Dual RNA-seq analysis reveals differences in defensive lncRNA expression in Pinus spp. with varying susceptibility to Fusarium circinatum
Cristina Zamora-Ballesteros, Julio J. Diez, Gloria Pinto, Artur Alves, Katrin Heer, Jorge Martín-García

TL;DR
This study uses dual RNA-seq to explore how lncRNAs in pine trees and a pathogen differ between resistant and susceptible species during infection.
Contribution
The first lncRNA catalogues for P. pinea and F. circinatum, revealing non-coding regulatory differences in resistance and susceptibility.
Findings
P. radiata and P. pinea showed distinct lncRNA expression patterns linked to defense and metabolic processes.
F. circinatum lncRNAs were associated with transcriptional control and toxin production during infection of resistant hosts.
Resistant pine lncRNAs clustered into defense-related modules, while susceptible ones were linked to photosynthesis.
Abstract
Long non-coding RNAs (lncRNAs) are emerging regulators of plant immunity, but their roles in conifer-pathogen interactions remain largely unexplored. We applied a dual RNA-seq approach to resistant Pinus pinea and susceptible P. radiata challenged with Fusarium circinatum at 4 dpi, and concurrently profiled fungal lncRNAs. Using a conservative multi-tool pipeline, we identified 8,783 lncRNAs in P. radiata, 5,255 in P. pinea, and 1,020 in F. circinatum. Pine lncRNAs displayed canonical features (shorter length, fewer/shorter exons, intergenic dominance) and limited primary-sequence