Elucidation of population-based bacterial adaptation to antimicrobial treatment by single-cell sequencing analysis of the gut microbiome of a hospital patient
Lianwei Ye, Yuchen Wu, Jiubiao Guo, Hanyu Wang, Jing Cai, Kaichao Chen, Ning Dong, Jiale Yu, Shan Chao, Hongwei Zhou, Gongxiang Chen, Sheng Chen, Rong Zhang

TL;DR
This study uses single-cell sequencing to track how gut bacteria adapt to antibiotics in a hospital patient, revealing new resistance genes and horizontal gene transfer.
Contribution
The study introduces single-cell sequencing as a novel method to detect and trace antibiotic resistance gene evolution and horizontal transfer in unclassified and known bacterial species.
Findings
Single-cell sequencing detected antibiotic resistance genes (ARGs) in 92 bacterial species, including unclassified strains.
The cfr(C) gene was found in 11 species, with distinct mutation patterns in Klebsiella pneumoniae and other taxa.
309 horizontal gene transfer events were identified, involving genes like folE and queE that aid DNA repair in antibiotic-treated bacteria.
Abstract
In this study, we used single-cell sequencing to analyze the gut microbiome of an adult male patient with acute cerebral hemorrhage undergoing antibiotic treatment. We identified 92 bacterial species, including 23 Firmicutes and one archaeon from Methanobacteriota, along with 69 unclassified strains. Single-cell sequencing effectively detected bacteria carrying antibiotic resistance genes (ARGs), particularly in unclassified species, and traced the evolution of these genes across diverse bacterial taxa. Notably, the cfr(C) gene was detected in 11 bacterial species following antimicrobial treatment, with mutation patterns characterized in Enterococcus faecalis, Klebsiella pneumoniae, Ruthenibacterium UN-1, and four unclassified species. In total, 29 ARG subtypes across eight types were identified in 13 known, five unknown, and 18 unclassified species, allowing us to trace their evolution…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
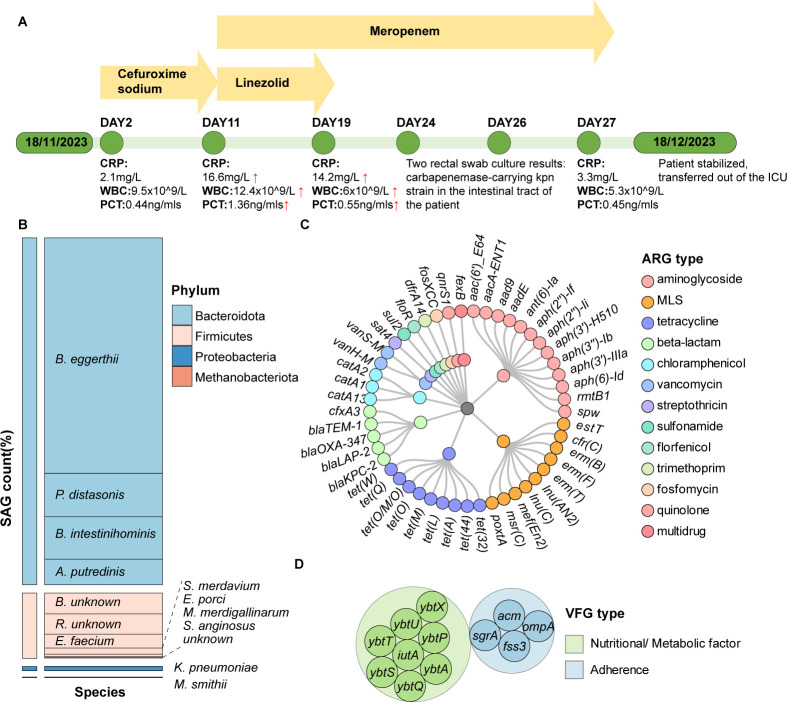 Fig 1
Fig 1 Fig 2
Fig 2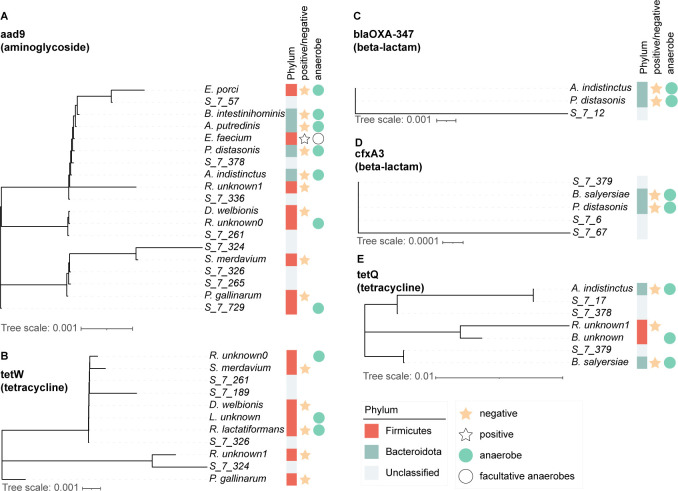 Fig 3
Fig 3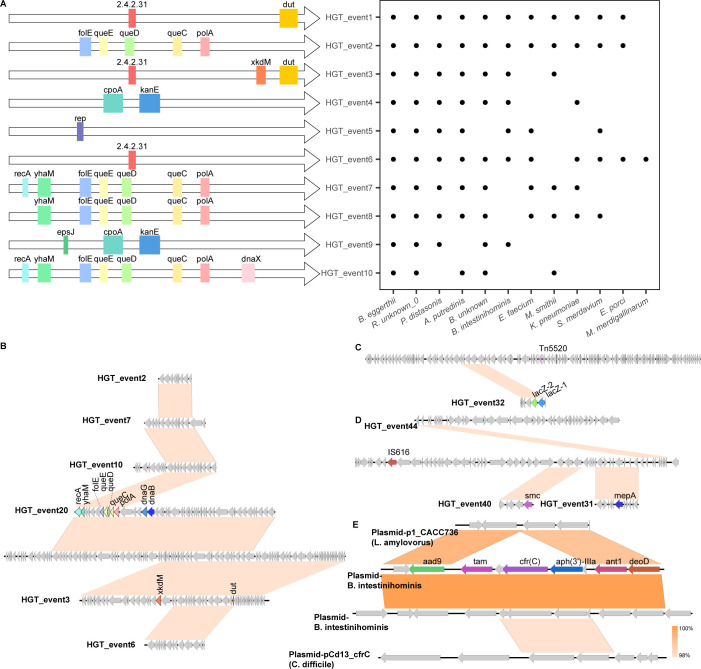 Fig 4
Fig 4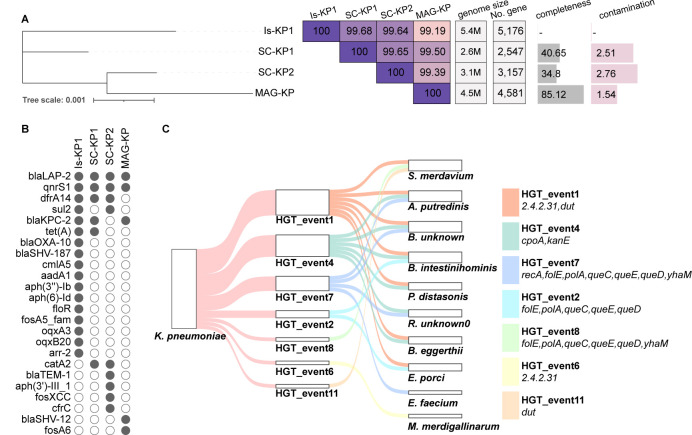 Fig 5
Fig 5- —National Key Research and Development Program of Chinahttp://dx.doi.org/10.13039/501100012166
- —National Natural Science Foundation of Chinahttp://dx.doi.org/10.13039/501100001809
- —Theme-base research scheme
- —general research fund of research grant councile of the Government of Hong Hong SAR
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGut microbiota and health · Bacterial biofilms and quorum sensing · Antibiotic Resistance in Bacteria
INTRODUCTION
The human intestine harbors a vast array of microorganisms, including bacteria, viruses, fungi, and archaea. These microorganisms play a significant role in human health, aiding digestion and warding off pathogenic invaders (1–3). On the flip side, the gut microbiota may harbor numerous genes, such as antibiotic resistance genes (ARGs) and virulence factor genes (VFGs), that enable potential bacterial pathogens to exhibit antibiotic resistance and the ability to cause infection. Experimental evidence gathered in recent studies suggests that significant changes occur in the gut microbiota following drug treatments, the extent of which requires further studies to ascertain their implications (4, 5). In simpler terms, the gut microbiota can carry genes that help bacteria resist antibiotics (ARGs) or cause infections (virulence genes). When people take antibiotic treatment, these treatments can disturb the balance of the gut microbes and increase the risk of drug-resistant or harmful bacteria growing. More studies are needed to understand how these changes affect human health. Hence, studying the gut microbiota holds great significance in devising new strategies for disease control through boosting immunity and mitigating dissemination of potential pathogens in the human body. On the other hand, there is increasing concern over settlement of pathogenic bacteria carrying drug-resistance genes within the gut microbiota. Not only do these pathogenic strains pose a threat to human health by causing opportunistic infection, but they also render commonly used antibiotics ineffective through carriage and dissemination of ARGs. Among them, CR-KP, or carbapenem-resistant K. pneumoniae, is a major pathogen known to cause a wide range of infections, including pneumonia, urinary tract infections, and bloodstream infections (6–8).
