Whole-genome characterization and molecular epidemiology of Feline coronavirus (FeCoV) circulating in domestic cats in Thailand: First report of FeCoV-II whole genomes
Yu Nandi Thaw, Kamonpan Charoenkul, Chanakarn Nasamran, Ekkapat Chamsai, Waleemas Jairak, Eaint Min Phyu, Hnin Wai Phyu, Supassama Chaiyawong, Somsak Pakpinyo, Alongkorn Amonsin

TL;DR
This study reports the first complete genome sequences of FeCoV-II in Thailand and finds that FeCoV-I is the dominant strain among domestic cats.
Contribution
The study provides the first whole-genome sequences of FeCoV-II in Thailand and highlights the predominance of FeCoV-I.
Findings
FeCoV positivity was 21.87% in domestic cats, with genotype I being overwhelmingly predominant (99.03%).
Thai FeCoV-I strains clustered closely with Chinese and Dutch strains, while FeCoV-II grouped with Chinese FeCoV-II.
No mutations were detected in the S1/S2 cleavage sites of FeCoV-I, and FeCoV-II showed characteristic deletion and insertion patterns.
Abstract
Feline coronavirus (FeCoV) is a widely circulating Alphacoronavirus that causes mild enteric infections and, in some cases, progresses to Feline infectious peritonitis, a fatal systemic disease. FeCoV consists of two genotypes (I and II) and two biotypes (FeCoV and feline infectious peritonitis virus [FIPV]). Despite its importance, whole-genome data, particularly for FeCoV genotype II, remain limited in Thailand. This study aimed to determine the prevalence of FeCoV in domestic cats and to genetically characterize circulating strains using whole-genome and S gene sequencing. A total of 471 rectal swabs were collected from domestic cats presented to private small animal hospitals in Bangkok and neighboring provinces from October 2022 to October 2023. FeCoV detection and genotyping were performed using one-step reverse transcription polymerase chain reaction targeting the 3′UTR and S…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
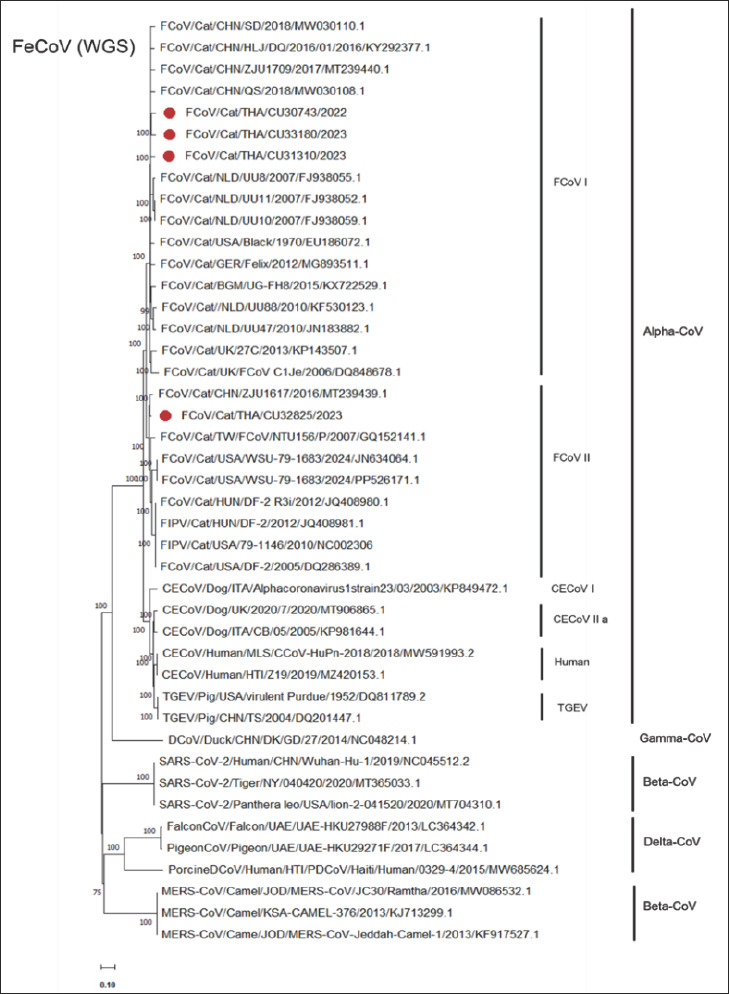 Figure 1
Figure 1 Figure 2
Figure 2| Demographic factors | No. of rectal swabs | FeCoV positive / samples tested (% FeCoV-positive) |
|---|---|---|
| Year | ||
| 2022 | 95 | 27/95 (28.42%) |
| 2023 | 376 | 76/376 (20.21%) |
| Total | 471 | 103/471 (21.87%) |
| Age of the animals | ||
| < 6 months | 123 | 35/123 (28.46%) |
| 6 months to 2 years | 151 | 30/151 (19.87%) |
| > 2 years | 185 | 38/185 (20.54%) |
| Unknown | 12 | 0/12 (0%) |
| Total | 471 | 103/471 (21.87%) |
| Sex | ||
| Male | 163 | 39/163 (23.93%) |
| Neutered male | 80 | 13/80 (16.25%) |
| Female | 158 | 37/158 (23.42%) |
| Neutered female | 59 | 12/59 (20.34%) |
| Unknown | 11 | 2/11 (18.18%) |
| Total | 471 | 103/471 (21.87%) |
| Breed | ||
| Pure breed | 229 | 33/229 (14.41%) |
| Mixed | 176 | 53/176 (30.11%) |
| Unknown | 66 | 17/66 (25.76%) |
| Total | 471 | 103/471 (21.87%) |
| Clinical status | ||
| Asymptomatic | 253 | 60/253 (23.72%) |
| Symptomatic | 218 | 43/218 (19.72%) |
| Total | 471 | 103/471 (21.87%) |
| Factors | FeCoV | |||
|---|---|---|---|---|
|
| ||||
| Positive (%) | Negative (%) | Chi-square | P-value | |
| Age | ||||
| Up to 6 months | 35 (28.46) | 88 (71.54) | 7.03 | 0.68 |
| Older than 6 months to 2 years | 30 (19.87) | 121 (80.13) | ||
| More than 2 years | 38 (20.54) | 147 (79.46) | ||
| Unknown | 0 (0.00) | 12 (100.00) | ||
| Total | 103 | 368 | ||
| Clinical Status | ||||
| Asymptomatic | 60 (23.72) | 193 (76.28) | 1.09 | 0.29 |
| Symptomatic | 43 (19.72) | 175 (80.28) | ||
| Total | 103 | 368 | ||
| Season | ||||
| Winter (November–January) | 31 (24.03) | 98 (75.97) | 1.31 | 0.52 |
| Summer (February– May) | 38 (23.17) | 126 (76.83) | ||
| Rainy (June–October) | 34 (19.10) | 144 (80.90) | ||
| Total | 103 | 368 | ||
| Virus | Year | Genotype | Total | ||
|---|---|---|---|---|---|
|
| |||||
| I | IIa | Mixed (I + IIa) | |||
| FeCoV | 2022 | 27 | 0 | 0 | 27 |
| 2023 | 75 | 1 | 0 | 76 | |
| Total | 103 | 1 | 0 | 103 | |
| No. | ID | Year | Location | Age | Sex | Breed | Clinical sign | Gene sequencing | Accession No. | |
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Genotype I | Genotype IIa | |||||||||
| 1 | CU30655 | Oct-2022 | Bangkok | 2 months | F | Scottish Fold | Healthy | S | ||
| 2 | CU30743 | Nov-2022 | Bangkok | 7 years | F | Domestic Short Hair | N/A | WGS | ||
| 3 | CU31310 | Dec-2022 | Bangkok | 2 years 5 months | NF | British Short Hair | Diarrhea | WGS | ||
| 4 | CU31327 | Dec-2022 | Bangkok | 4 months | F | N/A | Healthy | S | ||
| 5 | CU32238 | Mar-2023 | Bangkok | 5 months | F | Domestic Short Hair | Diarrhea | S | ||
