and deletions.
To identify the de novo mutations from each trio family, we then performed several filtering steps to remove false positives in R 4.3.1 using VariantAnnotation (Bioconductor 3.17) and vcfR 1.14.0 (Knaus and Grünwald 2017): (i) sites were kept only if each sample's depth lay within 0.5 to 2× its genome-wide mean; (ii) positions with greater than 1 alternate read in either parent were discarded; (iii) positions with more than 3 alternative reads in all individuals other than the focal trio family were removed; (iv) autosomal heterozygous calls were subjected to a binomial allelic-balance test (probability = 0.5; P ≥ 0.05 required); (v) for sex chromosome (chr6), the same criterion applied to females (XX), whereas male offspring (XO) had to be hemizygous with depth thresholds halved to match ploidy. The same depth filtering process was also applied to estimate the size of the callable genome size for each trio. The mutation rates were calculated as number of de novo mutations divided by two times callable genome size (as for a diploid genome). Our filtering strategy yields a conservative mutation-rate estimate because it excludes early germline mutations. When we also included de novo mutations shared among offspring from the same parents (include early germline mutations), the estimated mutation rate increased ∼3-fold. However, given the limited number of sequenced trios, we could not reliably distinguish these shared variants from false positives.
Mutation Validations
To estimate false positives, we performed Sanger sequencing on selected variants (Table S4). We used the same DNA that was used for whole-genome sequencing for PCR and Sanger sequencing. For each variant, primers at up and downstream regions were designed using Primer3. Only primer pairs that successfully amplified 300 to 800 bp products were kept. PCR was performed under following conditions: 98 °C for 30 s; 35 cycles of 98 °C for 10 s, 64 °C (or 61 °C for low-GC amplicons) for 30 s and 72 °C for 20 s; a final extension at 72 °C for 2 min. Each 50 µl reaction contained 10 µl 5 × Q5 Reaction Buffer, 1 µl 10 mM dNTP mix, 2.5 µl of each primer (10 µM; final 0.5 µM), 1 µl genomic DNA, 0.5 µl Q5 High-Fidelity DNA Polymerase (New England Biolabs) and 32.5 µl nuclease-free water. PCR products were purified using Macherey-Nagel™ NucleoSpin PCR Clean-up Kit. Sequencings were performed by using Sanger sequencing at StarSEQ (https://www.starseq.com/). The failed sequencing reactions were excluded from the analysis.
Phylogenetic Analysis
To perform phylogenetic analysis, we first constructed the phylogenetic tree using COI sequences from each species (Table S5) with Phylogeny.fr (Dereeper et al. 2008). The DNA sequences were aligned using MUSCLE and tree was built using PhyML with parameters: substitution model HKY85, gamma shape parameter equals 0.80, transition/transversion ratio equals 1.88, number of categories is 4, proportion of invariant is 0.24. The phylogenetic tree was then used to fit a linear model using PGLS function from the R-package caper v1.0.3, taking into account phylogenetic nonindependence between mutation rates and life history traits and genomic features (Table S5). Genome size and GC content information were retrieved from NCBI database (https://www.ncbi.nlm.nih.gov/datasets/genome/) and the life history traits were extracted from publicly available information (Table S5).
Supplementary Material
evag027_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bergeron LA, et al Evolution of the germline mutation rate across vertebrates. Nature. 2023:615:285–291. 10.1038/s 41586-023-05752-y.36859541 PMC 9995274 · doi ↗ · pubmed ↗
- 2Brown J, Pirrung M, Mc Cue LA. FQC dashboard: integrates Fast QC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics. 2017:33:3137–3139. 10.1093/bioinformatics/btx 373.28605449 PMC 5870778 · doi ↗ · pubmed ↗
- 3Cheng H, et al Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 2021:18:170–175. 10.1038/s 41592-020-01056-5.33526886 PMC 7961889 · doi ↗ · pubmed ↗
- 4Cohen ZP, et al Evidence of hard-selective sweeps suggests independent adaptation to insecticides in Colorado potato beetle (Coleoptera: chrysomelidae) populations. Evol Appl. 2022:15:1691–1705. 10.1111/eva.13498.36330305 PMC 9624080 · doi ↗ · pubmed ↗
- 5Dai TM, et al The complete mitochondrial genome of invasive insect Leptinotarsa decemlineata say 1824 (Coleoptera: chrysomelidae). Mitochondrial DNA B Resour. 2022:7:358–360. 10.1080/23802359.2022.2035280.35174290 PMC 8843170 · doi ↗ · pubmed ↗
- 6Dereeper A, et al Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008:36:W 465–W 469. 10.1093/nar/gkn 180.18424797 PMC 2447785 · doi ↗ · pubmed ↗
- 7Edison A, et al Evidence of active oviposition avoidance to systemically applied imidacloprid in the Colorado potato beetle. Insect Sci. 2024:31:1543–1554. 10.1111/1744-7917.13319.38282249 · doi ↗ · pubmed ↗
- 8Edison A, et al Bulk segregant analysis reveals genomic regions associated with imidacloprid resistance in the Colorado potato beetle. Ecol Evol. 2025:15:e 72527. 10.1002/ece 3.72527.41262162 PMC 12626429 · doi ↗ · pubmed ↗