Traditional research techniques such as bacterial culture and metagenomic sequencing have their own advantages and limitations in the analysis of the structure of the human gut microbiome. Bacterial culture allows for the isolation of specific strains and their genomes. The organisms can then be tested in in vitro and animal experiments to validate the functional characteristics of specific strains such as drug resistance and pathogenicity (9–12). This approach can generate valuable data that facilitate development of preventive and therapeutic strategies. However, the main drawback of this method lies in its limitation to study only a small number of cultivable microorganisms. On the other hand, metagenomic sequencing offers the advantage of studying the entire microbial community, not only identifying the range of bacteria present but also depicting the range of functional genes that exist within the entire microbiome, including ARGs, VFGs, and those that encode various metabolic functions. Moreover, through metagenome-assembled genomes (MAGs) (13–16), unculturable and unclassified genomes within the human gut microbiome can be identified. Nonetheless, a plethora of genetically uncharacterized microorganisms exist in the human microbiome. It is necessary to determine the types of ARGs, VFGs, and other functional genes harbored by different bacterial species and characterize the horizontal gene transfer (HGT) events that occur among members of the gut microbiome.
Recent advancements in single-cell sequencing technologies have revolutionized our ability to explore the genetic landscape of microbial communities at unprecedented resolution (17–19). By isolating and sequencing individual bacterial cells, researchers can now delve into the genetic makeup of previously unknown species that inhabit the gut environment. In addition, this approach enhances our understanding of HGT activities among strains of different bacterial species, including exchanges between unclassified and uncultivable bacteria or archaea.
In this study, we conducted an in-depth investigation of the composition and characteristics of a patient’s gut microbiome following antibiotic treatment. Our main scientific question was: How does antibiotic treatment influence the co-evolution and transfer of ARGs and virulence genes in the human gut microbiome at the single-cell level? To address this, we leveraged a combination of single-cell sequencing, metagenomics, and comparative genomics to reveal strain-level variations in ARGs, VFGs, and plasmid profiles. We also delineated the network of HGT events that occurred among known and previously unidentified bacterial taxa. Our findings highlight the power of single-cell analysis in uncovering novel bacterial species and the genomic mechanisms driving the emergence and spread of antibiotic resistance in clinically relevant microbiota.
RESULTS
Gut microbiome composition at the single-cell level
We conducted an analysis of the composition of the gut microbiome of an adult male patient presented with acute cerebral hemorrhage; initial antimicrobial treatment encompassed intravenous administration of cefuroxime sodium given to this patient. Subsequently, meropenem and linezolid were administered upon the manifestation of fever and productive cough with sputum. Concurrently, a fecal sample was obtained for single-cell sequencing on 28 November 2023 during the antimicrobial treatment period (Fig. 1). Analysis of the patient’s gut microbiome showed that it was predominantly composed of three bacterial phyla—Bacteroidetes, Firmicutes, and Proteobacteria—along with Archaea from the phylum Methanobacteriota. Among them, Bacteroidetes and Firmicutes were predominant, constituting 83.2% and 15.6% of the single-amplified genomes (SAGs), respectively (Fig. 1; Tables S1 and S2). At the species level, certain taxa exhibited notably high prevalence, with Bacteroides eggerthii (Bacteroidetes) emerging as the predominant strain; this species accounted for as much as 56.5% of the microbial population. Parabacteroides distasonis and B. intestinihominis (both Bacteroidetes) were also abundant, comprising 10.4% and 10.2% of the microbial community, respectively. We were able to identify less abundant yet potentially clinically relevant species in the data set, including Enterococcus faecium (Firmicutes, 3.3%), K. pneumoniae (Proteobacteria, 1.0%), Methanobrevibacter smithii (Methanobacteriota, 0.2%), and Streptococcus anginosus (Firmicutes, 0.1%). Comparison with the metagenomic data revealed that Methanobrevibacter smithii (Methanobacteriota) was exclusively detected in the single-cell data set. Other bacterial species uniquely identified through single-cell analysis included E. faecium, Scatomonas merdavium (Bacteroidetes), Eisenbergiella porci (Firmicutes), Massiliomicrobiota merdigallinarum (Firmicutes), and M. smithii. These findings underscore the value of single-cell analysis in uncovering previously undetectable taxonomic groups within the gut microbiome.
Analysis of variation in the composition of the gut microbiome of a patient subjected to serial antimicrobial treatment. (A) Timeline of treatment of the patient by various antibiotics. Key events, such as detection of carbapenem-resistant K. pneumoniae and stabilization of the condition of the patient, are highlighted. (B) Bar graph showing the abundance of different bacterial species in the patient’s microbiome, as determined by recording single amplified genome (SAG) counts. Each color represents a different bacterial species. (C) Circular diagram illustrating the diversity of antibiotic resistance gene (ARG) types and subtypes present in the microbiome. Each ARG type and subtype is color-coded for easy identification. (D) Representation of virulence factor genes (VFGs) identified in the microbiome, with a focus on genes associated with nutritional uptake, metabolic factors, and adherence functions.
A comprehensive investigation of ARGs within the gut microbiome revealed the presence of 13 distinct types, spanning various antibiotic classes (Fig. 1; Tables S3 and S4). Prominent among these ARGs were aminoglycoside resistance genes, which comprised 10 subtypes, along with prevalent beta-lactam resistance genes, including blaKPC-2 and blaTEM-1, and chloramphenicol resistance genes such as catA13 and catA14. Additional ARG classes, such as those that encode resistance to florfenicol, fosfomycin, macrolide–lincosamide–streptogramin (MLS), quinolone, streptothricin, sulfonamide, tetracycline, trimethoprim, and vancomycin, were found to involve a diverse array of resistance determinants.