| 6 | CU32640 | May-2023 | Nonthaburi | 2 months | F | Domestic Short Hair | Diarrhea | S | ||
| 7 | CU32825 | June-2023 | Nonthaburi | 1 year | F | Domestic Short Hair | Diarrhea | WGS | ||
| 8 | CU33011 | June-2023 | Nonthaburi | 25 days | M | British Short Hair | Diarrhea | S | ||
| 9 | CU33180 | July-2023 | Nonthaburi | 12 days | F | Domestic Short Hair | Diarrhea | WGS | ||
| 10 | CU33441 | Aug-2023 | Nonthaburi | 1 year 6 months | F | Scottish Fold | Diarrhea | S | ||
| Strain | Accession No. | Genotype | Spp. | Country | Year | WGS | ORF1ab | S | 3a | 3b | 3c | E | M | N | 7a | 7b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||
| n (aa%) | n (aa%) | n (aa%) | n (aa%) | n (aa%) | n (aa%) | n (aa%) | n (aa%) | n (aa%) | n (aa%) | n (aa%) | ||||||
| CU33180 | FeCoV-I | Feline | Thailand | 2023 | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | |
| CU30655 | FeCoV-I | Feline | Thailand | 2022 | (-) | (-) | 86.5 (91.6) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | |
| CU30743 | FeCoV-I | Feline | Thailand | 2022 | 92.3 (88.2) | 92.4 (89.2) | 90.8 (92.2) | 93.5 (93.5) | 92.9 (93.1) | 97.4 (97.7) | 90.3 (92.1) | 89.7 (93.2) | 94.0 (92.1) | 93.3 (92.0) | 90.8 (87.0) | |
| CU31310 | FeCoV-I | Feline | Thailand | 2023 | 87.8 (84.5) | 92.2 (90.4) | 87.4 (91.7) | 94.6 (95.2) | 92.9 (93.1) | 95.8 (96.5) | 93.0 (97.4) | 92.0 (93.6) | 92.3 (92.4) | 95.2 (93.4) | 90.1 (91.5) | |
| CU31327 | FeCoV-I | Feline | Thailand | 2023 | (-) | (-) | 85.5 (90.1) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | |
| CU32238 | FeCoV-I | Feline | Thailand | 2023 | (-) | (-) | 87.0 (92.3) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | |
| CU32640 | FeCoV-I | Feline | Thailand | 2023 | (-) | (-) | 86.0 (91.0) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | |
| CU33011 | FeCoV-I | Feline | Thailand | 2023 | (-) | (-) | 87.3 (91.7) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | |
| CU33441 | FeCoV-I | Feline | Thailand | 2023 | (-) | (-) | 84.9 (89.9) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | |
| CU32825 | FeCoV-II | Feline | Thailand | 2023 | 78.4 (74.5) | 92.1 (89.5) | 31.5 (28.4) | 60.1 (64.8) | 53.4 (59.5) | 85.1 (75.7) | 92.6 (96.1) | 92.3 (92.4) | 94.5 (95.7) | 95.6 (93.4) | 94.3 (94.1) | |
|
| ||||||||||||||||
|
| ||||||||||||||||
|
| ||||||||||||||||
| SD | FeCoV-I | Feline | China | 2018 | 91.6 (93.1) | 92.4 (90.8) | 86.4 (92.3) | 94.6 (95.2) | 92.9 (93.1) | 95.8 (96.5) | 91.7 (98.7) | 90.6 (93.2) | 92.8 (93.9) | 95.6 (94.7) | 90.1 (88.8) | |
| SMU-CD86 | FeCoV-I | Feline | China | 2020 | NA | NA | 87.5 (92.4) | NA | NA | NA | NA | NA | NA | NA | NA | |
| UCD1 | AB0882222.1 | FeCoV-I | Feline | USA | 1970 | NA | NA | 84.8 (90.1) | NA | NA | NA | NA | NA | NA | NA | NA |
| HLJ/DQ/2016/01 | FeCoV-I | Feline | China | 2016 | 91.0 (92.8) | 92.0 (90.2) | 84.9 (91.3) | 93.5 (93.5) | 92.0 (92.2) | 95.9 (96.5) | 91.7 (97.4) | 91.2 (94.0) | 91.8 (91.2) | 95.2 (94.7) | 90.7 (88.8) | |
| UG-FH8 | FeCoV-I | Feline | Belgium | 2105 | 90.9 (87.3) | 91.8 (90.1) | 84.3 (90.1) | 94.1 (95.2) | 95.7 (95.7) | 94.7 (96.5) | 92.6 (97.4) | 91.4 (93.2) | 91.7 (93.0) | 92.0 (90.6) | 89.8 (89.7) | |
| ZJU1617 | FeCoV-II | Feline | China | 2016 | 77.4 (72.8) | 91.0 (87.8) | 30.8 (27.6) | 60.1 (64.8) | 53.4 (59.5) | 84.2 (74.2) | 90.7 (92.1) | 89.7 (92.0) | 92.7 (93.0) | 95.6 (93.4) | 92.6 (91.5) | |
| NTU156/P | FeCoV-II | Feline | Taiwan | 2007 | 75.9 (70.0) | 89.4 (84.8) | 31.4 (28.3) | 62.0 (67.0) | 58.4 (63.1) | 47.1 (32.9) | 78.0 (77.4) | 83.5 (86.8) | 90.6 (91.2) | 95.2 (94.7) | 93.2 (93.3) | |
| SMU-CD14 | FeCoV-II | Feline | China | 2018 | NA | NA | 30.5 (27.1) | NA | NA | NA | NA | NA | NA | NA | NA | |
| SMU-CD59 | MW31685.1 | FeCoV-II | Feline | China | 2018 | NA | NA | 30.9 (28.6) | NA | NA | NA | NA | NA | NA | NA | NA |
|
| ||||||||||||||||
|
| ||||||||||||||||
|
| ||||||||||||||||
| Alphacoronavirus 1 strain 23/03 | CECoV I | Canine | Italy | 2003 | 62.0 (58.2) | 81.0 (75.8) | 29.6 (27.4) | 68.0 (67.0) | 68.0 (71.2) | 84.2 (75.7) | 80.8 (80.5) | 80.5 (85.0) | 75.9 (72.3) | 88.5 (86.3) | 61.3 (51.9) | |
| CB/05 | CECoV II | Canine | Italy | 2005 | 70.9 (64.2) | 80.8 (75.8) | 32.0 (29.8) | 63.0 (64.8) | 49.6 (40.1) | 81.7 (77.1) | 79.7 (77.4) | 78.6 (82.8) | 74.8 (74.5) | 82.7 (80.2) | 60.4 (53.2) | |
| CCoV-HuPn-2018 | CECoV II | Human | Malaysia | 2018 | 67.7 (61.2) | 80.4 (75.3) | 30.9 (27.1) | 63.0 (64.8) | 50.6 (58.2) | 83.2 (77.1) | 79.2 (75.8) | 78.4 (80.9) | 74.6 (74.2) | 82.2 (77.0) | 60.1 (56.0) | |
