Genetic Adaptation of Mesorhizobium Symbionts Associated With Caragana in Northern China Deserts
Xiaoxia Yuan, Hua Li, Xiumin Yu, Zhaojun Ji

TL;DR
This study explores how Mesorhizobium bacteria, which help Caragana plants in Chinese deserts, adapt genetically to harsh environments through specific genes.
Contribution
The study reveals that recombination, not mutation, drives adaptation in Mesorhizobium through key genes like cysW and uvrC.
Findings
Mesorhizobium strains show strong genetic cohesion within lineages despite geographic separation.
Purifying selection and recombination drive adaptation in membrane transporters and nucleotide repair genes.
Harsh desert environments weaken the distance-decay relationship among bacterial populations.
Abstract
Caragana, a keystone leguminous species dominating arid semi‐fixed deserts in northern China, forms specialized symbiotic nitrogen‐fixing partnerships with Mesorhizobium, which are indispensable for sustaining ecosystem function globally. However, the roles of membrane transporters and nucleotide repair genes in conferring survival advantages to desert‐dwelling Mesorhizobium across desert environments remained poorly elucidated. Therefore, a total of 68 representative Mesorhizobium strains associated with Caragana, isolated from five geographically distant areas (A to E) in the desert belt of northern China, were investigated to elucidate the pivotal roles of three membrane transporters (cysW, exoY, idhA) and two nucleotide repair genes (mutS, uvrC) in microbial adaptation to environmental stress. Phylogenetic analysis results revealed that strains assigned to the same genospecies…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
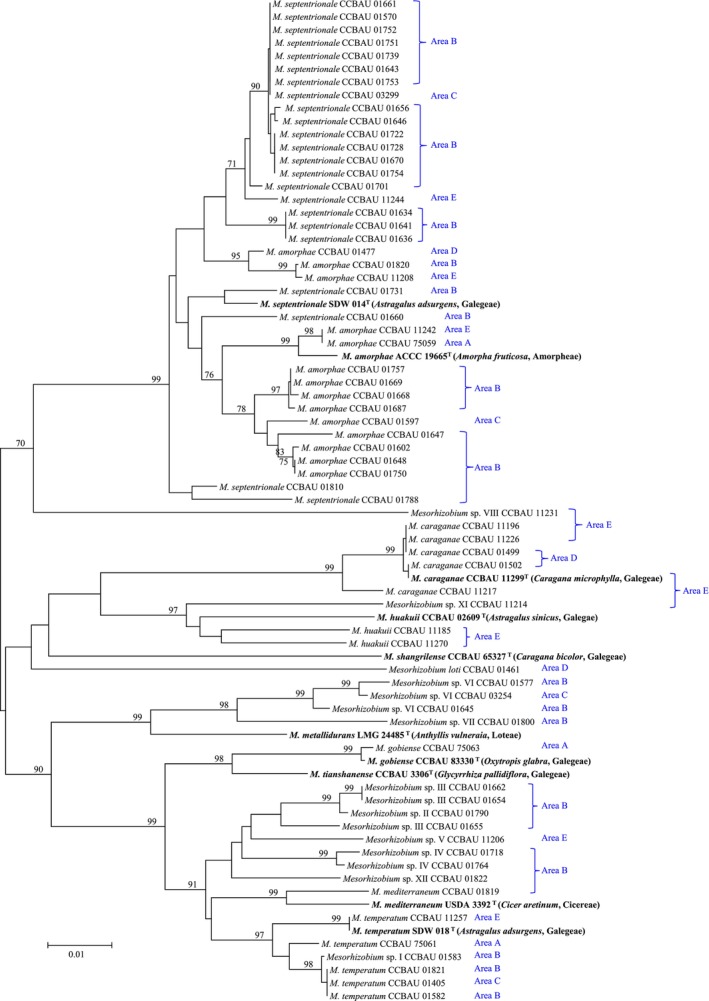 FIGURE 1
FIGURE 1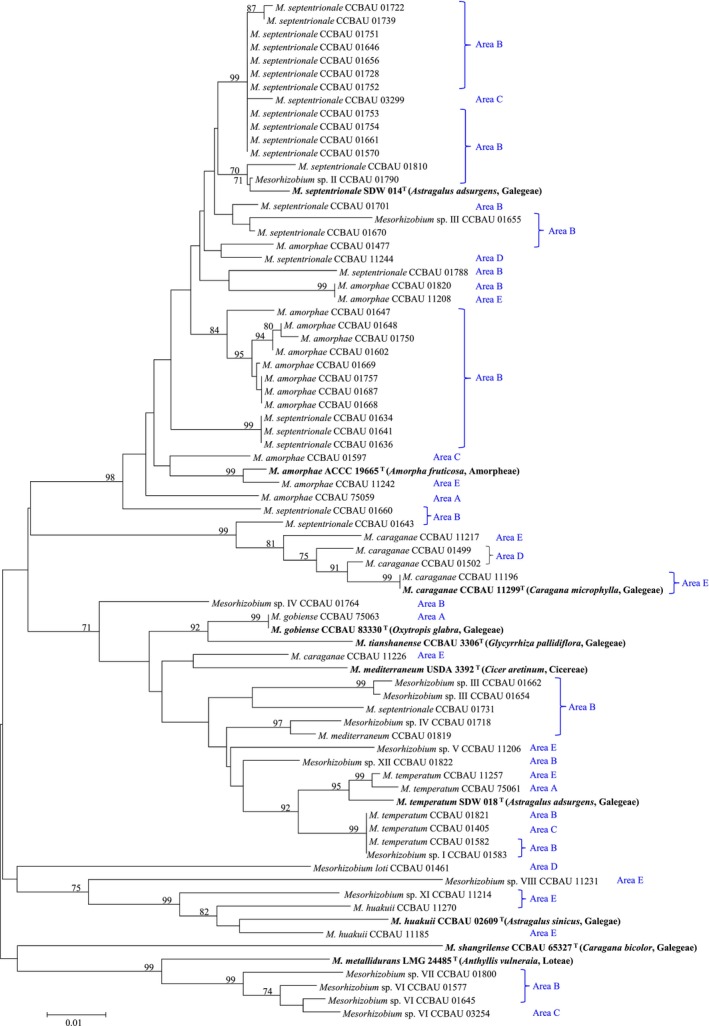 FIGURE 2
FIGURE 2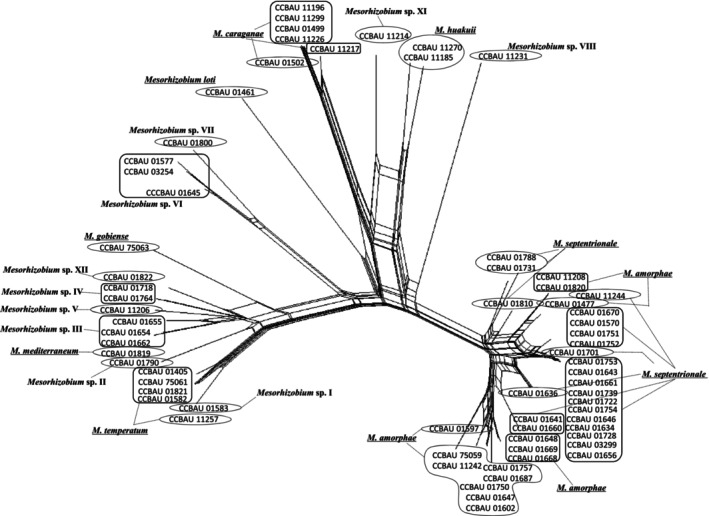 FIGURE 3
FIGURE 3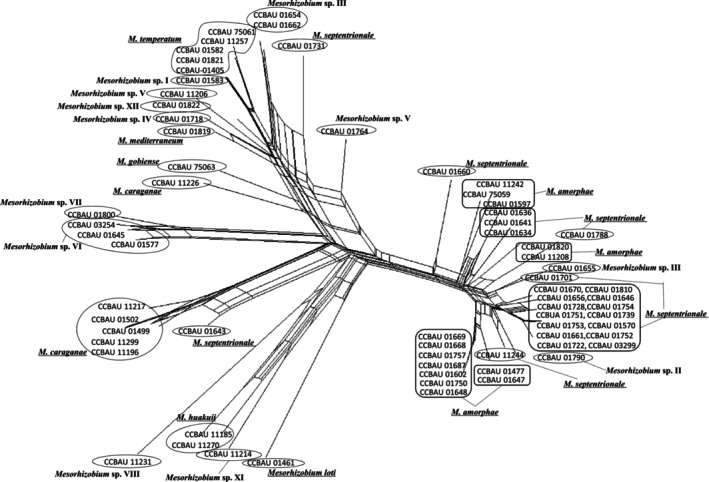 FIGURE 4
FIGURE 4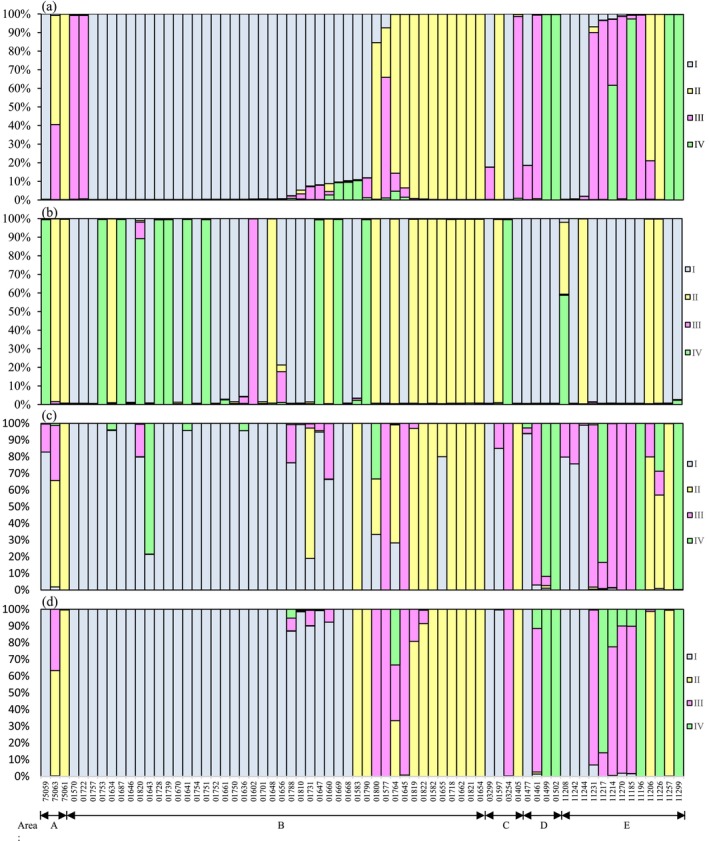 FIGURE 5
FIGURE 5|
| A | B | C | D | E |
|---|---|---|---|---|---|
| Concatenated membrane transporters | |||||
| A | 22.07 | 4.02 | 2.72 | 7.35 | |
| B | 0.07078e | 5.10 | 1.89 | 2.81 | |
| C | 0.07228e | 0.06215e | 0.82 | 11.21 | |
| D | 0.09687e | 0.08627e | 0.09222e | 9.92 | |
| E | 0.09052e | 0.08380e | 0.08823e | 0.07954e | |
| Concatenated nucleotide repair genes | |||||
| A | 18.17 | 4.10 | 8.55 | 18.84 | |
| B | 0.07013e | 6.76 | 4.30 | 3.82 | |
| C | 0.07317e | 0.06594e | 0.02 | 36.76 | |
| D | 0.08771e | 0.07896e | 0.08582e | 16.93 | |
| E | 0.08238e | 0.08199e | 0.08639e | 0.08453e | |
| Concatenated core genes | |||||
| A | 96.64 | −5.53 | 3.47 | 27.05 | |
| B | 0.05465e | −7.09 | 2.87 | 6.68 | |
| C | 0.05631e | 0.05097e | 9.63 | −22.15 | |