Further analysis uncovered a rich diversity of subtypes within each ARG type, which is indicative of the genetic complexity of gut bacteria and their adaptability to pressures associated with antimicrobial treatment. For instance, the aminoglycoside resistance genes were found to comprise 13 subtypes, including aph(2'')-If and aph (6)-Id, illustrating that gut bacteria utilize a diverse range of mechanisms to counteract the action of this class of antibiotics. Tetracycline resistance genes had nine subtypes, including tet(32) and tet(M). Among the beta-lactam resistance genes, a myriad of subtypes were also identified. Noteworthy beta-lactam subtypes included blaKPC-2, and blaLAP-2, each exhibiting distinct mechanisms of resistance but all being clinically important. Notable ARGs exclusively identified in the single-cell data set included aacA-ENT1, aph(2'')-If, aph(2'')-Ii, aph(3')-H510, floR, erm(B), erm(F), erm(G), estT, msr(C), tet(32), tet(40), tet(44), tet(L), tet(M), tet(O/M/O), tet(Q), vanH-M, and vanS-M (Table S5), indicating that single-cell analysis can help produce high-resolution genetic features of members of the human gut microbiome.
Bacterial hosts of ARGs and VFGs
Comparing different sets of metagenomic data, we found that the advantage of single-cell analysis lies in its capacity to directly identify bacterial strains harboring specific ARGs, VFGs, and plasmids. Within the human gut microbiome, a total of 13 types of ARGs were identified at the species level of SAGs; these ARGs were found to be carried by bacteria of 41 species, including 22 unknown species and 19 unclassified species (Fig. 2; Tables S3 to S6). Six species in the Bacteroidota bacterial group were commonly found in the human gut—Alistipes putredinis, Alistipes indistinctus, B. eggerthii, Bacteroides salyersiae, Bacteroides intestinihominis, and P. distasonis, all exhibiting aminoglycoside resistance genes such as aad9, aadE, and aph(2'')-Ii. Notably, certain species were sporadically reported to carry additional genes such as cfr(C), fosXCC, blaOXA-347, tet(Q), and cfxA3. For instance, B. eggerthii manifested a diverse repertoire of ARGs, including aminoglycoside, fosfomycin, MLS, sulfonamide, and tetracycline resistance genes, with a total of eleven subtypes of such genes being detected. Similarly, B. intestinihominis and P. distasonis exhibited a comparable diversity in ARGs.
Antibiotic resistance genes (ARGs) in different gut microbiome bacterial strains. This figure illustrates the ARG profiles of seven bacterial strains (A–G). Each panel (A–G) represents a different bacterial strain. The lines connecting each bacterium and different ARGs represent the specific resistance genes each strain carries. The types of antibiotics to which these genes confer resistance are indicated by different line styles. The ARGs include a variety of resistance genes such as aph(3’)IIIa, aac(6’)Ie-aph(2’‘)Ia, aad9, tet(O), tet(M), tet(W), cfr/cfr(C), erm(B), erm(F), msr(A), blaOXA-30, blaOXA-347, blaOXA-61, blaTEM1-B1, qnrS1, vanA, dfrA14, floR, catA13, aac(6’)IIc, mph(A), mef(A), aadE-sat4-aadA1, strAB, aadD2, mph(B), and fosXCC. The key on the right side of the figure depicts the color coding for each ARG type. Each panel has a unique set of connections, indicating specific relationships exist between each bacterial species and the ARGs.
The Firmicutes group was found to comprise 12 genera and six unknown species. Several taxa were infrequently reported in the human gut, including M. merdigallinarum, E. porci, S. merdavium, Borkfalkia, Ruthenibacterium, and Pararuminococcus gallinarum. Furthermore, Enterococcus faecium strains, which are prevalent in the human gut, were found to harbor five types of ARGs, including those which encoded resistance to aminoglycoside, MLS, tetracycline, vancomycin, and various other agents. These genes could be sub-divided into twelve subtypes, including aacA-ENT1, aph(2'')-Ii, aph(3')-IIIa, erm(T), fexB, msr(C), poxtA, spw, tet(L), tet(M), vanH*-M*, and vanS-M. Another common bacterium, S. anginosus, was found to carry the erm(B) gene, which conferred resistance to macrolides and lincosamides.
Uncommon bacteria in the gut, such as M. merdigallinarum, Borkfalkia, Eisenbergiella, Ruthenibacterium, and Scatomonas, also exhibited diverse profiles of ARGs. For example, M. merdigallinarum was found to harbor genes that encoded resistance to aminoglycosides, chloramphenicol, MLS (macrolide, lincosamide, and streptogramin), and tetracycline. Unknown species within the Borkfalkia genus were also found to harbor a range of ARGs, including those that conferred resistance to aminoglycosides, beta-lactams, chloramphenicol, MLS, fosfomycin, and tetracycline. The species E. porci was found to exhibit resistance to tetracycline through carriage of tet(32) and tet(Q). Similarly, the unknown species within the Ruthenibacterium genus carried multiple types of ARGs, and a total of nine subtypes of such genes were identified. The species S. merdavium exhibited resistance to six types of antibiotics, and as many as fourteen subtypes of ARGs could be detected; these included the aad9, aadE, ant (6)-Ia, aph(3')-IIIa, catA13, catA14, cfr(C), fosXCC, lnu(C), sat4, tet(O), tet(Q), and tet(W). At the Firmicutes level, a bacterium within the Lachnospiraceae family was found to carry the ARGs of aad9, aph(2'')-Ii, cfr(C), and tet(O/M/O).
Among the Proteobacteria, K. pneumoniae was found to carry various ARGs, including blaKPC-2, blaLAP-2, catA2, dfrA14, qnrS1, rmtB1, and tet(A). Within the Archaea domain, specifically the phylum Methanobacteriota, M. smithii was found not to carry any ARGs. The diversity of ARGs carried by these unclassified bacteria is a hallmark of the complexity of antimicrobial resistance-encoding genetic elements harbored by the microbial communities in the human intestine. These 19 unclassified bacteria collectively harbor 23 subtype ARGs, including aad9, ant (6)-Ia, aph(2'')-Ii, aph(3')-IIIa, cfr(C), fosXCC, lnu(C), npmA, sat4, sul2, tet(W), aadE, bla_OXA-347_, catA14, cfr(E), cfxA3, erm(B), erm(F), lnu(AN2), mef(En2), tet(40), tet(M), and tet(Q).
Analysis of VFGs harbored by the gut microbiome strains showed that only two bacterial strains harbored VFGs at the strain level. Enterococcus faecium was found to harbor fss3, acm, and sgrA, whereas K. pneumoniae carried a variety of VFGs including ybtS, ybtU, ybtT, ybtA, ybtP, ybtS, ybtA, iutA, ompA, ybtQ, ybtS, and ybtX.
Analysis of metagenome-assembled genomes (MAGs) offers insights into the unique genetic features of clinically important bacterial species and their associated ARGs, albeit only partially. In this study, a total of 19 MAGs were identified (Table S7), mostly belonging to Firmicutes, Bacteroidota, and Proteobacteria, with Firmicutes comprising the majority (14 out of 19). However, only 10 MAGs carried ARG genes, with 8 MAGs carrying fewer than 3 ARGs and 2 MAGs carrying 5 ARGs. The predominant types of ARGs identified in MAGs were mainly associated with tetracycline, aminoglycoside, and beta-lactamase resistance. This finding suggests that, although analysis of MAGs can help identify species harboring ARGs, their ability to comprehensively capture ARG data in the gut microbiome is limited.