| Z19 | CECoV II | Human | Hati | 2019 | 67.8 (61.4) | 80.4 (75.4) | 30.9 (26.9) | 63.0 (64.8) | 50.6 (58.2) | 83.2 (77.1) | 76.8 (75.8) | 79.3 (82.8) | 74.6 (75.3) | 78.0 (77.0) | 60.7 (55.7) | |
| virulent Purdue | TGEV | Pig | USA | 1952 | 67.2 (60.9) | 80.3 (75.4) | 30.4 (26.8) | 61.3 (64.8) | N/A | 81.3 (74.2) | 78.0 (74.1) | 77.2 (81.8) | 73.4 (73.5) | 78.6 (77.0) | NA | |
| TS | TGEV | Pig | China | 2004 | 66.9 (60.2) | 80.0 (74.6) | 29.9 (26.4) | 57.3 (60.2) | N/A | 80.4 (72.7) | 78.0 (74.1) | 76.8 (81.4) | 72.9 (73.8) | 76.8 (72.1) | NA | |
| Strain | Accession No. | Genotype | Spp. | Country | Year | WGS | ORF1ab | S | 3a | 3b | 3c | E | M | N | 7a | 7b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||
| n (aa%) | n (aa%) | n (aa%) | n (aa%) | n (aa%) | n (aa%) | n (aa%) | n (aa%) | n (aa%) | n (aa%) | n (aa%) | ||||||
| CU32825 | FeCoV-II | Feline | Thailand | 2023 | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | |
| CU30655 | FeCoV-I | Feline | Thailand | 2022 | (-) | (-) | 30.6 (28.8) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | |
| CU30743 | FeCoV-I | Feline | Thailand | 2022 | 78.8 (74.0) | 92.4 (88.8) | 32.2 (28.6) | 56.4 (64.8) | 59.2 (63.1) | 83.2 (74.2) | 91.3 (03.5) | 92.0 (94.0) | 95.1 (95.1) | 95.2 (96.1) | 92.7 (89.7) | |
| CU31310 | FeCoV-I | Feline | Thailand | 2023 | 83.9 (80.3) | 92.9 (90.5) | 32.5 (29.3) | 63.0 (64.8) | 59.2 (63.1) | 83.7 (74.2) | 94.4 (98.7) | 92.7 (94.4) | 94.9 (95.7) | 96.1 (97.4) | 92.1 (93.3) | |
| CU31327 | FeCoV-I | Feline | Thailand | 2023 | (-) | (-) | 32.6 (28.4) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | |
| CU32238 | FeCoV-I | Feline | Thailand | 2023 | (-) | (-) | 32.1 (29.3) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | |
| CU32640 | FeCoV-I | Feline | Thailand | 2023 | (-) | (-) | 31.4 (28.9) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | |
| CU33011 | FeCoV-I | Feline | Thailand | 2023 | (-) | (-) | 32.0 (28.3) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | |
| CU33180 | FeCoV-I | Feline | Thailand | 2023 | 78.4 (74.5) | 92.1 (89.5) | 31.5 (28.4) | 60.1 (64.8) | 53.4 (59.5) | 85.1 (74.2) | 92.6 (96.1) | 92.3 (92.4) | 94.5 (95.7) | 95.6 (93.4) | 94.3 (94.1) | |
| CU33441 | FeCoV-I | Feline | Thailand | 2023 | (-) | (-) | 32.6 (27.6) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | (-) | |
| Reference FeCoV | ||||||||||||||||
| SD | FeCoV-I | Feline | China | 2018 | 79.1 (75.7) | 92.9 (90.8) | 32.0 (28.6) | 63.0 (64.8) | 59.2 (63.1) | 83.7 (74.2) | 92.6 (96.1) | 92.0 (94.0) | 96.0 (97.4) | 96.5 (98.7) | 91.6 (90.6) | |
| SMU-CD86 | FeCoV-I | Feline | China | 2020 | NA | NA | 31.8 (27.8) | NA | NA | NA | NA | NA | NA | NA | NA | |
| UCD1 | AB0882222.1 | FeCoV-I | Feline | USA | 1970 | NA | NA | 30.9 (27.2) | NA | NA | NA | NA | NA | NA | NA | NA |
| HLJ/DQ/2016/01 | FeCoV-I | Feline | China | 2016 | 79.2 (75.8) | 93.0 (90.6) | 31.8 (28.4) | 57.4 (64.8) | 59.5 (63.1) | 83.8 (74.2) | 94.9 (98.7) | 93.1 (94.9) | 95.1 (94.2) | 96.1 (98.7) | 94.1 (91.5) | |
| UG-FH8 | FeCoV-I | Feline | Belgium | 2105 | 78.7 (75.4) | 92.3 (90.0) | 32.1 (27.7) | 59.3 (67.0) | 58.7 (63.1) | 85.6 (74.2) | 94.0 (98.7) | 93.2 (92.8) | 94.7 (96.0) | 95.7 (97.4) | 90.4 (89.7) | |
| ZJU1617 | FeCoV-II | Feline | China | 2016 | 95.6 (93.8) | 95.0 (93.5) | 98.0 (97.7) | 100.0 (100.0) | 100.0 (100.0) | 99.3 (98.9) | 93.6 (96.1) | 95.5 (96.9) | 96.1 (96.9) | 98.3 (100.0) | 96.4 (95.0) | |
| NTU156/P | FeCoV-II | Feline | Taiwan | 2007 | 92.4 (92.1) | 93.2 (92.5) | 95.9 (97.2) | 98.4 (98.4) | 97.5 (97.5) | 65.3 (63.4) | 79.7 (78.9) | 87.2 (89.8) | 94.0 (94.2) | 97.8 (98.7) | 94.6 (93.3) | |
| SMU-CD14 | FeCoV-II | Feline | China | 2018 | NA | NA | 98.1 (98.9) | NA | NA | NA | NA | NA | NA | NA | NA | |
| SMU-CD59 | MW31685.1 | FeCoV-II | Feline | China | 2018 | NA | NA | 97.1 (97.9) | NA | NA | NA | NA | NA | NA | NA | NA |
|
| ||||||||||||||||
| Alphacoronavirus 1 strain 23/03 | CECoV I | Canine | Italy | 2003 | 65.3 (63.9) | 83.2 (80.2) | 80. 2. (81.1) | 86.8 (91.9) | 73.6 (75.6) | 90.7 (87.9) | 82.4 (82.0) | 82.1 (85.5) | 76.9 (73.8) | 88.0 (86.3) | 61.7 (51.9) | |
| CB/05 | CECoV II | Canine | Italy | 2005 | 78.1 (73.4) | 83.2 (80.3) | 89.8 (91.8) | 95.7 (95.2) | 24.7 (29.0) | 93.8 (95.3) | 81.4 (78.9) | 80.1 (83.2) | 75.8 (75.3) | 83.3 (80.2) | 62.7 (54.5) | |
| CCoV-HuPn-2018 | CECoV II | Human | Malaysia | 2018 | 82.1 (78.2) | 83.0 (80.2) | 81.1 (82.8) | 95.7 (96.8) | 91.2 (91.3) | 94.3 (95.3) | 80.8 (77.4) | 81.3 (84.6) | 76.0 (75.3) | 81.6 (77.0) | 68.7 (58.9) | |
