Tutorial for variant interrogation in tumor samples
Riley J. Arseneau, Leah K. MacLean, Jeanette E. Boudreau, Daniel Gaston

TL;DR
This paper provides a step-by-step guide to help researchers analyze genetic mutations in cancer samples using next-generation sequencing.
Contribution
The paper introduces a practical, accessible framework for variant analysis in tumor samples, emphasizing reproducibility and clinical relevance.
Findings
The framework is structured into four phases: Planning, Gathering Resources, Filtering and Validation, and Dissemination and Storage.
It emphasizes the importance of reproducibility and transparency in variant analysis for translational research.
The guide is designed to lower the barrier for researchers new to the field of somatic mutation analysis.
Abstract
The increasing accessibility of next-generation sequencing has empowered researchers to investigate somatic mutations in cancer. The complexity of variant analysis pipelines, terminology, and tool selection remains a major barrier, especially for those new to the field or working in translational settings. To address this challenge, we present a practical framework that guides researchers through the critical steps of variant interrogation in tumor samples. This guide is broken into four phases: Planning—laying the foundation for thoughtful experimental design and a clear understanding of sequencing outputs; Gathering Resources—assembling the tools, reference data, and variant annotation sets required for analysis; Filtering and Validation—executing a systematic approach to prioritize meaningful variants; and Dissemination and Storage—ensuring findings are reproducible and accessible…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
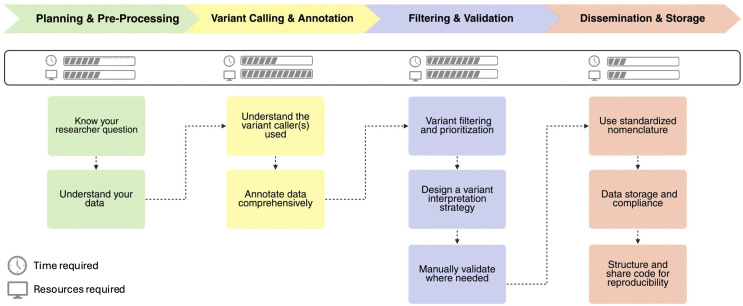 Fig 1
Fig 1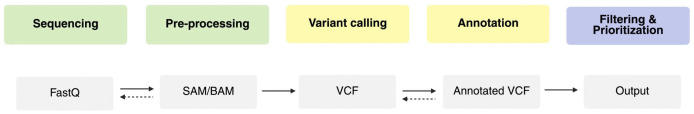 Fig 2
Fig 2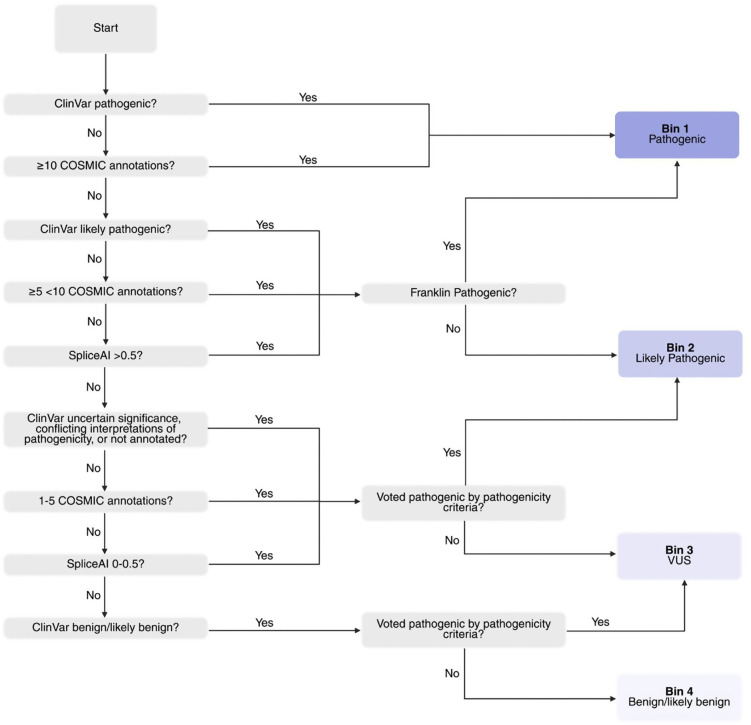 Fig 3
Fig 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer Genomics and Diagnostics · Genomics and Rare Diseases · Genomic variations and chromosomal abnormalities
Introduction
Next-generation sequencing (NGS) enables the investigation of somatic mutations in cancer. However, the concurrent proliferation of analysis pipelines, plugins, and programs [1] can be overwhelming for beginners. This tutorial for variant interrogation in tumor samples (Fig 1 provides a practical roadmap for analyzing sequencing data and disseminating findings. It is intended for researchers new to NGS or seeking greater confidence in variant analysis workflows.