Co-evolution of ARGs in different bacterial species
In the human gut microbiome, co-evolution of ARGs and transmission between different bacterial species, which primarily involve Bacteroidota, Firmicutes, and unclassified bacteria, is a common event and underscores the dynamic interplay shaping development of microbial resistance in major bacterial pathogens. Notably, the genera such as Ruthenibacterium, Enterococcus, and Alistipes emerge as key players in the co-evolutionary process. Phylogenetic analysis conducted for ARGs carried by strains of at least three species showed the existence of 8 types of ARGs and that these 8 types comprised a total of 25 sub-types (Fig. 3; Fig. S1 and S2). These subtypes exhibit distinct clustering patterns among various bacterial species, which are indicative of divergent evolutionary trajectories. Within the realm of aminoglycoside resistance genes, a spectrum of subtypes including AAC(6')-Ie-APH(2'')-Ia, AAC6, aad (6), aad9, ant(6)-Ia, ANT9, aph(2'')-Ii, aph(3')-III, APH(3')-IIIa, and APH2 have been implicated in co-evolutionary dynamics. This co-evolution event predominantly involves representative strains of the Bacteroidota, Firmicutes, and Terrabacteria groups, alongside numerous unidentified bacterial taxa. For instance, the aad9 gene was found in three discernible clusters across diverse bacterial hosts. Cluster 1 encompasses 13 species, including both anaerobic gram-negative (e.g., E. porci, B. intestinihominis, A. putredinis, P. distasonis, and A. indistinctus) and aerobic gram-positive (e.g., E. faecium and Dysosmobacter welbionis) bacteria from the Bacteroidota and Firmicutes phyla. Cluster 2 comprises a singular unknown species within the Lachnospiraceae family, andCluster 3 comprises P. gallinarum, S. merdavium, and three unidentified species. Similarly, although the aph(2'')-Ii gene was not detectable in the metagenomic data, it was found in three distinct clusters involving eight species, including E. faecium and various unidentified species within the genera Borkfalkia, Lawsonibacter, Ruthenibacterium, and Lachnospiraceae.
Co-evolution of antibiotic resistance genes (ARGs) in different bacterial species. This figure illustrates the co-evolution features of ARGs in various bacterial species. Each panel (A–E) represents a different ARG, with panel A specifically showing the aad9 gene that is related to aminoglycoside resistance. In panel A, the aad9 gene is found in 19 different bacterial species, which are further clustered into three distinct groups. The other panels (B–E) depict the evolutionary trees of tet(W), blaOXA-347, cfxA3, and tet(Q) genes, which encode tetracycline and beta-lactam antibiotic resistance. The color-coded phyla indicate the diversity of bacteria carrying these ARGs. Additionally, yellow stars indicate gram-negative bacteria, while white stars indicate gram-positive bacteria. This diagram provides a comprehensive overview of the distribution and co-evolution of ARGs among different bacterial species.
Co-evolutionary dynamics of the beta-lactamase genes primarily revolve around two elements: cfxA3 and blaOXA-347. The blaOXA-347 gene was detected in three distinct species, namely, A. indistinctus, P. distasonis, and species of unknown identity. Conversely, cfxA3 was mainly found in three discernible clusters: one cluster encompasses A. indistinctus and P. distasonis, whereas the other two clusters comprise individual unidentified species. Chloramphenicol resistance was encoded by a single gene, cat, which was detectable in strains of three distinct subgroups, namely, cluster 1 (Unclassified-7-324; Unclassified-7-326), cluster 2 (S. merdavium), and cluster 3 (Ruthenibacterium) (Fig. 3; Fig. S1 to S3).
The fosfomycin gene fosXCC exhibited a distinct clustering pattern. One cluster encompassed A. putredinis, K. pneumoniae, P. distasonis, and P. gallinarum, along with two unidentified species of bacteria. Another cluster was associated with the genus Ruthenibacterium, while the third cluster involved yet another unidentified bacterial species.
Regarding the MLS genes, which include cfr(C), cfr(E), erm(F), ErmB, lnu(AN2), and mef(En2), specific clustering patterns emerged. For the cfr(C) gene, three distinct clusters were identified: cluster 1 (comprising Unclassified-7-378, P. distasonis, A. indistinctus, and K. pneumoniae), cluster 2 (including Unclassified-7-265 and Unclassified-7-266), and cluster 3 (featuring Enterococcus faecalis, Ruthenibacterium of an unidentified species, A. putredinis, Unclassified-7-379, and B. intestinihominis). Similarly, for cfr(E), clustering revealed the presence of cluster 1 (associated with Lawsonibacter), cluster 2 (linked to R. lactatiformans), and cluster 3 (comprising Unclassified-7-324, Ruthenibacterium of an unidentified species, and Unclassified-7-326). For the lnu(AN2) and mef(En2) genes, three clusters were identified, all involving the same three species: K. pneumoniae, B. eggerthii, and an unidentified species classified as Unclassified-7-64. Moreover, it is noteworthy that the genes erm(F) and ermB were exclusively detected in single-cell data. For the erm(F) gene, clustering revealed the existence of cluster 1 (associated with Unclassified-7-326), cluster 2 (linked to Ruthenibacterium of an unidentified species and S. anginosus), and cluster 3 (comprising E. faecium). As for the ermB gene, clustering analysis revealed the presence of cluster 1 (associated with E. faecium), cluster 2 (linked to Ruthenibacterium), and cluster 3 (involving S. anginosus and Unclassified-7-326) (Fig. 3; Fig S1, S2 and S3a through c).
Among the tetracycline genes, particularly tet(Q) and tet(W), distinct clustering patterns were observed, shedding light on the co-evolution of ARGs within the human gut microbiome. The tet(Q) gene, which was exclusively detected in single-cell data, exhibited three clusters: cluster 1 comprised A. indistinctus, Unclassified-7-17, and Unclassified-7-378; cluster 2 contained Unclassified-7-379 and B. salyersiae; cluster 3 involved Ruthenibacterium of an unidentified species and Borkfalkia of an unidentified species. Similarly, tet(W) could be clustered into three clusters. Cluster 1 was associated with Ruthenibacterium of an unidentified species, S. merdavium, Unclassified-7-261, Unclassified-7-189, D. welbionis, Lawsonibacter of an unidentified species, and Ruthenibacterium lactatiformans; cluster 2 was linked to P. gallinarum; cluster 3 was associated with Ruthenibacterium of an unidentified species and Unclassified-7-324. These clustering patterns suggest intricate evolutionary relationships among tetracycline resistance genes and various bacterial species, including unclassified and rare bacteria such as E. porci, S. merdavium, and f_Lachnospiraceae; s_unknown_0, among others, which were the major players that shaped the ARG landscape within the gut microbiome (Fig. 3; Fig. S3d).
For the tet(M) gene, although only two species, E. faecium and an unclassified bacteria designated as unclassified bacteria123, carried this gene in the gut microbiome, notable differences in the gene sequence were observed. Specifically, the tet(M) gene in unclassified bacteria exhibited a 99.79% similarity to the reference sequence, while the tet(M) gene in E. faecium displayed a 99.32% similarity, indicating the presence of mutations that involved 19 nucleobase changes and 10 amino acid changes (Fig. 3; Fig. S3e).
HGT landscape in the gut microbiome
Contigs exceeding five kilobase pairs were chosen to study HGT events that occurred in Firmicutes, Bacteroidota, and Proteobacteria, with one case involving Archaea from the Methanobacteriota phylum. A total of 309 HGT events involving 14 species were identified; this phenomenon suggested that extensive genetic exchange occurred within the human gut microbiome (Fig. 4; Table S8).