| Z19 | CECoV II | Human | Hati | 2019 | 82.5 (78.4) | 83.1 (80.2) | 81.2 (82.9) | 95.7 (96.8) | 91.2 (91.3) | 94.3 (95.3) | 78.5 (77.4) | 82.2 (84.1) | 75.8 (76.0) | 78.6 (77.0) | 63.8 (57.0) | |
| Virulent Purdue | TGEV | Pig | USA | 1952 | 76.5 (72.3) | 82.8 (80.0) | 80.8 (81.2) | 94.6 (95.2) | N/A | 93.1 (92.9) | 80.8 (75.8) | 80.5 (84.6) | 74.3 (73.5) | 79.2 (77.0) | NA | |
| TS | TGEV | Pig | China | 2004 | 76.5 (71.6) | 82.5 (79.2) | 80.1 (80.3) | 92.2 (88.4) | N/A | 92.2 (91.7) | 80.8 (75.8) | 80.1 (84.6) | 74.2 (74.5) | 77.5 (72.1) | NA | |
| Virus | Country | Year | Genotype | Putative Pathotypes | Predicted proteolytic cleavage sites | |
|---|---|---|---|---|---|---|
|
| ||||||
| S1/S2 (828 - 830) | S2 (1034 - 1039) | |||||
| Ref sequences used in this study | ||||||
| HLJ/DQ/2016/01 | China | 2018 | I | FeCoV | RRSRRS | KR---S |
| SD | China | 2018 | I | FeCoV | RRSRRS | KR---S |
| FCoV/CD0610 | China | 2020 | I | FeCoV | RRARRS | KR---S |
| USD1 | USA | 1970 | I | FeCoV | RRSRGS | QR---S |
| UG-FH8 | Belgium | 2015 | I | FeCoV | KRLRRS | KR---S |
| UU10 | Netherlands | 2007 | I | FeCoV | KRSRRS | KR---S |
| UU11 | Netherlands | 2007 | I | FeCoV | KRSRRS | KR---S |
| UU34 | Netherlands | 2007 | I | FeCoV | RRSRRS | KR---S |
| UU8 | Netherlands | 2007 | I | FIPV | RRSRRS | KR---S |
| HLJ/HRB/2016/11 | China | 2016 | I | FIPV | RRSRRS | KR---S |
| HLJ/HRB/2016/10 | China | 2016 | I | FIPV | RRSRRS | KR---S |
| HLJ/HRB/2016/13 | China | 2016 | I | FIPV | RRSRRS | KR---S |
| QS | China | 2018 | I | FIPV | RRSRTS | KR---S |
| BLACK | USA | 1970 | I | FIPV | KRSRRP | VR---S |
| 26M | UK | 2013 | I | FIPV | RGARRS | KR---S |
| FCoVCC1Je | UK | 2006 | I | FIPV | RQSRRS | KR---S |
| 79-1146 | USA | 2010 | II | FIPV | DEL | KRKYGS |
| DF-2 | Hungary | 2012 | II | FIPV | DEL | KRKYGS |
| DF-2 | USA | 2005 | II | FeCoV | DEL | KRKYGS |
| DF-2R3i | Hungary | 2012 | II | FeCoV | DEL | KRKYGS |
| WSU79-1683 | USA | 2024 | II | FeCoV | DEL | KRKYRS |
| Tokyo/cat/130627 | Japan | 2014 | II | FeCoV | DEL | KRKYRS |
| NTU156/P | Taiwan | 2007 | II | FeCoV | DEL | KRKYRS |
| SMU-CQ14 | China | 2018 | II | FeCoV | DEL | KRKYRS |
| SMU-CQ59 | China | 2018 | II | FeCoV | DEL | KRKYRS |
|
| ||||||
| This study | ||||||
| THA/CU30655 | Thailand | 2022 | I | FeCoV | RRSRRS | KR---S |
| THA/CU30743 | Thailand | 2022 | I | FeCoV | RRSRRS | KR---S |
| THA/CU31310 | Thailand | 2023 | I | FeCoV | RRSRRS | KR---S |
| THA/CU31327 | Thailand | 2023 | I | FeCoV | RRSRRS | KR---S |
| THA/CU32238 | Thailand | 2023 | I | FeCoV | RRSRRS | KR---S |
| THA/CU32640 | Thailand | 2023 | I | FeCoV | RRSRRS | KR---S |
| THA/CU33011 | Thailand | 2023 | I | FeCoV | RRARRS | KR---S |
| THA/CU33180 | Thailand | 2023 | I | FeCoV | RRSRRS | KR---S |
| THA/CU33441 | Thailand | 2023 | I | FeCoV | RRSRRS | KR---S |
| THA/CU32825 | Thailand | 2023 | II | FeCoV | DEL | KRKYRS |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAnimal Virus Infections Studies · SARS-CoV-2 and COVID-19 Research · Viral Infections and Outbreaks Research
INTRODUCTION
Coronaviruses (CoVs) are enveloped, nonsegmented, single-stranded RNA viruses belonging to the family Coronaviridae. Their genomes contain 11 open reading frames (ORFs) arranged in the order 5′UTR–ORF1a–ORF1b–S–ORF3a/b/c–E–M–N–ORF7a/b–3′UTR [1, 2]. CoVs are classified into four genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus. Within the Alphacoronavirus genus, the Alphacoronavirus 1 species comprises canine enteric coronavirus (CECoV) and feline coronavirus (FeCoV), both of which primarily infect dogs and cats, respectively, and are commonly associated with gastroenteritis and diarrhea in affected animals [3].
FeCoV comprises two major genotypes, type I (FeCoV-I) and type II (FeCoV-II), which are differentiated by genetic divergence in the spike (S) gene [4, 5]. FeCoV is also divided into two biotypes: the feline enteric coronavirus (FeCoV) and the highly pathogenic Feline infectious peritonitis (FIP) virus (FIPV) [6]. FeCoV is widespread and typically causes mild to moderate gastroenteritis, whereas FIPV results in FIP, a fatal systemic disease of felids. FIP occurs in two clinical forms: the wet (effusive) form, characterized by peritonitis and/or pleuritis, and the dry (non-effusive) form, which features granulomatous lesions affecting organs such as the central nervous system and eyes. FIPV is believed to emerge from mutations in FeCoV within infected hosts; however, the precise genetic determinants driving this biotype transition remain incompletely understood [7, 8].