General workflow of variant interrogation in tumor samples.Please note that the steps are intended to be taken sequentially, and each should be completed before moving on to the next; however, depending on how data has been processed prior to your work, it may be necessary to start later in the pipeline. Created in BioRender. Arseneau, R. (2026) https://BioRender.com/armq9mn.
We aim to empower researchers to navigate variant interrogation in tumor samples using the tools available publicly. Inspired by clinical guidelines but adapted for translational research, this guide excludes clinical decision making, which requires clinical training, licensure, and stringent criteria [2,3]. Our glossary of key terms and concepts should be reviewed before reading the tutorial (Table 1). Most sequencing analysis, including many of the tools discussed in this article, requires familiarity with the command line interface (CLI); resources are available elsewhere [4]. CLI code examples are provided throughout this manuscript, and working through our demonstration dataset will reinforce key principles (S1 Fig, S1 Data).
Table 1: Key terms and concepts.
Phase 1: Planning and pre-processing
Tailor the sequencing approach or selection of existing datasets to the research question
Whether generating new data or analyzing existing datasets, the research question(s) determines the most appropriate sequencing type, as methods differ in variant detection [1,5]. An ill-suited method risks poor data [5], while the right approach maximizes relevant variant detection [5]. Key guiding questions include:
Are you characterizing alterations across the genome, or focusing on specific genes or mutation types (e.g., single nucleotide variants (SNVs), insertions/deletions (indels), structural variants (SVs), or copy number variations (CNVs))?Are you seeking novel or low-frequency variants?Are you interested in coding regions, non-coding regions, or both?
These questions clarify whether whole genome sequencing (WGS), whole exome sequencing (WES), or targeted sequencing (TS) best suits the study. Each involves trade-offs in breadth and depth of coverage, variant detection capability, and cost (Table 2). Higher depth of coverage, or read depth, increases confidence in detecting low-frequency variants [6], while breadth of coverage reflects how much of the genome is sequenced [7]. Depth is particularly relevant in highly heterogenous samples like tumors, where high variability and low-frequency mutations are expected [6]. Higher depth increases cost and computational requirements; WES/TS offer higher depth over smaller regions, while WGS provides broad coverage at lower per-region depth [1]. While each approach can detect the types of variants listed in Table 2, their sensitivity varies (e.g., CNV and SV detection with WES and TS is limited by capture biases and uneven coverage) [1,8,9].
Table 2: Summary of sequencing approaches for variant detection.
Long read sequencing technologies are increasingly incorporated into cancer genomic studies [10]. While they offer improved SV detection and resolution of complex regions, they have higher error rates, lower throughput, and greater cost compared to short-read platforms [10,11].
Even with a clear strategy, practical constraints may necessitate compromise. Considerations include:
Tumor content: Samples with <30% tumor cells require greater sequencing depth due to reduced sensitivity [14].Sample quality: Fresh frozen samples generally yield high-quality DNA, while formalin-fixed paraffin-embedded (FFPE) tissues often require higher depth to account for artifacts [15].Budget: WGS costs most per sample, despite having the lowest cost per base pair.Computational resources: requisite processing power is directly proportional to the amount of data; narrowing breadth of coverage reduces data burden.Control samples: Control samples (e.g., commercial samples [16], panel of normals, germline samples) [17–19] improve confidence of variant calling [20].Public dataset (e.g., The Cancer Genome Atlas [21]) availability may necessitate adaption of the research objectives.