| D | 0.06627e | 0.05970e | 0.06143e | −32.35 | |
| E | 0.06269e | 0.05856e | 0.06062e | 0.05715e | |
| Concatenated nodulation genes | |||||
| A | 94.64 | −5.53 | 3.47 | 27.05 | |
| B | 0.00029e | −7.09 | 2.87 | 6.68 | |
| C | 0.05631e | 0.05097e | 9.63 | −22.15 | |
| D | 0.06627e | 0.05970e | 0.06143e | −32.35 | |
| E | 0.06269e | 0.05970e | 0.06062e | 0.05715e | |
| Genes | Length (bp) |
|
|
|
|---|---|---|---|---|
| Concatenated membrane transport genes | 837 | 61 | 2.48 | 0.72 |
| Concatenated nucleotide repair genes | 705 | 57 | 1.15 | 0.33 |
| Concatenated core genes | 879 | 57 | 21.46 | 6.78 |
| Concatenated nodulation genes | 999 | 62 | 171.39 | 63.22 |
| Genes | Length (bp) |
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|---|
| Membrane transporters | ||||||||
|
| 327 | 100 | 129 | 35/0.944 | 0.06547 | 0.23925 | 0.01569 | 0.06558 |
|
| 300 | 99 | 141 | 40/0.970 | 0.09122 | 0.47662 | 0.01149 | 0.02411 |
|
| 210 | 52 | 69 | 35/0.942 | 0.04722 | 0.20279 | 0.00921 | 0.04542 |
| Avg. | 279 | 83.67 | 113.0 | 36.7/0.952 | 0.06797 | 0.30622 | 0.01213 | 0.04503 |
| Nucleotide repair genes | ||||||||
|
| 144 | 54 | 68 | 36/0.935 | 0.08291 | 0.35343 | 0.01814 | 0.05133 |
|
| 561 | 173 | 227 | 45/0.974 | 0.06607 | 0.25698 | 0.01782 | 0.06934 |
| Avg. | 352.5 | 113.5 | 147.5 | 40.5/0.955 | 0.07449 | 0.30521 | 0.01798 | 0.06033 |
| Core genes | ||||||||
|
| 288 | 75 | 98 | 49/0.983 | 0.05502 | 0.23468 | 0.00154 | 0.00656 |
|
| 591 | 179 | 219 | 46/0.958 | 0.05494 | 0.19047 | 0.02090 | 0.10973 |
| Avg. | 439.5 | 127 | 158.5 | 47.5/0.971 | 0.05498 | 0.21258 | 0.01122 | 0.05815 |
| Nodulation genes | ||||||||
|
| 507 | 131 | 149 | 30/0.912 | 0.02718 | 0.07595 | 0.01185 | 0.15602 |
|
| 492 | 119 | 132 | 71/1.000 | 0.04283 | 0.10580 | 0.02666 | 0.25198 |
| Avg. | 499.5 | 125 | 140.5 | 50.5/0.956 | 0.03501 | 0.09088 | 0.01926 | 0.20400 |
| Area (No. of strains) | Length (bp) |
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|---|
| Concatenated membrane transporters | ||||||||
| A (3) | 837 | 102 | 106 | 3/1.000 | 0.08284 | 0.37023 | 0.01139 | 0.03076 |
| B (44) | 837 | 184 | 224 | 29/0.964 | 0.05559 | 0.23325 | 0.00926 | 0.03970 |
| C (4) | 837 | 119 | 131 | 4/1.000 | 0.08224 | 0.34824 | 0.01523 | 0.04373 |
| D (4) | 837 | 116 | 127 | 4/1.000 | 0.08084 | 0.35292 | 0.01457 | 0.04128 |
| E (13) | 837 | 196 | 243 | 12/0.987 | 0.08668 | 0.38097 | 0.01543 | 0.04050 |
| Average | — | 143.4 | 166.2 | 10.4/0.990 | 0.07764 | 0.33712 | 0.01318 | 0.03920 |
| Concatenated nucleotide repair genes | ||||||||
| A (3) | 705 | 83 | 87 | 3/1.000 | 0.08038 | 0.30316 | 0.02337 | 0.07709 |
| B (44) | 705 | 171 | 206 | 30/0.952 | 0.05612 | 0.21654 | 0.01412 | 0.06521 |
| C (4) | 705 | 107 | 114 | 4/1.000 | 0.08629 | 0.34927 | 0.02238 | 0.06408 |