Previous studies conducted in Thailand have primarily focused on FeCoV genotype I (FeCoV-I), examining partial and complete genomic regions, yet no whole-genome sequencing (WGS) of FeCoV-II has been performed to date [9–12]. This represents a critical gap because FeCoV-II, although less frequently detected, plays an important role in viral evolution and may arise through recombination between FeCoV-I and canine coronaviruses. A similar pattern is observed across other Asian countries. Reports from China [13, 14], Indonesia [15–17], Malaysia [18], and Vietnam [19] have also emphasized FeCoV-I, with FeCoV-II characterized primarily through partial S gene sequencing rather than complete genomic analysis. As a result, the full genomic diversity, recombination potential, and evolutionary relationships of FeCoV-II remain poorly understood in the region. This lack of whole-genome data restricts our ability to track genotype shifts, detect recombination events, and identify genomic determinants associated with pathogenicity, including those distinguishing FeCoV from FIPV. Given the scarcity of comprehensive molecular data on circulating FeCoVs in Thailand, particularly FeCoV-II, there is a significant gap in understanding the epidemiology, molecular evolution, and genetic characteristics of FeCoVs in Thai domestic cats.
To address this gap, the present study aimed to conduct a cross-sectional survey and whole-genome characterization of FeCoVs circulating in domestic cats in Thailand. Building on the limitations identified in previous studies [9–19], this research sought to determine FeCoV occurrence using reverse transcription polymerase chain reaction (PCR) (RT-PCR), classify circulating strains into FeCoV-I and FeCoV-II genotypes through S gene–based genotyping, and generate WGS and complete S gene sequences for selected FeCoV-positive samples. By applying WGS, the study aimed to provide a more complete understanding of the genomic features, phylogenetic relationships, and molecular diversity of Thai FeCoVs. The resulting genomic data are intended to support improved surveillance, facilitate early detection of genotype shifts or recombination events, and contribute to the development of more effective prevention, diagnostic, and vaccination strategies for FeCoV infection in Thailand.
MATERIALS AND METHODS
Ethical approval
This study was conducted strictly in accordance with the ethical principles, guidelines, and institutional regulations governing the use of animals in research at Chulalongkorn University. All procedures involving animals were reviewed and approved by the Chulalongkorn University Animal Care and Use Committee (CU-ACUC) under protocol numbers CU-VET IACUC#2031025 and 2331076. The protocol covered sample collection, handling, transportation, and laboratory processing of biological materials.
As the sampling involved non-invasive rectal swabs collected from client-owned domestic cats during routine veterinary visits, no experimental manipulation, sedation, or procedures causing pain, distress, or behavioral alteration were performed. Sample collection was carried out exclusively by licensed veterinarians or trained veterinary personnel to ensure animal welfare and procedural compliance. The study adhered to all institutional and national standards for minimizing animal discomfort and ensuring responsible use of animals in research.
Prior to sample collection, verbal informed consent was obtained from all pet owners or guardians after providing clear information regarding the study purpose, sampling procedures, potential benefits, and assurance of confidentiality. Participation was entirely voluntary, and owners retained the right to withdraw their animals at any stage without affecting veterinary services. No incentives were offered for participation.
This research fully followed the Animal Research: Reporting of In Vivo Experiments 2.0 guidelines for reporting animal research and complied with the ethical frameworks outlined in the Guide for the Care and Use of Laboratory Animals, the Thai Animal Welfare Act, and the institutional policies of the Faculty of Veterinary Science, Chulalongkorn University. All samples were anonymized and coded to protect the identity of both owners and their animals.
Study period and location
A cross-sectional study was conducted from October 2022 to October 2023 at private small animal hospitals in Bangkok, Nonthaburi, and Samut Prakan.
Calculation of sample size and sample collection
The sample size was calculated as described by Cochran [20]. The sample size calculation was based on the parameters, including the prevalence (p-value) of CoV in cats (at 31%) [10]. The confidence interval (z value) was 95%, and the precision (d value) was 0.045. Thus, the minimum required sample size was 406. A total of 471 samples were collected to increase the reliability of the sample size, which exceeded the required number. In this study, 471 rectal swab samples were collected from cats (n = 471) using convenience sampling. The samples were collected regardless of the age, sex, breed, or clinical status of the cats. Demographic data, including age, sex, breed, and clinical status, were collected and recorded from both hospital patient records and pet owners while collecting the samples. The collected swabs were placed in viral transport media (Eagle’s Minimum Essential Medium) and temporarily stored in a refrigerator at 4°C at the animal hospital, then transported to the laboratory within 24 h.
RNA extraction
RNA was extracted from the rectal swab sample using the GeneAll® GENTiTM Viral DNA/RNA Extraction Kit (GeneAll®; Lisbon, Portugal) on a GENTiTM 32 (GeneAll) following the manufacturer’s instructions. The NanoDrop Spectrophotometer (Thermo Fisher Scientific, USA) was then used to quantify the RNA prior to FeCoV detection.
FeCoV detection by one-step RT-PCR
One-step RT-PCR was used to detect FeCoV by targeting the 3'UTR [21]. One-step RT-PCR was performed in a final volume of 50 µl consisting of 3 µl of template RNA, 25 µl of 2X Reaction Mix, 1 µl of 10 µM forward and reverse primers, 2 µl of SuperScript III RT (Invitrogen, Thermo Fisher, USA), and distilled water. The RT-PCR assay included a cDNA synthesis step at 55°C for 15 min, followed by 94°C for 2 min, and then 40 cycles of 94°C for 15 s, 55°C for 30 s, and 68°C for 1 min, with a final extension at 68°C for 5 min. PCR products were analyzed on a 1.5% agarose gel containing RedSafeTM (iNtRON Biotechnology, Inc., Korea) at 100 V for 45 min. The expected size of the FeCoV-positive amplified products was 223 bp. Positive and no-template controls were used in the RT-PCR reaction to ensure the reliable detection of FeCoV. Laboratory standards were implemented to prevent contamination, including the separation of workspaces for RNA extraction and PCR preparation and the use of sterilized equipment and utensils.
Genotyping of FeCoV using one-step RT-PCR
All FeCoV-positive samples were genotyped by one-step RT-PCR using primers specific for the S gene classification [9, 22]. One-step RT-PCR was performed using SuperScript™ III RT-PCR with Platinum™ Taq Mix (Invitrogen, Thermo Fisher Scientific). For genotyping, the PCR reaction and conditions were similar to those used for virus detection (FeCoV), except for the annealing temperature of 45°C for 30 s. PCR products were run on a 1.5% agarose gel mixed with RedSafeTM (iNtRON Biotechnology, Inc.) at 100 V for 45 min. The samples with 376- and 283-bp amplification products were classified as FeCoV genotypes I and II, respectively.
Whole-genome and S gene sequencing of the FeCoV strain
Four FeCoV-positive samples (n = 4) were subjected to WGS. In addition, complete S gene sequencing was performed for six FeCoVs (n = 6). The FeCoV samples were selected to represent different locations, collection dates, clinical histories, and high-quality RNA samples. Sample selection was also based on the quality of PCR amplicons visualized on agarose gels, with stronger, more distinct bands indicating a higher RNA yield. Each viral gene was amplified using newly designed primer sets from the Primer 3 Plus program, as well as previously described primer sets (Supplementary Table 1) [12, 23]. Nucleotide amplification for each gene was performed using one-step RT-PCR with a final total volume of 50 µl. This consisted of 3 µl of template RNA, 25 µl of 2X Reaction Mix, 10 µl of 10 µM forward and reverse primers, 2 µl of SuperScript III RT (Invitrogen), and distilled water. Under these conditions, a cDNA synthesis step was included at 55°C for 30 min, followed by an initial denaturation step at 94°C for 2 min, 40 cycles of denaturation at 94°C for 30 s, annealing at 45°C–48°C for 30 s, and extension at 68°C for 1–4 min, along with a final extension step at 68°C for 5–10 min. Agarose gel electrophoresis was performed to confirm positive PCR amplification. The PCR products were then purified using NucleoSpin® Gel and PCR Clean-up (MACHEREY-NAGELTM, Germany).