Understand the capabilities of the sequencing data used
Understand the sequencing data type and its processing history. Data may be received at any stage in the processing pipeline, with each step involving different files, tools, and assumptions. Incomplete or overprocessed data can lead to false positives, missed variants, and irreproducible results [22,23].
The typical workflow includes pre-processing/alignment, variant calling, annotation, filtering, and prioritization (Fig 2). Table 3 outlines common genomic file type structures (e.g., FASTQ, sequencing alignment map (SAM)/ binary alignment map (BAM), variant call format (VCF), and annotated VCFs).
Table 3: Common sequencing file types and their structure.
Flowchart of common sequencing file types and analysis stages.The diagram illustrates the typical file types encountered throughout the sequencing and analysis pipeline, progressing from raw data (left) to results (right). File types are grouped by processing phase: green (Phase 1: Planning and pre-processing), yellow (Phase 2: Variant calling and annotation), and blue (Phase 3: Filtering and validation). Solid arrows indicate the standard forward progression of file generation, while dotted arrows represent steps where data can be reverted to a previous file type. The objective is to complete the pipeline, transforming raw reads into interpretable variants. Created in BioRender. Arseneau, R. (2026) https://BioRender.com/wx6ql68.
Questions to understand previous processing:
FASTQ: Are the reads raw or processed (e.g., adaptor trimmed)? Which tools and/or thresholds were used?SAM/BAM: Has alignment been performed? Were they aligned to a modern reference genome?VCF: Which variant caller(s) was used? Were any filtering parameters applied?Annotated VCF: What annotations were applied? Are they suitable for your goals? Were any filters applied that could limit variant output?
CLI Example Code 1. FASTQ pre-processing.
Quality check raw FASTQ files using FastQC
INPUT: sample_R1.fastq.gz and sample_R2.fastq.gz (paired-end reads)
OUTPUT: HTML and.zip reports in qc_reports/ directory
fastqc sample_R1.fastq.gz sample_R2.fastq.gz -o qc_reports/
Align reads to GRCh38 reference genome using BWA-MEM
INPUT: FASTQ files (R1 and R2), reference genome GRCh38.fa
OUTPUT: unsorted SAM stream
bwa mem GRCh38.fa sample_R1.fastq.gz sample_R2.fastq.gz -o sample.sam
Convert SAM to BAM using samtools view
INPUT: sample.sam
OUTPUT: sample.bam
samtools view -@ 8 -bS -o sample.bam sample.sam
Sort BAM file by genomic coordinates
INPUT: sample.bam
OUTPUT: sample_sorted.bam
samtools sort -@ 8 -o sample_sorted.bam sample.bam
Index BAM for fast retrieval in downstream tools
INPUT: sample_sorted.bam
OUTPUT: sample_sorted.bam.bai (index file)
samtools index sample_sorted.bam
Phase 2: Variant calling and annotation
Understand the variant caller(s) used
Variant calling transforms sequencing data into a list of genetic changes and is typically the most computationally demanding step [27]. While DRAGEN [28], MuTect2 [29], and GATK HaplotypeCaller [30] are widely used callers for detecting SNVs and Indels [22,31], they are not optimal for all sample types or goals. Challenging samples or specialized analyses may require adjusting thresholds or selecting alternate callers [32]. Adjustable caller settings, such as minimum read depth, base quality score thresholds, variant allele frequency (VAF) cutoffs, and variant quality scores, should match the study’s goals; inappropriate thresholds risk false negatives or false positives [33]. Table 4 outlines several variant callers and their typical use cases.
Table 4: Variant Caller information.
CLI Example Code 2. SNV calling with MuTect2
Call somatic variants using GATK Mutect2
INPUT: tumor BAM, reference genome GRCh38.fa
OUTPUT: somatic.vcf.gz (compressed VCF of called variants)
gatk Mutect2 \
-R GRCh38.fa \ # reference genome
-I tumor.bam \ # tumor sample BAM
-O somatic.vcf.gz # output VCF file
Key considerations when selecting and configuring variant caller(s):
Variant type: Tools vary in sensitivity to detect different types of variants. Comparative studies [44,45] and documentation can guide selection. Note that SNV and CNV calling can be inconsistent between tools, so validation and cross-caller consensus may be necessary [22]. Consensus calling reduces false positives but may exclude true variants, so it is generally best used for validation or high-confidence reporting, rather than exploratory analyses.Sample heterogeneity: Highly heterogenous tumors may require lowering VAF or read depth thresholds to capture subclonal variants [46].Sample quality: FFPE DNA is prone to artifacts (e.g., cytosine deamination (C > T) transitions) [15]. Minimize false positives by increasing quality thresholds [15,46].Discovery vs. validation: For exploratory analysis, relaxing filtering parameters and/or using multiple variant callers can maximize sensitivity. For validation, stricter filters may be warranted.