| D (4) | 705 | 102 | 114 | 4/1.000 | 0.08534 | 0.35791 | 0.02154 | 0.06018 |
| E (13) | 705 | 168 | 204 | 12/0.987 | 0.08887 | 0.35717 | 0.02370 | 0.06635 |
| Average | — | 126.2 | 145 | 10.6/0.988 | 0.07940 | 0.31681 | 0.02102 | 0.06658 |
| Concatenated core genes | ||||||||
| A (3) | 879 | 79 | 83 | 3/1.000 | 0.06143 | 0.18865 | 0.01897 | 0.10055 |
| B (44) | 879 | 174 | 199 | 34/0.978 | 0.04729 | 0.17233 | 0.01297 | 0.07526 |
| C (4) | 879 | 96 | 101 | 4/1.000 | 0.06238 | 0.19755 | 0.01709 | 0.08651 |
| D (4) | 879 | 75 | 98 | 49/0.983 | 0.05502 | 0.17007 | 0.01436 | 0.08443 |
| E (13) | 879 | 174 | 193 | 13/1.000 | 0.06167 | 0.19369 | 0.01737 | 0.08967 |
| Average | — | 119.6 | 134.8 | 20.6/0.992 | 0.05756 | 0.18446 | 0.01615 | 0.08728 |
| Concatenated nodulation genes | ||||||||
| A (3) | 999 | 74 | 75 | 3/1.000 | 0.03313 | 0.08849 | 0.03634 | 0.41066 |
| B (44) | 999 | 182 | 196 | 44/1.000 | 0.02718 | 0.07962 | 0.01712 | 0.21502 |
| C (4) | 999 | 46 | 46 | 4/1.000 | 0.02536 | 0.05962 | 0.01348 | 0.22609 |
| D (4) | 999 | 20 | 20 | 4/1.000 | 0.01118 | 0.02938 | 0.00493 | 0.16780 |
| E (13) | 999 | 124 | 130 | 13.1/1.000 | 0.03722 | 0.08425 | 0.02106 | 0.24997 |
| Average | — | 89.2 | 93.4 | 13.62/1.000 | 0.02681 | 0.06827 | 0.01859 | 0.25391 |
- —Natural Science Foundation of Inner Mongolia Autonomous Region of China
- —National Natural Science Foundation of China10.13039/501100001809
- —Key Research and Development Program of Inner Mongolia Autonomous Region of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLegume Nitrogen Fixing Symbiosis · Bacterial Genetics and Biotechnology · Plant-Microbe Interactions and Immunity
Introduction
1
Caragana species, pivotal leguminous plants in barren semi‐fixed deserts of northern China, serve dual ecological roles in windbreak/sand fixation and watershed protection. Dominating the arid landscapes of Inner Mongolia, these areas were characterized by strong winds and extreme temperature fluctuations (−30°C to 50°C; Wu et al. 2015). Caragana's remarkable tolerance to the low‐nutrient conditions positioned it as a keystone species for desert ecosystem restoration (He et al. 2024; Li et al. 2014). This keystone role is owing to its symbiotic association with rhizosphere microbes, which enhance soil fertility and nutrient bioavailability, thereby bolstering Caragana's growth and survival while underpinning broader ecological recovery in degraded desert ecosystems (Liu et al. 2022). Among these microbes, Mesorhizobium stood out as the primary symbiotic microsymbiont, forming mutualistic relationships with Caragana to secure sufficient nitrogen nutrition (Ji et al. 2015). Notably, Mesorhizobium exhibited superior alkaline tolerance compared to other rhizobia, a trait attributed to its capacity to secrete organic acids, enabling colonization and N_2_ fixation in the alkaline soils typical of Caragana habitats (Chen et al. 2010). This adaptation was pivotal, enabling Mesorhizobium to thrive in Caragana's root zone and establish a robust symbiosis that enhanced plant resilience in harsh environments.