conservation. Differential expression analysis revealed 37 (P. radiata) and 34 (P. pinea) infection-responsive lncRNAs. Predicted cis targets in P. radiata were enriched for energy/redox and gibberellin-related functions, whereas P. pinea targets pointed to TCA/redox and translation control. Weighted gene…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
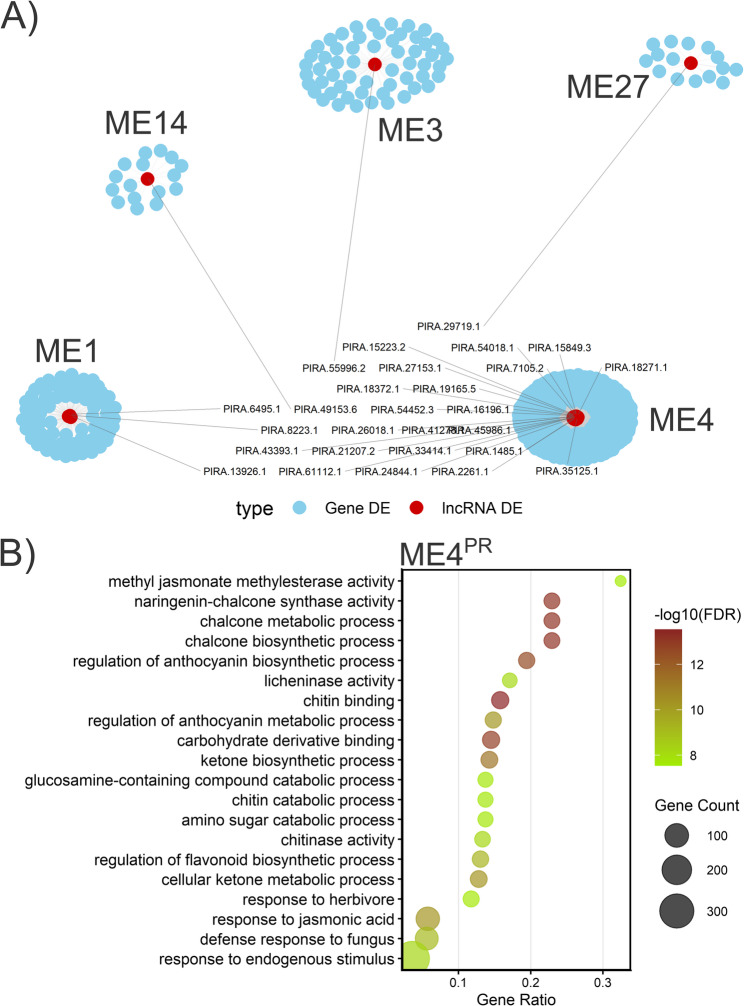 Figure 10
Figure 10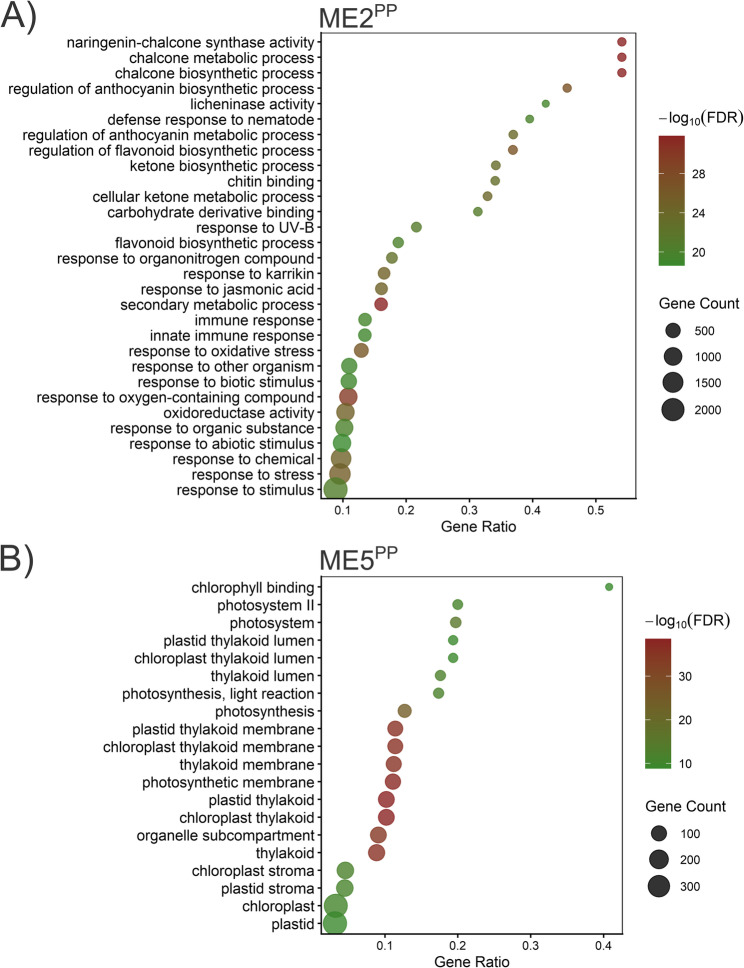 Figure 14
Figure 14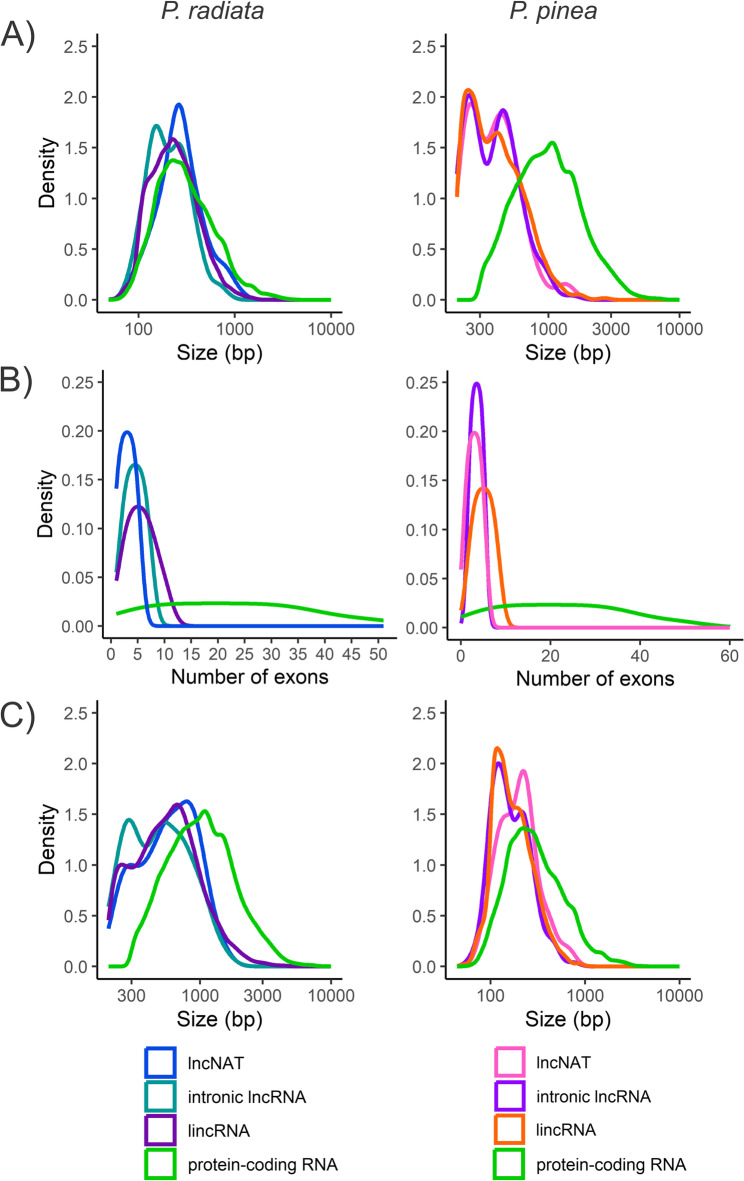 Figure 1