Variant callers require alignment to an up-to-date reference genome (e.g., National Library of Medicine [47] or Ensembl [48,49]. If SAM/BAM or FASTQ files are available, it’s best practice to re-align to a modern reference genome. If only VCF files are available, coordinates can be converted between assemblies with LiftOver tools (e.g., BCFtools/liftover, CrossMap [50,51]) for better annotation.
CLI Example Code 3. Cross Caller Consensus Using BCFtools
Intersect variants from two callers using bcftools isec
INPUT: VCF from caller 1 (c1.vcf) and caller 2 (c2.vcf)
Use bcftools isec to find variants detected by BOTH tools.
OUTPUT: consensus_output/ directory containing:
# 0000.vcf - > intersection of both callers
# 0001.vcf - > unique to first file (c1.vcf)
# 0002.vcf - > unique to second file (c2.vcf)
# sites.txt - > list of positions considered in the comparison
NOTE: -n = 2 ensures only variants present in both files are included in 0000.vcf.
bcftools isec -n=2 c1.vcf c2.vcf -p consensus_output/
CLI Example Code 4. LiftOver with CrossMap
Convert VCF coordinates from GRCh37 to GRCh38 using CrossMap
INPUT: chain file (GRCh37_to_GRCh38.chain), VCF file, reference genome
OUTPUT: somatic_lifted.vcf
CrossMap.py vcf GRCh37_to_GRCh38.chain somatic_filtered.vcf.gz \
GRCh38.fa somatic_lifted.vcf
Annotate data comprehensively
Annotation adds biological context, enables prioritization of meaningful variants, and reduces the need for manual review. Variant annotations can be associated with a specific variant (e.g., KRAS c.34G > T, p.G12C), groups of related variants at the same codon (e.g., KRAS codon 12 mutations: G12C, G12D, G12V), gene (e.g., all pathogenic variants in the KRAS gene*)*, or broader regions of the genome (e.g., SVs or amplifications spanning the KRAS locus on 12p12.1) [52].
Annotations are obtained through variant annotators, commonly via the CLI or alternatively, web-based platforms. Ensembl’s Variant Effect Predictor (VEP) [53] is widely used, offering annotations like population frequencies, clinical significance, and predicted pathogenicity. ANNOVAR [54] and SnpEff [55] are popular alternatives.
Several annotation sources are particularly relevant for somatic cancer analysis. Population allele frequency databases (e.g., gnomAD [56] and TOPmed [57]) help exclude common germline polymorphisms. Pathogenicity predictors estimate the impact of variants on protein function or gene regulation (Table 5). Curated databases, including ClinVar [58], VarSome [59], Franklin by GenoOx [60], OncoKB [61,62], Genomenon Cancer Knowledgebase (Formerly JaxKB) [63], and the Catalogue of Somatic Mutations in Cancer (COSMIC) [64] consolidate expert-reviewed literature, functional data, and clinical annotations. COSMIC data are freely accessible for academic use following registration. In the demo dataset provided with this tutorial, the Genome Screens Mutant dataset was used. Beyond these resources, specialized annotations from the literature or pathway databases can offer insight into drug response, regulatory impact, or broader pathways.
Table 5: Commonly used pathogenicity predictors.
Comprehensive annotation is important; however, excessive annotations can inflate file sizes and complicate variant filtering or interpretation. Select complementary resources that align with your research objectives [52] using recent literature and tool or database documentation [52,65].