Horizontal Gene Transfer (HGT) events in different bacterial species. This figure illustrates the relationship between HGT events and the bacterial species involved. The left panel shows different genes involved in HGT events, each represented by a colored block and labeled accordingly (e.g., folE, polA, and queC). Each row corresponds to a specific HGT event (e.g., HGT_event1 and HGT_event2). The right panel displays a dot plot representing the presence (indicated by black dots) of these specific genes in various bacterial strains (e.g., E. porci). (B) Shows the largest contigs’ HGT associated with HGT events 2, 7, 10, 20, 3, and 6, carrying the most genes, including folE, polA, queC. C and D depict the HGT with MGEs. C carries Tn5520 and also carries lacZ-1 and lacZ-2, while D suggests that HGT events 40 and 44 may have occurred through IS616 and also carries smc and mepA. E shows two plasmid HGT events that occurred in bacteria B. intestinihominis and A. putredinis, with reference plasmids from L. amylovorus and C. difficile.
HGT1, the most prevalent HGT event, was observed 28 times. This event involved the transfer of the gene dut (deoxyuridine triphosphatase) and the gene that encoded ADP-ribosyltransferase (EC 2.4.2.31), among other hypothetical protein genes. This event was observed in 11 bacterial species, including A. putredinis, B. eggerthii, B. intestinihominis, Borkfalkia unknown_0, E. porci, E. faecium, K. pneumoniae, M. smithii, P. distasonis, Ruthenibacterium unknown_0, and S. merdavium. HGT3 and HGT11 shared similarities with HGT1, with HGT3 involving the transfer of only the dut gene, while HGT11 involved an additional gene, xkdM. The dut gene is essential for DNA replication and repair in bacteria. These HGT events indicate the exchange of genetic material involved in crucial cellular functions among diverse bacterial species within the gut microbiome.
HGT2, the second-most prevalent HGT event, involved the transfer of the genes folE, polA, queC, queD, and queE among 11 bacterial species. This event excluded M. merdigallinarum, S. anginosus, and one unidentified species within the Lachnospiraceae family. Other HGT events, such as HGT7 and HGT10, were similar to HGT2 but included additional genes. HGT7 involved transfer of the genes recA and yhaM, while HGT10 incorporated even more genes, including dnaX, recA, and yhaM. The presence of DNA repair and replication genes like polA, recA, and dnaX in these HGT events suggests the potential enhancement of cellular mechanisms related to DNA maintenance and genomic stability in the recipient bacteria. Improved DNA repair and replication capabilities can contribute to the overall fitness and survival of bacteria, especially under prolonged treatment with antibiotics or other environmental stresses.
HGT4 involved transfer of the gene pair cpoA and kanE, along with other hypothetical protein genes, among six bacterial species: A. putredinis, Barnesiella intestinihominis, B. eggerthii, an unidentified species of Borkfalkia, K. pneumoniae, an unidentified species of Ruthenibacterium, and P. distasonis. Similarly, HGT9 shared similarities with HGT4 but included an additional gene, epsJ. On the other hand, HGT5 was characterized by the presence of a single gene named rep, which was often accompanied by other hypothetical protein genes. This HGT event was observable in seven bacterial species: A. putredinis, B. eggerthii, B. intestinihominis, E. faecium, P. distasonis, an unidentified species of Ruthenibacterium, and S. merdavium.
Furthermore, there were 54 other HGT events, each occurring fewer than 10 times between different species, which involved functionally important genes. Despite their lower frequency, these events were still regarded as being significant in terms of enhancing bacterial survival fitness under adverse environmental conditions. For instance, in HGT 25, genes such as baeS, saeR, bceB, and yxdL were identified. These genes have been observed to undergo HGT across different bacterial species, contributing to cellular responses to environmental stress, biofilm formation, antibiotic sensing and resistance, and functions of the ABC transport systems. Another notable example is HGT 35, which involved the genes ant1, cfr, deoD, bioC, aadK, and aphA. This particular HGT event was mediated by plasmids harbored by B. intestinihominis and A. putredinis, indicating that transmission of resistance genes through this event is common. It is worth mentioning that P. distasonis exhibited 76 HGT events, while E. porci had 52, and A. putredinis had 38 events. Additionally, Acrea M. smithii participated in 11 HGT events, along with K. pneumoniae, S. merdavium, B. eggerthii, an unidentified species of Borkfalkia, and an unidentified species of Ruthenibacterium.
Strain-level characterization of K. pneumoniae
The IS-KP1 strain isolated from the patient’s gut exhibited a concerning array of ARGs, indicating that this strain possessed high-level resistance to various antibiotics commonly used in clinical settings. These ARGs included aadA1, aph(3'')-Ib, aph (6)-Id, arr-2, bla_KPC-2_, bla_LAP-2_, bla_OXA-10_, bla_SHV-187_, cmlA5, dfrA14, floR, fosA5_fam, oqxA3, oqxB20, qnrS1, sul2, and tet(A) (Fig. 5; Table S10). Each of these genes confers resistance to specific classes of antibiotics, suggesting that the IS-KP1 strain was multidrug- resistant. Moreover, the presence of several VFGs in the IS-KP1 strain indicates that this strain also exhibited high-level virulence. The VFGs, including iucA, iucB, and ecpR, are known to play crucial roles in several virulence mechanisms in major bacterial pathogens.
Phylogenetic analysis, antibiotic resistance genes (ARGs), and horizontal gene transfer (HGT) events in major gut microbiome bacterial strains. A displays a phylogenetic tree representing the evolutionary relationships among different bacterial strains, along with ANI comparison values, genome sizes, gene completeness, and contamination levels. B illustrates the ARGs each strain carries. C shows the HGT events that occurred between K. pneumoniae (KP) and other bacterial species.
The utilization of single-cell analysis allowed us to discover two distinct strains of K. pneumoniae, demonstrating that this method is highly efficient in delineating strain-level characteristics of clinically relevant pathogens. Furthermore, metagenomic analysis led to the identification of one MAG that could be assigned to K. pneumoniae (denoted as MAG-KP1). Phylogenetic tree construction and average nucleotide identity (ANI) comparison revealed that one strain, SC-KP1, exhibited a high degree of similarity to the isolate IS-KP1, with a 99.56% ANI. Conversely, the other strain, SC-KP2, displayed a slightly lower similarity, with a 99.51% ANI to IS-KP1. However, MAG-KP1 showed a lower similarity, with a 99.19% ANI to IS-KP1, despite having a higher completeness level at 85%. Interestingly, SC-KP1 harbored only six ARG genes, which was the same as that of IS-KP1. In contrast, SC-KP2 carried nine ARGs, including three genes (fosXCC, cfrC, and aph(3')-III_1), which were absent in IS-KP1.
The finding that K. pneumoniae was involved in numerous ARG co-evolution events underscores its role in shaping the resistance landscape within the gut microbiome. Several key ARGs, including APH(3')-IIIa, cfr(C), FosXCC, and sul2, have exhibited notable co-evolution patterns. APH(3')-IIIa was linked with a diverse range of bacteria including P. gallinarum, S. merdavium, Enterococcus faecalis, B. intestinihominis, A. putredinis, E. faecium, A. indistinctus, and various unknown species. These associations formed three distinct clusters, highlighting the diversity and interconnectedness of ARGs across different bacterial taxa. Similarly, cfr(C) was found to have evolved into three clusters, involving bacteria such as P. distasonis, A. indistinctus, K. pneumoniae, Enterococcus faecalis, B. intestinihominis, and Ruthenibacterium unknown-1. Notably, certain strains of unknown bacterial species formed unique clusters, indicating that such strains also exhibited evolutionary trajectories in ARG acquisition and dissemination. Furthermore, evidence of evolution of the fosXCC gene was obtained in bacteria such as A. putredinis, K. pneumoniae, P. distasonis, an unknown species in Massilistercora, P. gallinarum, Ruthenibacterium unknown-1, and an unclassified group. These findings showed that the fosXCC gene could be found in bacteria of diverse genetic backgrounds. The sul2 gene was identified in K. pneumoniae, B. eggerthii, and an unknown species (Unclassified-7-64), indicating its presence across different bacterial taxa. Importantly, these ARGs were commonly associated with SC-KP2, suggesting that they play a crucial role in conferring phenotypic antibiotic resistance in this strain.