The Caragana‐Mesorhizobium symbiosis balanced mutual benefits (Caragana gains nitrogen, Mesorhizobium gains carbon) with energetic costs to the plant. Nevertheless, the fitness gains for both organisms justified this mutualism as the evolutionarily stable strategy (Carlson and Frederickson 2023; Mathesius 2022). Various Mesorhizobium species, including M. amorphae , M. septentrionale , M. caraganae , M. gobiense , M. temperatum , M. mediterraneum , M. shangrilense , M. tianshanense , M. huakuii , and M. metallidurans , have been documented to nodulate Caragana in extreme arid/semi‐arid deserts with fluctuating temperatures (Liu et al. 2022; Yan et al. 2017).
The ecological persistence and environmental adaptation of Mesorhizobium symbionts associated with Caragana in northern China deserts depend on pivotal genetic systems. Specifically, membrane transporter genes such as cysW (encoding a cysteine ABC transporter subunit, critical for sulfur assimilation and cysteine uptake to support redox balance; Chakraborty et al. 2021; Li et al. 2025), exoY (involved in exopolysaccharide biosynthesis, facilitating biofilm formation and adhesion to root surfaces; Jiang et al. 2001; Sakthivel et al. 2025), and idhA (encoding isocitrate dehydrogenase, a key enzyme in the tricarboxylic acid cycle that sustains energy production under nutrient limitation; Yang et al. 2022) were essential for adapting to nutrient‐poor and alkaline soils. Concurrently, nucleotide repair genes like mutS (mediating mismatch repair to correct replication errors and maintain genomic integrity; Margara et al. 2016; Martín‐Blecua et al. 2025) and uvrC (participating in nucleotide excision repair to counteract DNA damage from UV radiation or reactive oxygen species; Najjari et al. 2023; Santoyo et al. 2005) ensured genetic stability in harsh environments. Together, these genes underpinned the adaptive plasticity of Mesorhizobium, enabling it to thrive in extreme desert conditions and sustain its symbiotic relationship with Caragana. Therefore, adaptive strategies in Caragana‐nodulating Mesorhizobium would be investigated in this study, with a focus on genetic diversity, recombination, and gene flow mediated by membrane transporter genes (cysW, exoY, idhA) and nucleotide repair genes (mutS, uvrC) for clarifying the adaptation of symbiotic rhizobia to extreme environments.