Example CLI Code 5. Annotation with VEP
Annotate variants using Ensembl VEP
INPUT: somatic.vcf.gz
OUTPUT: somatic_annotated.vcf with annotations
vep \
--input_file somatic.vcf.gz \
--output_file somatic_annotated.vcf \
--cache \
--assembly GRCh38 \
--vcf
Phase 3: Variant filtering and validation
Filter and prioritize candidate variants
After variant annotation, reduce the variant list by quality filtering (remove unreliable variants [73,74]), and functional prioritization (elevate those most likely to be biologically or clinically relevant [75]).
Quality filtering.
Quality filtering uses caller metrics and sequencing parameters to remove artifacts. Here we discuss filtering considerations; however, thresholds will vary by dataset. During variant calling, variants receive a “PASS” FILTER flag if they meet all the caller’s quality requirements. Alternative FILTER field flags are defined by individual variant callers, described in the output VCF header or in the software documentation. Retaining only PASS flags may exclude true variants, but including non-PASS variants risks admitting artifacts. Publicly available VCFs are often pre-filtered.
VAF often informs PASS criteria. Depending on the assay’s detection limit, additional VAF-specific filtering may be necessary. Typical somatic cancer minimum VAF thresholds are 5%–10% of total reads [76–78]; however, dynamic thresholds can be used to account for variability in depth of coverage. Variants observed at highly similar VAFs across many samples may indicate run-specific artifacts [79]. Control samples with known VAFs can help empirically define the lower limit of detection for the sequencing run.
Variant callers aggregate base quality scores (Phred-scaled, 30 = 99.9% confidence [80]) and other signals to estimate confidence in the variant as a variant quality score. Minimum scores of 30 are commonly used to balance sensitivity and specificity [81,82].
Functional prioritization.
Functional prioritization ranks variants by biological or clinical relevance using annotations, either within annotation tools (e.g., Ensembl’s VEP) [53] or post hoc. Functional prioritization follows either clinical-grade binning or research-focused prioritization [2,3,75].
Clinical frameworks from organizations like the American Society of Clinical Oncology (ASCO) [2] and the American College of Medical Genetics and Genomics (ACMG) [3] classify variants into tiers or pathogenicity categories. These strategies are aimed at clinical decision-making, as their high stringency may omit variants that could be of interest in research.
Research prioritization strategies weigh features like predicted functional impact, evolutionary conservation, presence in known cancer gene lists or curated databases, and occurrence within your cohort or in public datasets [75]. Fig 3 illustrates an example prioritization scheme.
Example variant prioritization scheme.The flowchart illustrates a strategy for prioritizing variants into four bins based on predicted pathogenicity. Variants are initially assigned to a bin using criteria including ClinVar annotations, COSMIC frequency, and SpliceAI scores. Variants may then be reclassified to a different bin based on additional pathogenicity criteria, such as Franklin classifications (for promotion from Bin 2 to Bin 1), or other computational predictors including CADD, REVEL, and phastCons conservation scores (for promotion from Bin 4 to Bin 3, or Bin 3 to Bin 2). Bin 1 contains pathogenic variants, Bin 2 contains likely pathogenic variants, Bin 3 contains variants of uncertain significance (VUS), and Bin 4 represents likely benign or benign variants. Created in BioRender. Arseneau, R. (2026) https://BioRender.com/o4pbmzu.
During prioritization, common germline polymorphisms are excluded using population databases (e.g., gnomAD [56] and TopMED [57] with typical cutoffs of 0.01%–1% [83], while curated tumor lists can be used to help identify expected versus novel variants [75]. Most prioritization strategies emphasize protein-coding variants; however, adjust prioritization of non-coding or regulatory variants if of interest.
Employ a robust variant interpretation strategy
Interpretation integrates annotations, literature, databases, and prior knowledge to generate biologically meaningful hypotheses. Passing filters does not make a variant meaningful; a variant must relate to pathology by impacting gene expression, protein structure or function, regulatory mechanisms, or downstream molecular pathways [75].