In the gut microbiome analyzed in this work, K. pneumoniae was found to actively engage in HGT with 11 bacterial species. One notable event, namely, HGT2 as mentioned above, involved seven species, including B. eggerthii, Borkfalkia unknown_0, Ruthenibacterium unknown_0, S. merdavium, E. faecium, A. putredinis, and B. intestinihominis. This event involved the transfer of genes such as folE, polA, queC, queD, and queE, which encoded various physiological functions such as folate biosynthesis, DNA polymerization, and quorum sensing. Another significant HGT event involved the dut gene and was found to involve eight bacterial species: B. eggerthii, Borkfalkia unknown_0, S. merdavium, E. porci, M. merdigallinarum, P. distasonis, A. putredinis, and B. intestinihominis. In addition, an HGT event involves transfer of the genes cpoA and kanE among B. eggerthii, Borkfalkia unknown_0, Ruthenibacterium unknown_0, P. distasonis, A. putredinis, B. intestinihominis, and K. pneumoniae.
DISCUSSION
Single-cell analysis of the patient’s gut microbiome provides a powerful means to investigate the interaction between pathogenic and commensal bacteria during infection and antibiotic treatment. In this study, the patient was initially treated with cefuroxime sodium, followed by meropenem and linezolid as symptoms evolved. This sequential antibiotic regimen illustrates the adaptive clinical management required to control infection. Through single-cell analysis, we aimed to uncover microbiome composition shifts, explore resistance mechanisms, and assess treatment effectiveness.
The single-cell analysis in this study identified a diverse range of bacterial and a few archaeal genomes in the gut microbiome of the patient. Compared with a previous single-cell study of the normal gut microbiome that reported 100 bacterial species (19), our analysis revealed 11 species, nine of which were genera exclusively found in this patient. Notably, K. pneumoniae and Enterococcus faecium, both recognized as pathogens, were among the identified species. Species such as M. merdigallinarum, Borkfalkia, E. porci, S. merdavium, and M. smithii are infrequently reported in the human gut, and their genomes were rarely documented. However, such organisms could be identified in the single-cell SAGs in this work when compared to the metagenomic data. These species were also found to be predominantly involved in the evolution of ARGs and HGT events. In addition, a substantial number of unclassified and unknown bacteria were also detected, and these organisms also contributed to ARG evolution and HGT. Following antimicrobial treatment, the microbiome appeared to have undergone significant changes in the composition, favoring the proliferation of resistant bacteria while suppressing sensitive strains. Single-cell technology proved invaluable in detecting these pathogens, as well as uncommon and unclassified bacterial species, showcasing its application potential in clinical research.
Single-cell analysis offers the opportunity to directly detect ARGs harbored by specific bacterial strains in the human gut microbiome. Such capability is unmatched by other methods such as metagenomics analysis. Notably, this study revealed the presence of rare bacteria carrying ARGs, such as M. merdigallinarum, which harbored cfr(C), a gene typically associated with coagulase-negative Staphylococci, Staphylococcus aureus, and Enterobacteriaceae (20). This study uncovered 25 ARG subtypes, including tet(Q) and cfr(C), in multiple clusters. Notably, some genes like aph(2'')-Ii, ErmB, and tet(Q) were not detectable in metagenomic data, indicating that the resolution of single-cell analysis was much higher. Our data showed that uncommon and unknown bacteria played significant roles in ARG evolution. Examples include S. merdavium, which carried 9 ARG subtypes, and Ruthenibacterium, which harbored 15 ARG subtypes. In addition, the ARG subtypes of strains of these species often exhibited mutations when compared to the reference genes. Single-cell analysis not only allowed detection of specific or uncommon bacteria but also provided significant insights into their involvement in ARG evolution, which was not achievable by conventional metagenomic methods. Moreover, single-cell analysis allows direct detection of VFGs and plasmids harbored by specific bacterial strains within the human gut microbiome.
Our analysis highlights the involvement of 14 bacterial species in HGT events, with noteworthy contributions from less-studied taxa like E. porci, S. anginosus, and M. merdigallinarum. Our data, therefore, showed that bacterial strains of broad taxonomic diversity participated in genetic exchange within the gut microbiome. Specific genes like folE, polA, queC, queD, and queE were implicated in these HGT events, suggesting that acquisition of these genes is a key step in antibiotic resistance development in various gut microbiome strains and that these events contributed to the dissemination of functional traits among bacterial populations. Distinct clustering patterns of HGT events among different species indicate varying preferences for genetic exchange and existence of ecological niches that underlie the HGT dynamics. Notably, genes associated with plasmid transfer, such as mbeA, mbeC, and relG, exhibited specific associations with certain bacterial species, emphasizing the role of mobile genetic elements in facilitating HGT within microbial communities. Compared to a healthy human gut microbiome (19) using single-cell sequencing, our study reveals differences in HGT dynamics. Specific genes like traC, comB2, topB, radC, and wbpO were found to be prominent in healthy human cases. However, the main HGT genes identified in this study were folE, polA, queC, queE, queD, and dut, involving 13 bacterial species. These genes play a crucial role in enhancing the ability of the recipient strains to repair DNA damage and maintain genomic stability, which contributes to their overall fitness, especially after prolonged treatment with antibiotics.
Comparison between isolated genomes, such as that of the strain IS-KP1 and other single-cell genomes, offers detailed insights into the co-evolutionary dynamics and HGT mechanisms of K. pneumoniae within the human gut microbiome. Isolated genomes of K. pneumoniae exhibited a diverse array of ARGs and VFGs, confirming that this species is a clinically important pathogen. On the other hand, single-cell analysis distinguished two distinct K. pneumoniae strains, SC-KP1 and SC-KP2, each with unique genomic features. While SC-KP1 shares a similar ARG profile with IS-KP1, SC-KP2 carries additional ARGs, highlighting strain-specific differences in resistance. The co-evolution of these strains within the gut microbiome is reflected in the ARG transfer events involving key genes such as APH(3')-IIIa, cfr(C), FosXCC, and sul2, which are predominantly found in SC-KP2. These genes were found to be broadly distributed across diverse bacterial species, including the less explored gut inhabitants such as P. distasonis and P. gallinarum, highlighting the complex ecological dynamics in the gut microbiome that drive resistance gene dissemination and bacterial evolution. In addition, studies of HGT events between K. pneumoniae and other gut-associated species reveal the importance of genetic exchange in shaping bacterial adaptation to environmental changes and pathogenicity. Notable HGT events, such as those involving genes like cpoA and kanE, exhibit the potential of conferring antimicrobial resistance and enhancing bacterial fitness.
However, it is important to note that these findings are derived from a single patient, which limits the ability to generalize the results to broader populations. The gut microbiome is highly individual-specific and shaped by numerous factors, including genetics, diet, health status, and treatment history. Therefore, the microbial dynamics, ARG profiles, and HGT patterns observed in this study may not be representative of those in other individuals or patient groups. While this study provides a high-resolution view of microbial adaptation, it is inherently limited by its focus on a single patient. The findings, including the extensive HGT events and the micro-diversity of resistance strains, are illustrative of the processes that can occur within a complex gut ecosystem under strong selective pressure. However, individual factors such as the host’s unique native microbiota, genetics, and exact medical history undoubtedly influence these dynamics. Therefore, the generalizability of these specific results may be limited. Future studies with larger patient cohorts, stratified by disease state and treatment regimen, will be essential to confirm the prevalence and patterns of the adaptation mechanisms reported here. This work serves as a critical proof of concept, demonstrating the power of single-cell genomics to uncover these complex within-host evolutionary dynamics.