Oxford Nanopore sequencing technology was used for WGS and S gene sequencing. To perform Oxford Nanopore sequencing, the pooled PCR products of each gene were prepared and sequenced using the Oxford Nanopore sequencing device and MinION flow cells with the Oxford Nanopore rapid sequencing kit V14 (SQK-RAD114) according to the rapid sequencing protocol (ONT, UK). For WGS and S gene sequencing, the DNA library and the flow cell priming mix were prepared according to the manufacturer’s instructions and sequenced using a sequencing device (MinION Mk1b device) and MinION flow cells (FLO-FLG114, R10.4.1). MinKNOW software (version 24.11.8; ONT, UK) was used for sequencing control and raw signal acquisition. Basecalling was performed using the GPU-enabled Guppy basecaller (version 6.5.7) with a minimum Q-score threshold of 7. The sequencing duration was set to 12 h. The EPI2ME platform (version 5.2.3) was used to identify potential references for sequencing data using the wf-alignment workflow, with the reference set as all FeCoV references in the National Center for Biotechnology Information (NCBI) database. The reads were aligned to the references using Minimap2 (version 2.28). The resulting alignments were then polished using Racon (version 0.5.0), and Medaka (version 2.0.0) was used to refine the consensus sequence. This study employed reference-guided annotation for genome annotation. ORF and gene features were inferred based on the reference genomes of FeCoV-I (UU88; JN183882.1) and FeCoV-II (WSU-79-1683; JN634064) and subsequently confirmed by nucleotide Basic Local Alignment Search Tool (BLAST) searches against the NCBI database (Supplementary Table 2). The nucleotide sequences of each FeCoV gene segment were retrieved in a FASTA file format and compared to the NCBI database using BLAST to identify the closest matching reference sequence for each gene segment. Finally, the consensus sequence of each viral gene segment was exported in the FASTA format for further analysis.
Phylogenetic and genetic analysis of the FeCoV gene
Phylogenetic analysis of the S gene and whole-genome sequences (WGS) of FeCoV was performed by comparing the nucleotide sequences of Thai FeCoVs with reference CoVs from the coronavirus family available in the GenBank database, representing various geographical, host, and species origins. The coronavirus reference sequences from the Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus genera were used to root the trees and differentiate the host origin and coronavirus species. Phylogenetic trees of the S gene and WGS of FeCoVs were constructed using Molecular Evolutionary Genetics Analysis (MEGA) version 11.0 (Tempe, AZ, USA) with the neighbor-joining method applying the Kimura 2-parameter and 1,000 bootstrap replicates [24]. The Kimura 2-parameter (K2P) model was chosen because of its ability to effectively account for transition/transversion rate differences and provide reliable phylogenetic analysis of similar viral sequences. A pairwise comparison of the nucleotides and amino acids of FeCoV was conducted with FeCoVs from both the same and different genotypes. Genetic analysis was performed by aligning and comparing the S gene amino acid sequences of FeCoV with those of reference FeCoVs from the same and different genotypes using MegAlign software V.5.03 (DNASTAR Inc., Wisconsin, USA). We evaluated the unique and variable amino acids associated with receptor binding and viral fusion for cell entry, as well as host preferences.
Statistical analysis
The association between FeCoV occurrence in domestic cats and factors such as animal age, season, and clinical status was analyzed using the chi-square test (SPSS Statistics, version 29.0.1.0, IBM Corp., NY, USA). A p-value of < 0.05 was considered statistically significant. Categorical variables, including age, were classified based on animals; clinical status was grouped as symptomatic or asymptomatic; and season was categorized according to Thailand’s climate pattern. These groupings ensured adequate sample sizes per category and facilitated meaningful comparisons. The assumptions for chi-square testing were verified prior to analysis, including the assumption of categorical data, independent observations, and expected frequencies in contingency table cells that were generally greater than 5.
RESULTS
FeCoV occurrence in domestic cats
We conducted a cross-sectional survey of FeCoV in domestic cats (n = 471) at private small animal hospitals in Bangkok, Nonthaburi, and Samut Prakan, Thailand, from October 2022 to October 2023. FeCoV positivity was 21.87% (103/471) in domestic cats (Table 1). The FeCoV positivity was 28.42% (27/95) in 2022 and 20.21% (76/376) in 2023. By age, we found the highest FeCoV detection in younger cats (up to 6 months) at 28.46% (35/123), but this was not statistically significant. FeCoVs were most frequently detected in the winter (November to January) (24.03%) and summer (February to May) (23.17%) seasons, but no statistical significance was observed. By clinical status, FeCoV can be detected in both symptomatic and asymptomatic cats, with the highest positivity in asymptomatic cats (23.72%), but this difference was not statistically significant (Table 2). FeCoV genotype I (99.03%, 102/103) was more prevalent than FeCoV genotype II (0.97%, 1/103) (Table 3).
Whole-genome and S gene sequencing output
The WGS of FeCoV (n = 4) were successfully sequenced for phylogenetic and genetic analysis. The complete S gene sequences of FeCoV (n = 6) were also available. All Thai FeCoV nucleotide sequences were submitted to the database under the GenBank accession numbers (PV797400–PV797409). Table 4 and Supplemental Tables 2 and 3 provide detailed information on the FeCoVs characterized in this study.
In the phylogenetic analysis of the S gene, one FeCoV-I strain (CU33441) was closely related to Chinese FeCoV-I (HLJ/DQ/2016/01; SMU-CD60). Another Thai FeCoV-I strain (CU31327) was closely related to FeCoV-I from China (HLJ/HRB/2016/13). Thai FeCoV-I (CU33011) was closely related to US-derived FeCoV-I (UCD1). The other six Thai FeCoV-I strains were closely related to FeCoV-I from the Netherlands (UU10 and UU11). The Thai FeCoV-II strain (CU32825) was closely related to the Chinese FeCoV-II strain (ZJU1617; SMU-CD59). Additionally, all Thai FeCoV genotypes I and II were grouped with FeCoVs described as FeCoV biotypes from other countries (Figure 2).
Phylogenetic relationships based on whole-genome and S gene analysis
The phylogenetic analysis of whole-genome sequences and complete S gene of FeCoVs showed that all Thai FeCoV genotype I (FeCoV-I) were closely related to FeCoV-I from China (ZJU1709; QS; HLJ/DQ/2016/01) and the Netherlands (UU8; UU11; UU10). Similarly, Thai FeCoV-II (CU32825) was closely related to Chinese-FeCoV-II (ZJU1617) (Figure 1).