Materials and Methods
2
Rhizobial Strains and Gene Sequencing
2.1
A total of 708 mesorhizobial strains isolated from the roots of Caragana distributed across five desert areas (Areas A–E, three sampling sites in Areas A, C, D; four in Area B; and six in Area E) along the semi‐fixed desert belt in northern China (data not shown). And 68 representatives were selected based on the phylogenetic tree of the recA gene for analysis (Table S1). Nine type strains including Mesorhizobium septentrionale SDW 014^T^, M. gobiense CCBAU 83330^T^, M. amorphae ACCC 19665^T^, M. mediterraneum USDA 3392^T^, M. tianshanense CCBAU 3306^T^, M. metallidurans LMG 24485^T^, M. temperatum SDW018^T^ and M. huakuii CCBAU 2609^T^ and M. caraganae CCBAU 11299^T^ were employed as references to establish the phylogenetic relationship of these Caragana‐nodulating mesorhizobia. All strains were cultured in YMA medium at 28°C and preserved in 30% glycerol at −80°C following established protocols.
DNA Extraction and Gene Sequencing
2.2
The total template DNA of these Caragana mesorhizobia was extracted using the method as described previously (Terefework et al. 2001). The biological function and PCR amplification protocols were shown in Table S2 for nine genes, including three membrane transporter genes cysW (Sirko et al. 1995), exoY (Reed et al. 1991), idhA (Jiang et al. 2001), and two nucleotide repair genes mutS (Martínez‐Salazar et al. 2009), uvrC (Cubo et al. 1992), two core genes recA, rpoB, and two nodulation genes nodC, nodD. The primers were designed by using Premier 5.0 software based on conserved regions of cysW, exoY, idhA, mutS and uvrC, identified from whole‐genome sequences of the reference Mesorhizobium strains (CCBAU01583, CCBAU01570, CCBAU01502, CCBAU01399), as detailed in Table S2. And the PCR products were purified and commercially sequenced by ABI 3730xl sequenator in Beijing, China. Then, the sequences were manually checked using Chromas Pro (Ver. 2.1.3) and edited manually using DNAMAN (Ver. 9.0). Total 340 nucleotide sequences of 5 genes cysW, exoY, idhA, mutS and uvrC were newly obtained in this study which were deposited in the GenBank database under accession numbers MK252304 through MK252643 (Table S4).
Phylogenetic Analyses
2.3
These nucleotide sequences were aligned and the redundant bases removed using the ClustalW program. MEGA11.0 program was used for constructing the Neighbor‐Joining (NJ) phylogenetic trees for membrane transporters, nucleotide repair, core, and nodulation genes with the Kimura‐2‐parameter model (1000 bootstrap replicates) (Tamura et al. 2011), respectively. Then, reticulate evolutionary events, including potential hybridization and recombination, were inferred by quantifying phylogenetic network complexity using SPLITSTREE 4.0 software with the Neighbor‐Net algorithm (1000 bootstrap replicates; Huson and Bryant 2006).
Nucleotide Polymorphism and Population Genetics Analyses
2.4
Nucleotide polymorphisms including the haplotype diversity (Hd), number of haplotypes (h), nucleotide diversity (π), the π _ N _/π _ S _ ratios (where πS denotes synonymous substitutions per synonymous site and πN denotes non‐synonymous substitutions per non‐synonymous site) (Nei and Gojobori 1986), and two additional metrics: Dxy (average nucleotide divergence) and Nm (gene flow estimated as migrants) were estimated using DnaSP v5 software (Librado and Rozas 2009). STRUCTURE software was employed to analyze admixture levels in Mesorhizobium genospecies under the LOCPRIOR model (100,000 burn‐in and 1,000,000 iterations), with K (number of ancestral subpopulations) estimated separately for membrane transporters and nucleotide repair genes (Hubisz et al. 2009; Jakobsson and Rosenberg 2007).
Recombination Estimation
2.5
The minimal recombination events (Rm) within the populations were estimated using the DnaSP v5 (Hudson and Kaplan 1985). Two recombination rates—r/m (the relative impact of recombination compared with that of point mutation in lineage genetic diversification) and ρ/θ (the relative frequency of recombination occurrence compared with that of point mutation in lineage history), were calculated. The effect of recombination on evolutionary history was deduced using CLONALFRAME with proper parameters (100,000 burn‐in iterations plus 1,000,000 sampling iterations per run) for three independent runs (Didelot and Falush 2007).
Results
3
Phylogenies and Nucleotide Polymorphisms of Caragana Mesorhizobia
3.1
These representative Caragana‐associated mesorhizobia, isolated from different areas (A–E), were distributed across different branches, without geographical isolation across five distinct areas of the semi‐fixed desert belt in northern China. The dominant mesorhizobial communities of areas (A–E) comprised genospecies M. amorphae and M. septentrionale , which also exhibited a closely genetic relationship on the phylogenetic trees of membrane transporters (Figure 1), nucleotide repair genes (Figure 2), and core genes (Figure S1). Notably, the genospecies M. amorphae encompassed representative strains of Mesorhizobium sampled from all five desert areas (A–E) with an extensive genetic admixture and cross‐regional dispersal. This adaptation was primarily mediated by the convergent evolution of key genes, including membrane transporters (cysW, exoY, idhA) and nucleotide repair genes (mutS, uvrC), which overcame geographic barriers. In addition, the topological structures of the phylogenetic trees were similar to each other based on the concatenated sequences of membrane transporters, nucleotide repair genes, and core genes, although some variations were observed in specific cases (e.g., CCBAU 01790, CCBAU 01655, CCBAU 11226, and CCBAU 01731). However, the phylogenetic tree based on nodulation genes (nodC, nodD) exhibited a markedly different topological structure (Figure S2), although these mesorhizobial strains cross‐dispersed across five areas branched into multiple clades.
Neighbor‐Joining (NJ) tree constructed based upon the concatenated sequences of membrane transporters. Three membrane transporter genes (cysW, exoY, idhA) were used. Bootstrap values greater than 70% are indicated at the branch points. The type strains (Bold Fonts) are shown in parentheses after the strain numbers. The scale bar represents 1% nucleotide substitutions.