Cancer interpretation often focuses on oncogenes with activating mutations or amplifications [84], or tumor suppressor genes, which exhibit deletions, truncations, or inactivating mutations [85]. ClinVar [58] and COSMIC [64] remain central repositories for variant-level information. Online databases (e.g., OncoKB [62], VarSome [59], Franklin [60], and the Clinical Interpretation of Variants in Cancer [86]) provide additional information, including clinical significance, relevant publications, ACMG/ASCO classifications, pharmacogenomic associations, and community-submitted interpretation. Pathway analysis and Gene Set Enrichment Analysis [87,88] can reveal broader relevance for variants that may appear marginal in isolation. The list of molecular changes relevant to cancer continues to expand; thus, a comprehensive literature review is essential [89]. Ultimately, your scientific judgement is essential for variant interpretation.
Manually validate variants where appropriate
Pipeline quality controls may miss false positives, so manual review is essential for confirming variants. Tools like Integrative Genomics Viewer (IGV) [90] allow inspection of read alignment, the variants position within reads, and local CNV [91]. IGV is essential for novel or unexpected findings, and guidelines are available elsewhere [91].
For paired normal-tumor sequencing, both sequencing alignments (tumor and normal) should be evaluated to confirm somatic status [22,91]. Similarly, when a control sample has been sequenced, it should be compared with the sample of interest.
Phase 4: Disseminating and storage
Use standardized nomenclature
When disseminating results, standardized nomenclature ensures variants are universally understandable, traceable to reference data, and correctly interpreted [2,92] (Table 6).
Table 6: Variant reporting checklist.
Gene and protein nomenclature.
Use gene and protein symbols from the Human Genome Organization (HUGO) Gene Nomenclature Committee (HGNC) [92], maintaining one consistent name and introducing aliases at first mention (e.g., CD274, a.k.a. PDL1 or B7H1) [92]. Specify the reference transcript used for annotation, preferably the Matched Annotation from NCBI and EMBL-EBI (MANE) [93].
Variant reporting.
Report variants at the DNA level following Human Genome Variation Society (HGVS) guidelines [94]. Present designations in both the manuscript text and a table that includes DNA, RNA, and protein nomenclature where applicable [95]. Verify variant descriptions with tools like Mutalyzer [96] for HGVS compliance and formatting [97].
CLI Example Code 6. Verifying variant descriptions with Mutalyzer
Normalize an HGVS description (use canonical form for reporting)
INPUT: Genomic or transcript HGVS (e.g., “GRCh38 (chr<CHR>):g.<POS><REF>><ALT>” or “NM_<TRANSCRIPT>:c.<...>”)
OUTPUT: JSON with normalized_description (report this)
curl -sS “https://v3.mutalyzer.nl/api/normalize/<YOUR_HGVS_DESCRIPTION>”
Map a normalized description to a specific transcript (e.g., MANE Select)
INPUT: description=<YOUR_NORMALIZED_HGVS > ; target_selector=<TRANSCRIPT_ACCESSION> (e.g., NM_########.#)
OUTPUT: JSON with c.-notation for the requested selector
curl -sS --data-urlencode “description=<YOUR_NORMALIZED_HGVS>” \
--data-urlencode “target_selector=<TRANSCRIPT_ACCESSION>” \
“https://v3.mutalyzer.nl/api/map/”
List available selectors (transcripts) for a reference sequence
INPUT: reference_id=<REFERENCE_ACCESSION> (e.g., NC_########.##)
OUTPUT: JSON array of selector IDs (choose MANE when available)
curl -sS https://v3.mutalyzer.nl/api/get_selectors/<REFERENCE_ACCESSION>
Convert reference positions to selector-oriented coordinates (genome to c.-notation)
INPUT: Genomic HGVS (e.g., “GRCh38 (chr<CHR>):g.<POS><REF>><ALT>” or “NC_########.##:g.<...>”)
OUTPUT: JSON with c.-level coordinates aligned to the chosen selector
curl -sS --data-urlencode “description=<YOUR_GENOMIC_HGVS>” \
“https://v3.mutalyzer.nl/api/position_convert/”
Genomic data storage and compliance
Genomic data management should follow Findable, Accessible, Interoperable, and Reusable principles (FAIR) [98].
Working directory storage.
During active analysis, use hierarchical directory system for raw data, intermediate files, results, and metadata [99]. Apply a version control system to track changes in scripts and metadata [98].
Long-term storage.