Phylogenetic tree of whole-genome sequences of Thai feline coronavirus (FeCoV) using the neighboring method with the Kimura-2 model and 1,000 bootstrapping replicates. The red circle represents the Thai FeCoV.
Phylogenetic tree of S gene nucleotide sequences of Thai feline coronavirus (FeCoV) using the neighboring method with the Kimura-2 model and 1,000 bootstrapping replicates. The red circle represents the Thai FeCoV.
Pairwise nucleotide and amino acid identity analysis
Pairwise comparison of the WGS of the Thai FeCoV-I (CU33180) showed high nucleotide (nt%, 87.8% and 92.3%) and amino acid identities (aa%; 84.5% and 88.2%), respectively, compared with the other Thai FeCoV-I, CU31310 and CU30743. Thai FeCoV-I (CU33180) has a high degree of similarity to FeCoV-I from China (SD) (91.6% nt; 93.1% aa identities). Similarly, the WGS of the Thai FeCoV-I (CU33180) showed high nucleotide (% nt identities, 90.9% – 91.6%) and amino acid (% aa identities, 87.3% – 92.8%) when compared to other reference FeCoV-I (Table 5). The Thai FeCoV-II (CU32825) showed high nucleotide (95.6%) and amino acid (93.8%) identities with the Chinese FeCoV-II (ZJU1617) (Table 6). The WGS of the Thai FeCoV-II (CU32825) showed low nucleotide identities (nt%, 78.4% – 83.9%) and amino acid identities (aa%; 74.0% – 80.3%) compared to the Thai FeCoV-Is (Table 6).
Genetic characterization of S gene regions
For the genetic analysis of FeCoVs, the deduced amino acids of the S gene of Thai FeCoVs were aligned to those of reference FeCoVs from the same and different genotypes. Our results showed that the amino acids in the S1/S2 and S2 regions of Thai FeCoV-I and reference strains were identical (no mutation). Thai FeCoV-II (CU32825) contained a six-amino-acid deletion (positions 828–830) at the S1/S2 cleavage site and a three-amino-acid insertion (positions 1036–1038) at the S2 (Table 7).
DISCUSSION
Occurrence of FeCoV in domestic cats
In this study, the occurrence of FeCoV in domestic cats in Bangkok and its vicinity from October 2022 to October 2023 was 21.87%. This occurrence is lower than those reported in earlier studies in Thailand, where FeCoV was found at 30.97% (25). This discrepancy might be due to differences in sample size, geographic factors, and improvements in infection control measures. Our results showed that FeCoVs were highly detected in young cats (up to 6 months of age). This observation, in agreement with previous reports from Australia, China, and Japan, demonstrated that a younger age is a significant factor in FeCoV positivity (26–28). Similarly, our findings were consistent with the current report of FeCoVs in Thailand (12). In contrast, other studies from Australia, Hungary, and Malaysia reported that FeCoV infection was not associated with the age of the cat (26,29). The susceptibility and resistance to FeCoV associated with age remain unknown and vary across different geographic locations, as indicated by Li et al. (28).
Seasonal and clinical distribution of FeCoV
The highest positivity for FeCoV was found in summer and winter, compared to the rainy seasons. This finding was consistent with previous studies in Korea and Thailand, which found that FeCoV was the most prevalent during winter [12, 30]. However, some studies have reported that FeCoV is ubiquitous and mostly subclinical in all cats, occurring consistently throughout the year without any seasonal variation [30]. In this study, a high FeCoV positivity rate was observed among asymptomatic cats, consistent with previous studies from Thailand [9, 12]. The results suggested that asymptomatic cats may serve as potential reservoirs for FeCoV, contributing to viral transmission to susceptible populations. This consistent association between FeCoV infection and the health status of cats in Thailand highlights the importance of including asymptomatic cats in the development of effective prevention and control strategies for FeCoV infection in feline populations. Additionally, the proportion of FeCoV-infected cats that develop clinical signs, such as diarrhea, remains unclear [31].
Distribution and implications of FeCoV genotypes
Regarding genotyping results, FeCoV genotype I was identified as the predominant genotype in Thai cat populations. Other studies from Europe and the Americas reported a high prevalence of FeCoV-I, ranging from 80% to 95%, while FeCoV-II is less common [32]. This finding suggested that FeCoV-I may circulate more widely, possibly because current vaccines target only the FeCoV genotype II [33]. The prevalence of FeCoV-I in Thailand has implications for the development of epidemiological and translational vaccines. Most currently available vaccines are based on FeCoV-II strains, which may limit their effectiveness in regions where FeCoV-I is dominant. The high prevalence of FeCoV-I highlights the need for vaccine strategies that incorporate FeCoV-I epitopes or develop broadly protective formulations for both genotypes. From an epidemiological perspective, given the predominance of FeCoV-I, surveillance and diagnostic assays should prioritize It to avoid underestimating infection rates. Continued monitoring of genotype distribution is essential to detect potential shifts in FeCoV genotype prevalence, which could impact both control strategies and vaccine design in Thailand and the region.
Phylogenetic and molecular characterization of Thai FeCoVs
Phylogenetic analysis of whole-genome sequences and the S gene revealed that all Thai FeCoV genotypes I and II were closely related to FeCoV-I and FeCoV-II. Moreover, all Thai FeCoVs were grouped with all reference FeCoV biotypes, suggesting that Thai FeCoVs could be classified as FeCoV biotypes. This finding highlights the predominant biotypes of FeCoV infection in Thailand’s cat populations. In the S gene, Thai FeCoV-I contained no amino acid mutations in the S1/S2 or S2 regions compared to reference FeCoVs. Similarly, Thai FeCoV-IIs were consistent with reference FeCoV-IIs. Notably, the highly conserved regions in the S1/S2 region could be used to facilitate molecular diagnostic assays, vaccine development, and differentiation between FeCoV and FIPV biotypes. However, the association between pathogenicity and insertion/deletion at the S1/S2 and S2 sites remains unclear. These changes could impact the pathogenicity of FeCoVs (both FeCoV and FIPV biotypes). For example, FeCoVs consist of an S1/S2 cleavage site with the R-R-S-R-R-S motif, whereas FIPV exhibits several substitutions at this site [34, 35]. Thai FeCoVs were highly conserved (no amino acid change) at the conserved furin cleavage motif (R-R-S/A-R-R-S), which is typical for FeCoVs. These findings were consistent with those of a recent study from Vietnam [19]. Additionally, Thai FeCoVs were detected in fecal samples from non-FIP cats, and no clinical cases of FIP were observed during the sampling period. Therefore, the conserved amino acid changes and subclinical presentation suggest that Thai FeCoVs are the predominant FeCoV biotype.