Neighbor‐Joining (NJ) tree constructed based upon the concatenated sequences of nucleotide repair genes. Two nucleotide repair genes (mutS, uvrC) were used. Bootstrap values greater than 70% are indicated at the branch points. The type strains (Bold Fonts) are shown in parentheses after the strain numbers. The scale bar represents 1% nucleotide substitutions.
Nucleotide polymorphisms for each gene were presented in Table 3. No significant differences were detected in haplotype number (h) and haplotype diversity (Hd) values across the tested genes. Notably, the average nucleotide diversity (π) value of nodulation genes (0.03501) was markedly lower than that of membrane transporters (0.06797), nucleotide repair genes (0.07449), and core genes (0.05498), highlighting the particularity of symbiotic adaptation. It is noted that the πS values (synonymous substitutions per site) were significantly higher than πN values (non‐synonymous substitutions per site) for all tested genes. The πN/πS values were significantly higher for nodC and nodD than for other genes. In addition, π and πN/πS values of these strains from all five areas (A–E) were consistent for core, membrane transporter, and nucleotide repair genes, respectively (Table 4).
Gene Flow and Genetic Divergence of Caragana‐Associated Mesorhizobia
3.2
NeighborNet phylogenetic network analysis revealed a complex, reticulate recombination network architecture in both membrane transporter and nucleotide repair gene datasets (Figures 3 and 4), indicative of pervasive recombination‐driven gene flow and genetic exchange among Caragana‐associated mesorhizobia across the five desert areas (A–E) in northern China. In addition, four distinct genetic lineages (I, II, III, and IV) were identified by STRUCTURE analysis (Figure 5). Notably, lineage I was ubiquitously distributed across all five areas and served as the primary ancestral source in Area B. Lineages II–IV, by contrast, displayed irregular, patchy distributions, likely reflecting localized adaptation or stochastic recombination events. Critically, most strains (e.g., CCBAU 75063 and CCBAU 01764) showed evidence of mixed ancestry, inheriting genetic material from two or more lineages (Figure 5).
The Neighbor‐Nets generated using SplitsTree4 with the Hamming distance option. The trees are constructed based upon the concatenated membrane transporter genes (cysW, exoY, idhA). The scale bar represents 1% nucleotide substitutions.
The Neighbor‐Nets generated using SplitsTree4 with the Hamming distance option. The trees are constructed based upon the concatenated nucleotide repair genes (mutS, uvrC). The scale bar represents 1% nucleotide substitutions.
STRUCTURE analyses of Caragana‐associated Mesorhizobium populations from different areas. Concatenated core (a), nodulation (b), membrane transporter (c) and nucleotide repair (d) genes were analyzed. The inferred ancestries are designated sequentially as I, II, III, and IV, shown in bars and filled with different grayness or dots. The horizontal axis represents current Mesorhizobium individuals (in the same order in all panels), and the bar for each individual is filled according to the inferred proportions of single‐nucleotide alleles that were derived from each of the ancestries. The vertical axis represents the percentage of the strains.
Genetic differentiation (quantified as pairwise Dxy) among rhizobial populations isolated from the five desert areas (A–E) exhibited no statistically significant variation (Table 1). Notably, both the mean genetic distance (Dxy) and nucleotide diversity (π) values remained comparable across all areas (Tables 1 and 4), reflecting consistent levels of intra‐population genetic variation despite geographic dispersion. Dxy values, calculated from the concatenated membrane transporter genes, ranged from a minimum of 0.06215 (between areas B and C) to a maximum of 0.09687 (between areas A and D).
Recombination Events Among the Caragana Mesorhizobial Lineages
3.3
A total of 61 and 57 minimal recombination events (Rm) were inferred using DnaSP v5 software for the concatenated membrane transporter and nucleotide repair genes, respectively (Table 2). Subsequent analysis with CLONALFRAME revealed contrasting evolutionary dynamics: membrane transporter genes exhibited a higher recombination impact (r/m = 2.48) and a higher frequency (ρ/θ = 0.72) compared to nucleotide repair genes (r/m = 1.15; ρ/θ = 0.33). Notably, r/m values were > 1.0 for both gene categories, showing that recombination, not mutation, was the dominant driver of genetic diversification. This evolutionary process was also corroborated by SH tests, which showed significant topological incongruence between gene trees of membrane transporter/nucleotide repair loci and the core gene‐derived species tree (p < 0.05; Table S3).
Discussion
4
Phylogenies and Genetic Divergence of Caragana Mesorhizobia Based on Membrane Transporters and Nucleotide Repair Genes
4.1
The phylogenetic congruence observed across membrane transporters and nucleotide repair genes (Figures 1 and 2) revealed an evolutionary conservatism pattern of Caragana‐symbiotic Mesorhizobium, with parallel adaptive trajectories in response to shared environmental pressures (Ament‐Velásquez et al. 2022; Yamamoto et al. 2021), despite geographic isolation across northern China's semi‐fixed deserts (spanning > 1000 km). The intertwined phylogenetic patterns indicated that environmental selection pressures likely dominated the genetic adaptation of these strains (Ji et al. 2020). The contrasting topologies observed in nodulation gene trees highlighted the divergent evolutionary trajectories of symbiosis‐related traits, which are likely shaped by host‐specific selection pressures (Provorov et al. 2022). In contrast, the conserved topologies of membrane‐transporter and nucleotide repair gene trees (Figures 1 and 2) suggested that these loci were under strong purifying selection to maintain basal physiological functions critical for desert survival (Karasev et al. 2023). This phylogenetic cohesion contrasted sharply with the divergence observed in nodulation genes (nodC, nodD), suggesting that membrane transporters and DNA repair enzymes operated under distinct selective regimes (Karasev et al. 2023; Liu et al. 2023). Therefore, the strong selection pressure (pH > 8.0, K^+^ content shown in Table S1) in the semi‐fixed desert belt enhanced the compositional similarity index of these Mesorhizobium strains and greatly weakened their distance‐decay relationship, one of the most well‐documented universal biogeographic patterns in natural ecosystems (Chen et al. 2016; Ji et al. 2015; Zhang et al. 2020). Therefore, in the dynamic environmental conditions of northern China's semi‐fixed deserts, Mesorhizobium strains exhibit a striking evolutionary signature characterized by strong purifying selection alongside rapid adaptive evolution, reflecting their dual response to abiotic stress homogenization and niche‐specific innovation (Teng et al. 2023).