This study has several limitations. First, the analysis was confined to the gut microbiome. While the patient was treated for a suspected pulmonary infection, no corresponding respiratory samples were available for culture or sequencing. Therefore, we cannot establish a direct link between the resistant pathogens found in the gut and the initial cause of the lung inflammation. However, our findings provide a detailed map of the ARG landscape that emerged in the gut as a consequence of therapy, highlighting its role as a potential reservoir for resistance. The resistance genes identified in the gut microbiome, such as blaOXA-347, likely originated from environmental exposure—particularly the hospital setting—which serves as a common reservoir for antibiotic-resistant bacteria. These genes were probably acquired by the patient and subsequently established within the gut microbial community. Under the potent selective pressure of antimicrobial treatment, these resistance determinants were enriched and maintained, potentially through HGT to other commensal bacteria, allowing them to persist and evolve as a functional component of the patient’s adaptive gut resistome. The detection of high-risk resistance genes like blaOXA-347 likely reflects initial acquisition from the hospital environment, where cross-colonization with multidrug-resistant organisms is common. However, our single-cell data demonstrate these genes were not merely transient contaminants but were functionally established within the gut microbiome. The presence of these genes within multiple Single Amplified Genomes (SAGs), evidence of in situ mutation, and widespread HGT confirm they underwent active selection and evolution under antibiotic pressure, transforming the gut into a reservoir of adapted resistance.
While the resistance phenotype of the isolated K. pneumoniae strain was confirmed through antimicrobial susceptibility testing, the functional expression of resistance genes identified via single-cell sequencing in uncultured taxa warrants further investigation. Future studies should prioritize developing methods for phenotypic validation—such as heterologous expression or advanced culturing techniques—to directly link these genotypic profiles to observable resistance traits in diverse gut microbiota.
Future studies involving larger and more diverse cohorts are needed to validate these observations and to further elucidate the roles of rare and uncharacterized bacterial species in antimicrobial resistance evolution and pathogen ecology. Integrating single-cell approaches with longitudinal sampling and complementary techniques will also be critical for expanding our understanding of microbiome dynamics in clinical settings.
Conclusion
In this study, single-cell analysis was found to be highly effective in unraveling the intricate dynamics of bacterial colonization, antibiotic resistance development, and HGT events within the human gut microbiome. Through this approach, we identified a diverse array of bacterial species, including uncommon taxa rarely found in human gut microbiome, and directly detected ARGs and VFGs harbored by specific strains. The study revealed a complex landscape of ARG evolution that involved both common and uncommon bacteria and highlighted the role of HGT in disseminating functional traits across bacterial populations. Notably, specific genes and gene clusters implicated in HGT events were identified, shedding light on their diverse roles in bacterial adaptation to changes in environmental conditions and niche specialization. By juxtaposing isolated genomes with single-cell genomes, we gained insights into the co-evolutionary dynamics and strain-specific variations in antimicrobial resistance development, particularly in pathogens like K. pneumoniae. Despite the limitations of single-cell analysis, including relatively low sample representation, our findings underscore its significant potential in advancing our understanding of the ecology of the human gut microbiome and facilitating development of strategies for combating antibiotic resistance and infectious diseases in clinical settings.
MATERIALS AND METHODS
Patient case
An adult male was admitted on 18 November 2023 with "acute cerebral hemorrhage." Cefuroxime sodium was given on Day 2. Treatment changed to meropenem and linezolid on Day 11 due to fever and high inflammation. Linezolid was stopped on Day 19. By Day 21, the patient had diarrhea and constipation; rectal swabs showed carbapenemase-producing K. pneumoniae. Meropenem was stopped on Day 27 after inflammation normalized. The patient left the ICU on Day 31. A rectal sample for single-cell analysis was taken on 12 December 2023. See supplementary methods for details.
Isolation and lysis
The specimens were mixed with 25% glycerin and frozen at −80°C. For each experiment, 1-3 μL aliquots were incubated at 37°C for 30 minutes, centrifuged at 13,400 rpm for 1 minute, and 900 µL of the supernatant was discarded. This was repeated three times with 900 µL of PBS. The final bacterial concentration was adjusted to 50 × 10^^6^ cells/mL and then mixed with OptiPrep. A lysis reagent mix (240 µL) included green buffer, lysozyme, prepgem, lysostaphin, BSA, random hexamer, and water. The microbial suspension was connected to a device via a syringe and polyethylene tubing, with flow rates set for microbial suspension, lysis reagents, and oil. Droplets were collected and transferred to a PCR tube, with mineral oil added to prevent evaporation. The lysis program involved incubation at 37°C for 30 minutes, 75°C for 15 minutes, and 95°C for 5 minutes, followed by storage at 4°C. See supplementary methods for details.
Whole-genome amplification
In whole-genome amplification, a 100 µL MDA mix included phi29 DNA Polymerase Buffer, random hexamers, dNTPs, phi29 DNA Polymerase, BSA, Tween-20, and T2, adjusted with water. Droplet emulsion was injected into an M1 device, paired with droplets containing MDA reagents. Electric fields merged droplet pairs, initiating genome amplification. Incubation at 30°C for 8 hours, followed by 65°C for 10 minutes, was conducted, with samples stored at 4°C for analysis. See supplementary methods for details.
Droplet-Based tagmentation and PCR barcoding in DNA sequencing library preparation
In the experimental workflow, tagmentation involved preparing a 90 µL Nextera mix with Tagment Buffer L, Tagment DNA Enzymes B and C, BSA, and water. Droplets containing sample and tagmentation reagents were merged using the M1 device, followed by incubation at 55°C for 10 minutes and storage at 10°C. For PCR barcoding, a 420 µL PCR mix was prepared with water, reverse primer, BSA, and Tween-20. Sample droplets were merged with PCR reagents and barcoding beads using the M1 device. After UV exposure to release bar code oligos, PCR included denaturation, annealing, and extension cycles, with final storage at 4°C. Following purification with PFO and AMPure beads, DNA samples were pre-amplified using a mix of pre-amplification reagents. Post-purification steps involved ethanol washing and resuspension in water. Fragmentation and linking were performed to prepare samples for sequencing. See supplementary methods for further details.
Purification and amplification strategy for library fragment
After adding 69 µL of NF-H2O and 0.3X AMPure beads to the amplification product, incubate the mixture at room temperature for 5 minutes. Transfer the supernatant to a new centrifuge tube after placing it on a magnetic rack for 5 minutes. Add 0.3X AMPure beads to the tube and incubate again at room temperature for 5 minutes. Discard the supernatant after placing the tube on a magnetic rack for 5 minutes. Clean the AMPure beads twice with 1 mL of freshly prepared 80% ethanol. Allow the beads to dry, add 22 µL of ultrapure water, and incubate at room temperature for 5 minutes. Transfer 20 µL of the sample to a 200 µL centrifuge tube. For library tag amplification, prepare a 30 µL mix with 25 µL of amplification mixes, 2.5 µL of amplification primer 1 (Illumina S50X), and 2.5 µL of amplification primer 2 (Illumina N70X). Mix with the sample, and incubate at 98°C for 30 seconds, followed by 11 cycles of denaturation at 98°C for 10 seconds, annealing/extension at 65°C for 75 seconds, and a final extension at 65°C for 5 minutes. Store samples at 4°C for further analysis. Following amplification, add 50 µL of NF-H2O to the product and introduce 0.5X AMPure beads. Mix thoroughly, and incubate at room temperature for 5 minutes. Transfer the supernatant to a new tube after placing it on a magnetic rack for 5 minutes. Add 0.3X AMPure beads to the tube with the supernatant, mix thoroughly, and incubate at room temperature for 5 minutes. Discard the supernatant after placing the tube on a magnetic rack for 5 minutes. Clean the AMPure beads twice with 1 mL of freshly prepared 80% ethanol. Allow the beads to dry, add 22 µL of ultra-pure water, and incubate at room temperature for 5 minutes. Transfer 20 µL of the sample to a 200 µL centrifuge tube. See supplementary methods for further details.