Genomic features and recombination insights of FeCoV-II
This study revealed that the Thai FeCoV-II exhibited unique genomic features, but there was no evidence of recombination with other coronaviruses from different hosts. It has been reported that FeCoV-II originated from homologous recombination between FeCoV-I and canine coronavirus (CCoV), in which the common recombination hotspots are at the S gene and open reading frame 1ab (ORF1ab) regions [4, 36]. Pairwise comparison of the WGS of the Thai FeCoV-II showed high nucleotide and amino acid identities with FeCoV-II reference strains, but lower nucleotide and amino acid identities to FeCoV-I reference strains and Canine CoVs from dogs. Similarly, phylogenetic analysis of both the WGS and S genes revealed that Thai FeCoV-II was grouped within the FeCoV-II cluster and clearly distinct from other coronaviruses from different hosts. Although recombination analysis (e.g., with SimPlot or RDP4) was not conducted in this study, the phylogenetic analysis of Thai FeCoV-II showed no evidence of recent recombination with other coronaviruses. Future studies should apply recombination detection tools to confirm potential recombination hotspots, particularly in the S gene and ORF1ab, and further clarify the evolutionary history of FeCoV-II in Thailand. In addition, cross-species investigations should be conducted, particularly involving canine coronavirus (CCoV), to understand the recombination events and the potential for spillover between cats and dogs in Thailand.
Study limitations
This study has some limitations, including time constraints that restricted study sites, sample collection, and clinical follow-up. Moreover, only few FeCoVs were sequenced, which may underrepresent the genetic diversity of circulating FeCoVs in Thailand. Potential confounding factors, such as clinic-level clustering, owner socioeconomic status, and housing conditions, were not assessed due to limited information available in the outpatient data and from pet owners. These factors influence viral transmission and may explain the variability in the occurrence of FeCoV. Additionally, the statistical analysis was limited to bivariate comparisons of FeCoV occurrence with individual categorical factors (age, clinical status, and season), yielding no statistically significant results. Therefore, corrections for multiple testing were not applied. Despite these limitations, this study provided the first FeCoV-II WGS in Thailand and established valuable baseline data for future multi-regional and longitudinal investigations of FeCoVs.
CONCLUSION
This study provides the most updated molecular epidemiological overview of FeCoV circulating in domestic cats in Bangkok and surrounding provinces, with an overall FeCoV occurrence of 21.87%, the predominance of FeCoV genotype I (99.03%), and the detection of only one FeCoV-II strain. Younger cats (≤6 months) showed the highest positivity, and FeCoV was detected in both summer and winter, although none of the factors showed statistical significance. Importantly, this study generated the first FeCoV-II whole-genome sequence from Thailand, along with whole-genome and complete S gene sequences for multiple FeCoV-I strains. Phylogenetic analysis demonstrated that Thai FeCoV-I strains were closely related to those from China, the Netherlands, and the United States, while the Thai FeCoV-II clustered tightly with Chinese FeCoV-II. Genetic characterization revealed high conservation in the S1/S2 and S2 cleavage regions among Thai FeCoVs, consistent with previously described FeCoV biotypes, suggesting circulation of predominantly low-virulence strains in the sampled population.
The findings have several practical implications. First, the dominance of FeCoV-I highlights the urgent need to reconsider current vaccine formulations, which are largely based on FeCoV-II strains and may not provide optimal protection in regions where FeCoV-I prevails. Second, the high positivity rate in asymptomatic cats underscores their potential as silent reservoirs, underscoring the importance of routine surveillance, improved outbreak monitoring, and targeted prevention strategies. The genomic data generated in this study also enhance diagnostic assay design, particularly by identifying conserved spike regions relevant for molecular differentiation between FeCoV and FIPV.
A key strength of this study is the integration of epidemiological data with whole-genome and S gene sequencing, enabling a more comprehensive understanding of circulating FeCoV genotypes and their evolutionary relationships. Additionally, the multi-hospital sampling approach improved representativeness across the Bangkok metropolitan area.
However, this study has limitations, including the restriction of sampling to specific provinces, limited clinical follow-up, and sequencing of only a subset of positive samples, which may not capture the full genetic diversity of circulating FeCoVs. The lack of recombination analysis (e.g., SimPlot, RDP4) also limits conclusions about historical recombination events.
Future studies should expand sampling to additional regions of Thailand, incorporate longitudinal monitoring to track genotype shifts, and perform detailed recombination analyses—particularly focusing on the S gene and ORF1ab regions. Cross-species investigations involving canine coronavirus (CCoV) would also help elucidate potential recombination pathways and interspecies transmission risks.
In conclusion, this study establishes essential baseline genomic evidence for FeCoV in Thailand, enhances understanding of circulating biotypes and their evolutionary context, and provides foundational data to improve diagnostic tools, support vaccine development, and inform long-term surveillance strategies. These findings contribute significantly to regional and global efforts aimed at controlling FeCoV infections and mitigating their impact on feline health.
DATA AVAILABILITY
The nucleotide sequence data are available in the GenBank database under accession numbers # PV797400–409. The supplementary data can be available from the corresponding author upon request.
AUTHORS’ CONTRIBUTIONS
YNT: Drafted and revised the manuscript. YNT, KC, CN, and EC: Collected the samples. YNT, WJ, EMP, HWP, and SC: Performed virus detection, whole-genome characterization, and phylogenetic analysis. YNT, KC, CN, EC, SP, and SC: Participated in the phylogenetic and genetic analyses. AA: Designed the study, performed data analysis, and drafted, and revised the manuscript. All authors have read and approved the final version of the manuscript.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Le Poder S Feline and canine coronaviruses:Common genetic and pathobiological features Adv. Virol 201120116094652231234710.1155/2011/609465 PMC 3265309 · doi ↗ · pubmed ↗
- 2Chen Y Liu Q Guo D Emerging coronaviruses:Genome structure, replication, and pathogenesis J. Med. Virol 20209222493288101310.1002/jmv.26234 PMC 7435528 · doi ↗ · pubmed ↗
- 3Groot R. J Baker S. C Baric R. S Brown C. S Drosten C Enjuanes L Fouchier R. A. M Galiano M Gorbalenya A. E Memish Z. A Perlman S Poon L. L. M Snijder E. J Family Coronaviridae. In:King, A. M. Q., Adams, M. J., Carstens, E. B., and Lefkowitz, E. J. (eds.) Virus Taxonomy:Ninth Report of the International Committee on Taxonomy of Viruses.Elsevier Academic Press 20112011
- 4Herrewegh A. A Smeenk I Horzinek M. C Rottier P. Jde Groot R. J Feline coronavirus type II strains 79-1683 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus J Virol 19987245084514955775010.1128/jvi.72.5.4508-4514.1998 PMC 109693 · doi ↗ · pubmed ↗
- 5Cook S Castillo D Williams S Haake C Murphy B Serotype I and II feline coronavirus replication and gene expression patterns of feline cells:Building a better understanding of serotype I FIPV biology Viruses 20221413563589133810.3390/v 14071356 PMC 9320447 · doi ↗ · pubmed ↗
- 6Jaimes J. A Millet J. K Stout A. E AndréN. M Whittaker G. RA tale of two viruses:The distinct spike glycoproteins of feline coronaviruses Viruses 202012833193674910.3390/v 12010083 PMC 7019228 · doi ↗ · pubmed ↗
- 7Tekes G Hofmann-Lehmann R Stallkamp I Thiel V Thiel H.-J Genome organization and reverse genetic analysis of a type I feline coronavirus J Virol 200882185118591807772010.1128/JVI.02339-07PMC 2258703 · doi ↗ · pubmed ↗
- 8Gao Y. Y Chang X Wei Y Yu X He Y Ma J Huang T An updated review of feline coronavirus:Mind the two biotypes Virus Res 20233261990593673162910.1016/j.virusres.2023.199059 PMC 10194308 · doi ↗ · pubmed ↗