The uniform nucleotide diversity (π) and low πN/πS ratios (< 1.0) across all populations (Table 4) indicated that pervasive purifying selection might act on membrane transporters and nucleotide repair genes. These genes encode critical functions for desert survival: cysW mediates sulfate uptake for osmotic regulation (Srivastava et al. 2025), uvrC repairs UV‐induced DNA lesions (Selby 2017), while idhA modulates carbon flux under nutrient limitation (Lin et al. 2022). No significant difference in genetic divergence (Dxy) values among Caragana‐symbiotic Mesorhizobium from geographically distinct areas (Table 1) was observed, inferring that these strains might be canalized by abiotic stressors, such as extreme temperatures (−30°C to 50°C) (Peters et al. 2026; Rychel et al. 2025), alkaline soils (pH 8.5–10.2) (Shah et al. 2022), and saline‐alkali gradients (Fall et al. 2019), to maintain physiological homeostasis, and environmental selection might be the dominant driver.
In contrast to the divergence observed in nodulation genes (Wendlandt et al. 2021), the high gene flow and recombination rates among membrane transporter genes (Table 2) indicated that these genes were subject to host selection for desert‐adapted Mesorhizobium. This was exemplified by lineage I, which dominated in all five areas (Figure 5) and carried a suite of broadly adaptive genes. Such genomic fluidity, driven by recombination, allowed rapid dissemination of beneficial mutations, and ensured metabolic robustness across fragmented desert habitats, confirming that recombination was an essential agent of adaptive homogenization for Caragana‐symbiotic Mesorhizobium (Good et al. 2025; Mazzamurro et al. 2025).
While symbiosis‐related genes (nodC, nodD) exhibited elevated πN/πS ratios, reflecting host‐driven diversification (Singh and Valdés‐López 2023), the stoichiometric stability of membrane transporters and DNA repair enzymes underscored their role as evolutionary “buffer zones”(Moger‐Reischer et al. 2023). By maintaining core physiological functions, these genes relieved selective constraints on symbiosis loci, allowing niche‐specific innovations to evolve independently (Forni et al. 2025). This modular evolutionary architecture, where housekeeping (core) genes prioritized environmental adaptation and symbiosis genes prioritized host specialization, might underpin the resilience of desert rhizobia‐legume mutualisms, reflecting the implications for symbiosis‐environment coevolution.
Gene Flow, Recombination, and Environmental Adaptation of Caragana Mesorhizobia
4.2
The gene flow among all mesorhizobia, especially between the dominant genospecies M. septentrionale and M. amorphae associated with Caragana, was frequent for all tested genes (Figures S1 and S2), indicating that genetic diversity between coexisting strains of the same genospecies or closely related genospecies is an important ecological mechanism explaining the diversity of Caragana mesorhizobia (Fields et al. 2022). Extensive gene flow, characterized by elevated Nm values (Table 1), was observed across Caragana‐associated Mesorhizobium populations. Notably, membrane transporters exhibited particularly high gene flow between areas A and B (Nm = 22.07), whereas nucleotide repair genes showed pronounced gene flow between areas C and E (Nm = 36.76). These patterns underscored the dominant role of gene recombination in shaping the evolutionary trajectories of these symbionts. Complex gene interweaving, clearly found among many strains isolated from areas A to E (Figure 5), also indicated that Caragana‐associated Mesorhizobium populations experienced similar selection pressures, although they alternated between semi‐fixed desert soil and host environments (Burghardt et al. 2019). The high relative impact (r/m) and frequency (ρ/θ) of recombination for concatenated membrane transporter genes (Table 2) also indicated that gene recombination had played an important role in the evolutionary history. The strains tended to acquire special transporters in membranes for nutrient acquisition via recombination in various hostile environments, enhancing the evolutionary stability of nodulation on their hosts (de Faria et al. 2022). This pattern was particularly evident in membrane transporters (cysW, exoY, idhA), which exhibited the highest recombination frequencies (r/m = 2.48), underscoring their adaptive function across desert landscapes. In contrast, nucleotide repair genes (mutS, uvrC) maintained stricter purifying selection (πN/πS < 1), ensuring genome integrity under extreme abiotic stress (Lin et al. 2007). A selection‐driven recombination paradigm revealed that membrane transporters prioritized functional stability over sequence divergence. For example, recombination hotspots in the cysW gene spanned substrate‐binding domains, optimizing sulfate uptake efficiency under fluctuating salinity (Xamxidin et al. 2025), and nucleotide repair genes preserved symbiotic fidelity by suppressing non‐synonymous substitutions, enabling host‐specific nodulation traits to evolve independently, such as uvrC assisting in the adaptive piecing‐together of UV repair modules (Springall et al. 2018). This dual strategy, functional optimization via recombination for membrane genes and functional optimization via purifying selection for repair genes, enabled simultaneous adaptation to abiotic stress and maintenance of symbiotic precision. The congruent phylogenetic patterns of membrane transporters and nucleotide repair genes (Figures 1 and 2) further contrasted with the divergence of symbiosis‐related loci (nodC, nodD), revealing a modular evolutionary architecture where core genes buffer physiological stress.