Genomic analysis pipeline for single amplified genomes (SAGs)
Raw sequencing data underwent initial preprocessing with Fastp v0.24.1 (21) and Cutadapt v5.0 (22) to remove adapters and low-quality sequences. Barcoded reads were then segregated into separate files using a custom Python script. SAGs were individually *de novo-*assembled using SPAdes v4.2.0 (23), followed by quality assessment using CheckM v1.2.3 (24) and genome comparison via Sourmash v4.9.0 (25). Hierarchical clustering grouped SAGs into bins based on genome similarities. Bins were refined iteratively, combining genomes exceeding 95% ANI and filtering contigs based on the length and coverage. Genomes meeting quality thresholds were taxonomically classified using GTDB-Tk 2.4.1 (26). See supplementary methods for further details.
Phylogeny analysis of genomes
The phylogeny of high- or medium-quality bins was constructed using Anvi'o. Amino acid sequences of six ribosomal proteins (Ribosomal_L1, Ribosomal_L2, Ribosomal_L3, Ribosomal_L4, Ribosomal_L5, and Ribosomal_L6) were extracted, concatenated, and utilized in the analysis. The resulting phylogenomic tree was visualized using ggtree V3.16.0 (27).
Differentiating strains of the same species
Using Bcftools V1.21 (28), SNPs were identified in high- and medium-quality genomes. Hierarchical clustering based on SNP vectors revealed clusters of SAGs sharing similar SNP profiles, indicating strain proximity. UMAP V0.5.8 (29) plots further visualized SNP data dimensionally. Consensus genotyping of strains involved comparing SNP vectors, excluding ambiguous SAGs. Finally, strain-resolved genomes were co-assembled using SPAdes from SAG reads assigned to each strain. See supplementary methods for further details.
Horizontal gene transfer analysis
To detect HGT events, we identified DNA blocks > 5,000 bp with > 99.98% identity using blastp. SAGs were filtered based on read alignment ratios (>90%) to reduce contamination. Validation involved examining SAG reads covering HGT sequences between species A and B. Statistical modeling assessed the likelihood of contamination, assuming a 20% worst-case contamination rate. Gene prediction on HGT sequences was performed using prokka (version 1.14.5) (30). See supplementary methods for further details.
Sequencing of metagenomic DNA
Metagenomic sequencing was applied to the same sample used for single-cell sequencing (sampled on 12 December 2023) to validate DNA consistency and minimize batch effects. The genomic DNA was fragmented using the Frag enzyme, resulting in 350 bp-sized fragments. These fragments underwent end-polishing, A-tailing, and adapter ligation for DNB sequencing, followed by PCR amplification. The PCR product was heat-denatured with a complementary molecule and then ligated using DNA ligase. The remaining linear molecule was digested with exonuclease, producing a single-strand circular DNA library. Library quality was assessed using Qubit for quantification, real-time PCR, and a bioanalyzer for size distribution analysis. Quantified libraries were evenly pooled to form DNA Nanoballs (DNB), which were subsequently sequenced on a DNBseq-T7 platform with a PE150 read length, yielding 20 G of raw data per sample.
Assembly of metagenomes, prediction of genes, taxonomic assignments, and statistical analysis
Raw sequences underwent initial preprocessing with Trimmomatic (v0.39) to remove adapters, reads < 36 bases, and those with quality scores < 15. KneadData (v0.7.7) eliminated host contamination. Cleaned reads were aligned to microbial marker genes using Bowtie2 (v2.5.1) via MetaPhlAn4. Assembly used megahit v1.2.9; metaSPAdes assembled NGS reads. Binning employed Maxbin 2.0, MetaBAT2, and CONCOCT within metaWRAP v1.3, refined with bin_refinement. MAG quality was assessed by CheckM, assigned to SGBs by GTDB-Tk, and phylogenetic trees constructed with PhyloPhlAn 3.0 in iTOL. See supplementary methods for further details.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ghosh S, Pramanik S. 2021. Structural diversity, functional aspects and future therapeutic applications of human gut microbiome. Arch Microbiol 203:5281–5308. doi:10.1007/s 00203-021-02516-y 34405262 PMC 8370661 · doi ↗ · pubmed ↗
- 2Olivares M, Benítez-Páez A, de Palma G, Capilla A, Nova E, Castillejo G, Varea V, Marcos A, Garrote JA, Polanco I, Donat E, Ribes-Koninckx C, Calvo C, Ortigosa L, Palau F, Sanz Y. 2018. Increased prevalence of pathogenic bacteria in the gut microbiota of infants at risk of developing celiac disease: the PROFICEL study. Gut Microbes 9:551–558. doi:10.1080/19490976.2018.145127629672211 PMC 6287676 · doi ↗ · pubmed ↗
- 3Ruigrok R, Weersma RK, Vich Vila A. 2023. The emerging role of the small intestinal microbiota in human health and disease. Gut Microbes 15:2201155. doi:10.1080/19490976.2023.220115537074215 PMC 10120449 · doi ↗ · pubmed ↗
- 4Feng W, Liu J, Ao H, Yue S, Peng C. 2020. Targeting gut microbiota for precision medicine: focusing on the efficacy and toxicity of drugs. Theranostics 10:11278–11301. doi:10.7150/thno.4728933042283 PMC 7532689 · doi ↗ · pubmed ↗
- 5Wuethrich I, W Pelzer B, Khodamoradi Y, Vehreschild MJGT. 2021. The role of the human gut microbiota in colonization and infection with multidrug-resistant bacteria. Gut Microbes 13:1–13. doi:10.1080/19490976.2021.1911279 PMC 807874633870869 · doi ↗ · pubmed ↗
- 6Arato V, Raso MM, Gasperini G, Berlanda Scorza F, Micoli F. 2021. Prophylaxis and treatment against Klebsiella pneumoniae: current insights on this emerging anti-microbial resistant global threat. Int J Mol Sci 22:4042. doi:10.3390/ijms 2208404233919847 PMC 8070759 · doi ↗ · pubmed ↗
- 7Krapp F, Morris AR, Ozer EA, Hauser AR. 2017. Virulence characteristics of carbapenem-resistant Klebsiella pneumoniae strains from patients with necrotizing skin and soft tissue infections. Sci Rep 7:13533. doi:10.1038/s 41598-017-13524-829051525 PMC 5648777 · doi ↗ · pubmed ↗
- 8Faria NA, Touret T, Simões AS, Palos C, Bispo S, Cristino JM, Ramirez M, Carriço J, Pinto M, Toscano C, Gonçalves E, Gonçalves ML, Costa A, Araújo M, Duarte A, de Lencastre H, Serrano M, Sá-Leão R, Miragaia M. 2024. Genomic insights into the expansion of carbapenem-resistant Klebsiella pneumoniae within Portuguese hospitals. J Hosp Infect 148:62–76. doi:10.1016/j.jhin.2024.02.02838554808 · doi ↗ · pubmed ↗