Retain files essential for future auditing, reanalysis, or validation [100], including raw data, analysis scripts, auxiliary files, and selected results. Genomic files are large, making compression essential for storage. Two primary types of compression are available: lossless and lossy. Lossless formats like FASTQ.gz and BAM are preferred for permanent storage. When lossy compression is used, its impact on downstream analyses should be considered [101]. All transformations should be logged with details on software, versions, and parameters [98].
Storage scalability, redundancy, and security.
Combine institutional servers, cloud storage, and external drives for redundancy [99,100,102] and ensure compliance with ethical, legal, and institutional standards [103], including encryption and secure transfer protocols [100,104]. Deposit data in secure external repositories when possible [98].
Structure and share code for reproducibility
Genomic analyses rely on complex workflows that must be documented for reproducibility, validation, and reuse [105] (Table 7). Share code when possible [98,105] via repositories such as GitHub [106] or GitLab [107]. Use workflow managers (e.g., Snakemake [108], Nextflow [109]) and package/container managers (e.g., Conda [110], Docker [111]) to standardize environments and automate pipelines [112]. Include README files detailing file structures, workflows, and expected outputs [100,113]. Code availability statements can be referenced from The American Journal of Human Genetics [114] or Oxford Academic [115].
Table 7: Code reproducibility and sharing checklist.
Conclusions
This framework provides practical guidance for tumor sequencing analysis, covering the full workflow—from study design to data interpretation and dissemination—while emphasizing code sharing to foster reproducibility and collaborative science. By promoting a structured, reproducible approach, these guidelines support consistency in variant interpretation and reporting, contributing to greater clarity, transparency, and comparability across studies in cancer genomics.
Supporting information
S1 FigVariant interrogation in practice: step-by-step questions using demo data.Guiding questions for demo data used to demonstrate variant filtering and prioritization in tumor and control samples. Box 1 focuses on interrogating the dataset in question. Box 2 focuses on variant annotations and their use cases. Box 3 and Box 4 encompass quality filtering and functional prioritization of variants. Box 5 guides the identification and reporting of final variants of interest. Box colors correspond to processing phase: green (Planning and pre-processing), yellow (Variant calling and annotation), and blue (Filtering and validation). Created in BioRender. Arseneau, R. (2026) https://BioRender.com/gexi4i4.(TIF)
S1 DataExample data and associated code for working through variant annotation and interrogation following the guidelines laid out in this manuscript.(ZIP)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Satam H, Joshi K, Mangrolia U, Waghoo S, Zaidi G, Rawool S, et al. Next-generation sequencing technology: current trends and advancements. Biology (Basel). 2023;12(7):997. doi: 10.3390/biology 12070997 37508427 PMC 10376292 · doi ↗ · pubmed ↗
- 2Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer. J Mol Diagn. 2017;19(1):4–23.27993330 10.1016/j.jmoldx.2016.10.002PMC 5707196 · doi ↗ · pubmed ↗
- 3Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. doi: 10.1038/gim.2015.30 25741868 PMC 4544753 · doi ↗ · pubmed ↗
- 4The Biostar Handbook. 2nd ed [Internet]. [cited 2025 May 25]. Available from: https://www.biostarhandbook.com/index.html
- 5Abbasi A, Alexandrov LB. Significance and limitations of the use of next-generation sequencing technologies for detecting mutational signatures. DNA Repair (Amst). 2021;107:103200. doi: 10.1016/j.dnarep.2021.103200 34411908 PMC 9478565 · doi ↗ · pubmed ↗
- 6Williams MJ, Sottoriva A, Graham TA. Measuring clonal evolution in cancer with genomics. Annu Rev Genom Hum Genet. 2019;20(1):309–29.10.1146/annurev-genom-083117-02171231059289 · doi ↗ · pubmed ↗
- 7Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 2014;15(2):121–32. doi: 10.1038/nrg 3642 24434847 · doi ↗ · pubmed ↗
- 8Zare F, Dow M, Monteleone N, Hosny A, Nabavi S. An evaluation of copy number variation detection tools for cancer using whole exome sequencing data. BMC Bioinformatics. 2017;18(1):286. doi: 10.1186/s 12859-017-1705-x 28569140 PMC 5452530 · doi ↗ · pubmed ↗