Collectively, these findings established that recombination, rather than geographic isolation, represented the predominant evolutionary force shaping the adaptive trajectories of membrane transporters and nucleotide repair genes, thereby conferring ecological resilience to Caragana‐associated Mesorhizobium across heterogeneous desert landscapes. However, quantitative integration of environmental variables (e.g., soil pH, salinity) with genetic data lacked here. Future studies should employ systematic sampling and eco‐genomic approaches to disentangle abiotic drivers of adaptation.
Conclusion
5
The evolutionary trajectories of Caragana‐associated Mesorhizobium populations exhibited striking congruence in membrane transporters and nucleotide repair genes, reflecting adaptive convergence shaped by the harsh desert environment. Despite originating from geographically distinct areas (A–E) across the semi‐fixed desert belt in northern China, these rhizobia demonstrated synchronized evolutionary patterns, likely driven by homogenizing selective pressures, highlighting the dominance of environmental homogeneity over geographic isolation in structuring microbial communities. Genetic divergence among populations arose primarily via recombination, rather than mutation, with extensive recombination disseminating adaptive loci. Our findings illuminated the concerted evolutionary dynamics and shared selective pressures of microbial resilience in extreme ecosystems. Future studies should integrate functional genomics to unravel the adaptive roles of these genes and explore synthetic biology approaches to engineer stress‐resilient microbial populations.
Author Contributions
Xiaoxia Yuan: investigation (lead), writing – original draft (lead). Hua Li: funding acquisition (supporting), project administration (supporting). Xiumin Yu: data curation (equal). Zhaojun Ji: funding acquisition (supporting), project administration (equal), writing – review and editing (lead).
Funding
This study was financially supported by Natural Science Foundation of Inner Mongolia Autonomous Region of China (2024JQ12), National Natural Science Foundation of China (32170020), and Key Research and Development Program of Inner Mongolia Autonomous Region of China (2025YFDZ0116).
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
FIGURE S1: Neighbor‐Joining (NJ) tree constructed based upon the concatenated sequences of core. FIGURE S2: Neighbor‐Joining (NJ) tree constructed based upon the concatenated sequences of nodulation genes. TABLE S1: A total of 68 representative strains isolated from Caragana‐associated Mesorhizobium used in this study. TABLE S2: Primers designed for the genes and the annealing temperature of them (Tm, °C) in PCR amplification. TABLE S3: Shimodaira‐Hasegaewa (SH) test of each test gene locus in comparison with the concatenated core genes. TABLE S4: Accession numbers of the genes obtained in this study and deposited in GenBank.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ament‐Velásquez, S. L. , C. Gilchrist , A. Rêgo , et al. 2022. “The Dynamics of Adaptation to Stress From Standing Genetic Variation and de Novo Mutations.” Molecular Biology and Evolution 39, no. 11: msac 242. 10.1093/molbev/msac 242.36334099 PMC 9703598 · doi ↗ · pubmed ↗
- 2Burghardt, L. T. , B. Epstein , and P. Tiffin . 2019. “Legacy of Prior Host and Soil Selection on Rhizobial Fitness in Planta.” Evolution 73, no. 9: 2013–2023. 10.1111/evo.13807.31334838 · doi ↗ · pubmed ↗
- 3Carlson, C. , and M. E. Frederickson . 2023. “An Emerging View of Coevolution in the Legume‐Rhizobium Mutualism.” Molecular Ecology 32, no. 14: 3793–3797. 10.1111/mec.17055.37350376 · doi ↗ · pubmed ↗
- 4Chakraborty, S. , H. E. Driscoll , J. E. Abrahante , F. Zhang , R. F. Fisher , and J. M. Harris . 2021. “Salt Stress Enhances Early Symbiotic Gene Expression in Medicago Truncatula and Induces a Stress‐Specific Set of Rhizobium‐Responsive Genes.” Molecular Plant‐Microbe Interactions (MPMI) 34, no. 8: 904–921. 10.1094/mpmi-01-21-0019-r.33819071 PMC 8578154 · doi ↗ · pubmed ↗
- 5Chen, J. , M. Hu , H. Ma , et al. 2016. “Genetic Diversity and Distribution of Bradyrhizobia Nodulating Peanut in Acid‐Neutral Soils in Guangdong Province.” Systematic and Applied Microbiology 39, no. 6: 418–427. 10.1016/j.syapm.2016.06.002.27499533 · doi ↗ · pubmed ↗
- 6Chen, W. M. , W. F. Zhu , C. Bontemps , J. P. Young , and G. H. Wei . 2010. “ Mesorhizobium alhagi sp. Nov., Isolated From Wild Alhagi sparsifolia in North‐Western China.” International Journal of Systematic and Evolutionary Microbiology 60, no. Pt 4: 958–962. 10.1099/ijs.0.014043-0.19661514 · doi ↗ · pubmed ↗
- 7Cubo, M. T. , A. Economou , G. Murphy , A. W. Johnston , and J. A. Downie . 1992. “Molecular Characterization and Regulation of the Rhizosphere‐Expressed Genes Rhiabcr That Can Influence Nodulation by Rhizobium leguminosarum Biovar Viciae .” Journal of Bacteriology 174, no. 12: 4026–4035. 10.1128/jb.174.12.4026-4035.1992.1597418 PMC 206112 · doi ↗ · pubmed ↗
- 8de Faria, S. M. , J. J. Ringelberg , E. Gross , et al. 2022. “The Innovation of the Symbiosome Has Enhanced the Evolutionary Stability of Nitrogen Fixation in Legumes.” New Phytologist 235, no. 6: 2365–2377. 10.1111/nph.18321.35901264 PMC 9541511 · doi ↗ · pubmed ↗