Figure 1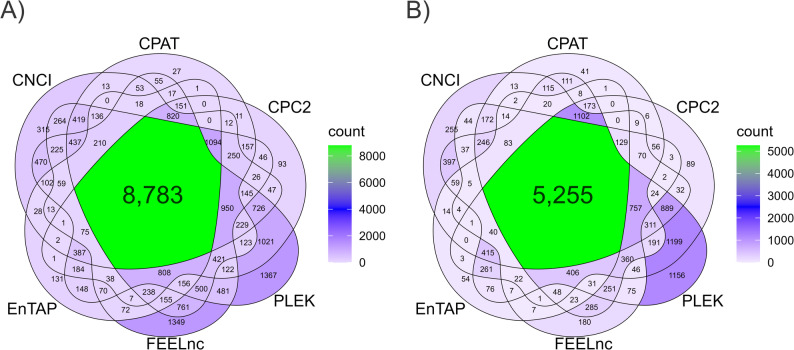 Figure 20
Figure 20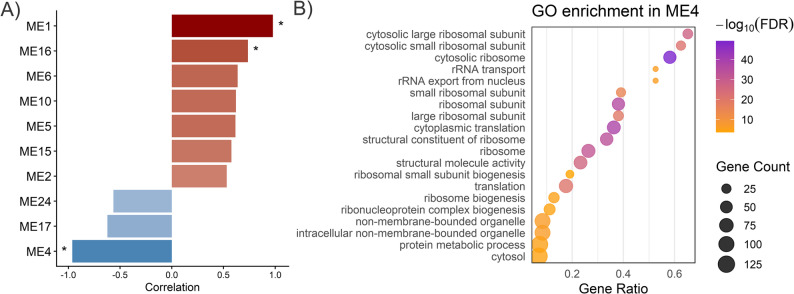 Figure 21
Figure 21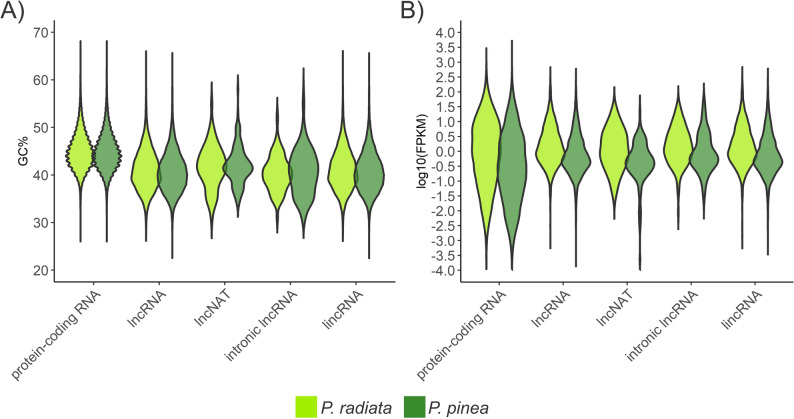 Figure 2
Figure 2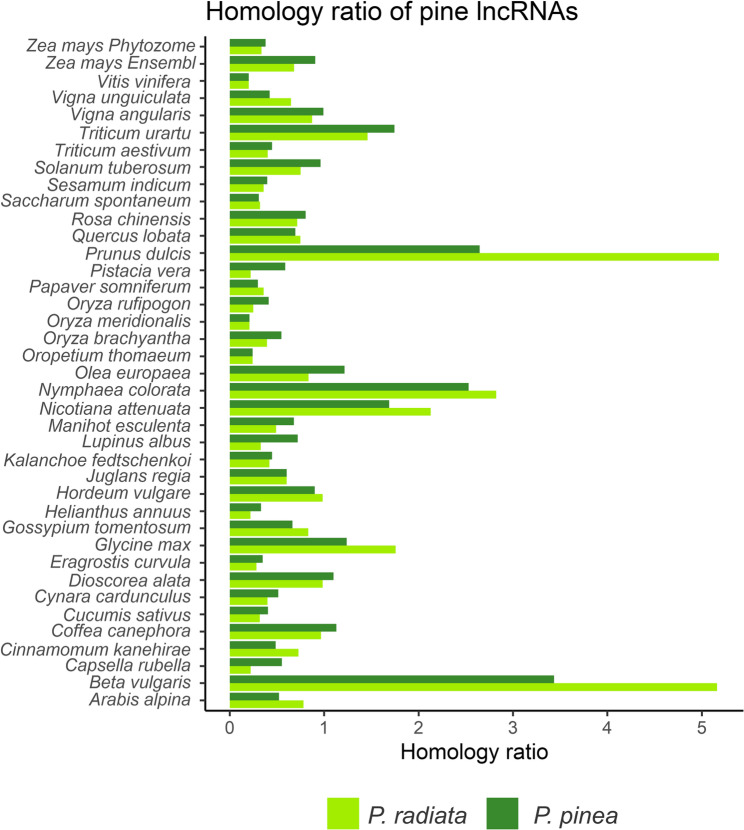 Figure 3
Figure 3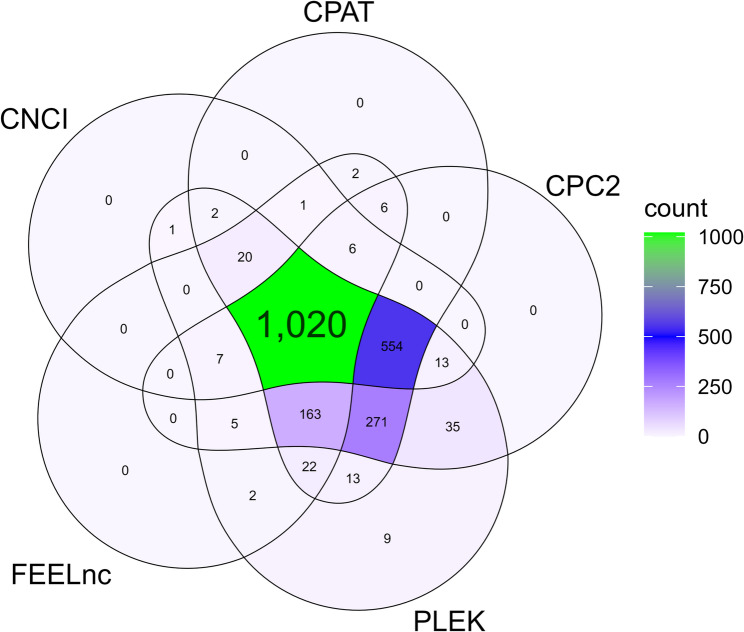 Figure 4
Figure 4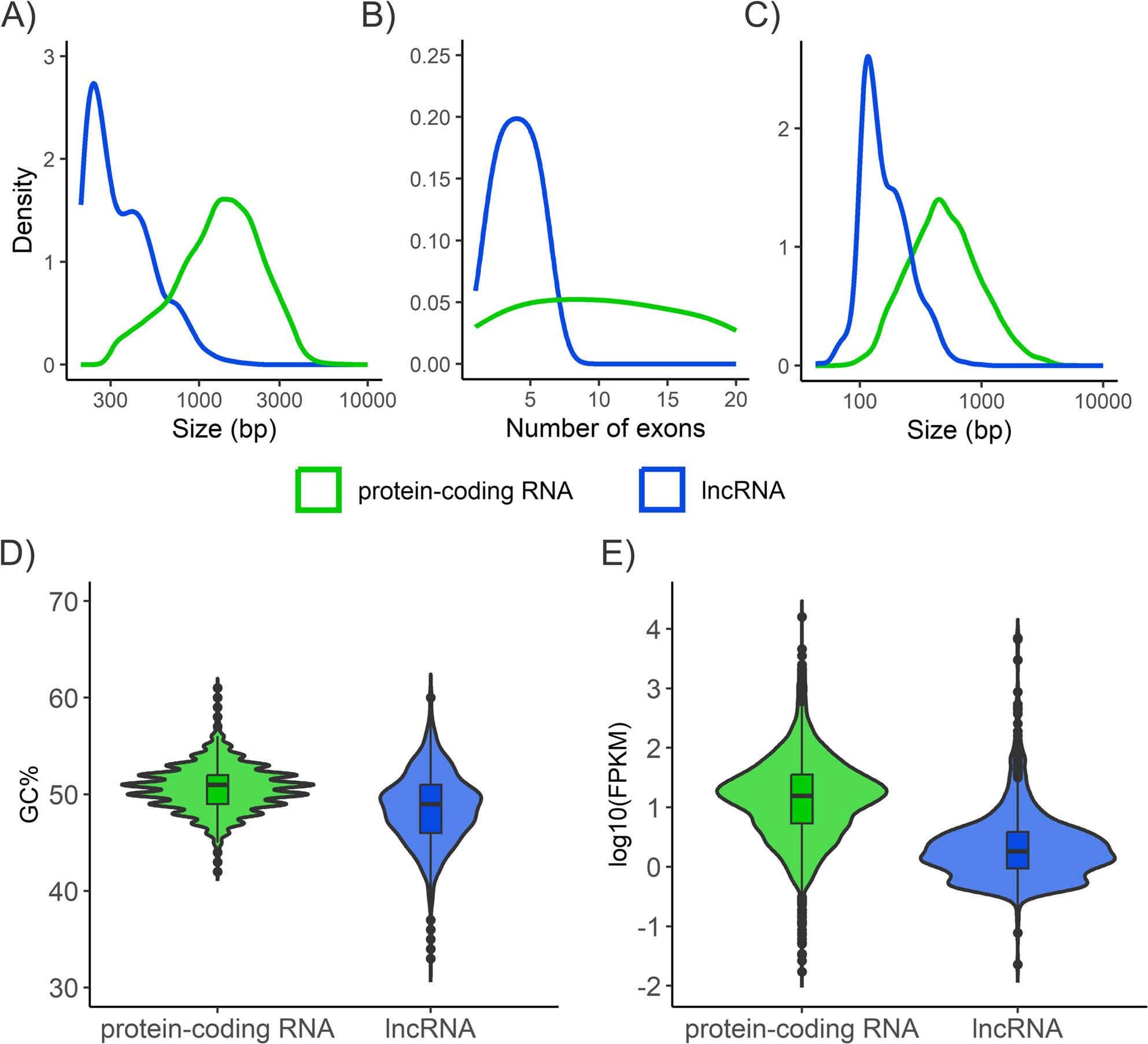 Figure 5
Figure 5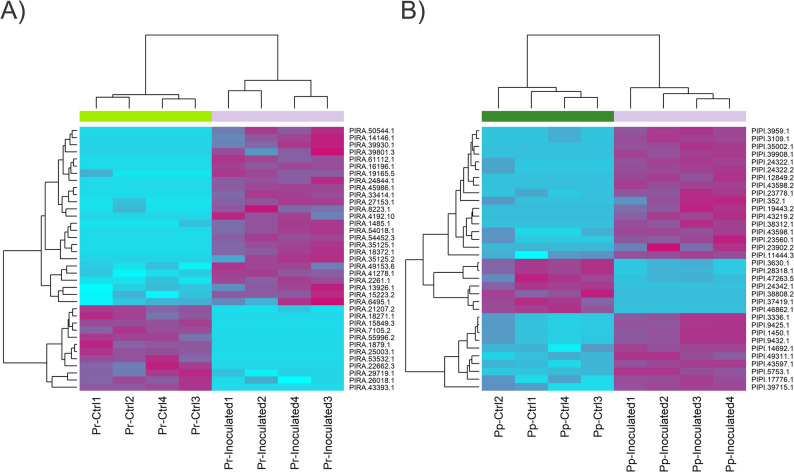 Figure 6
Figure 6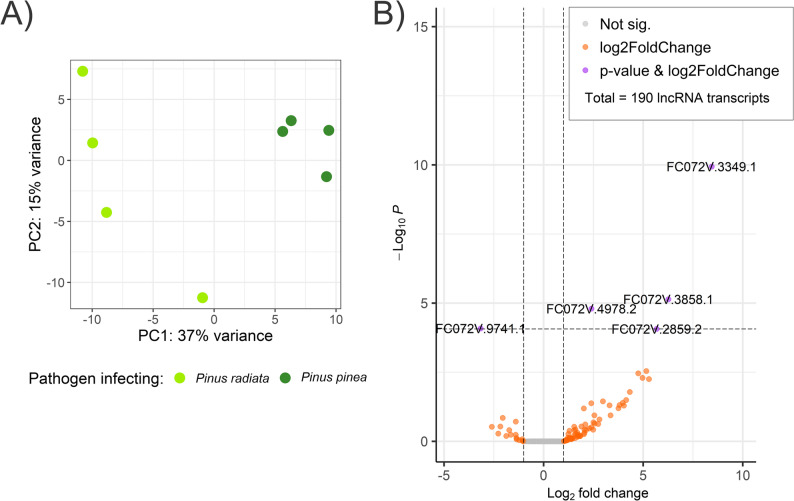 Figure 7
Figure 7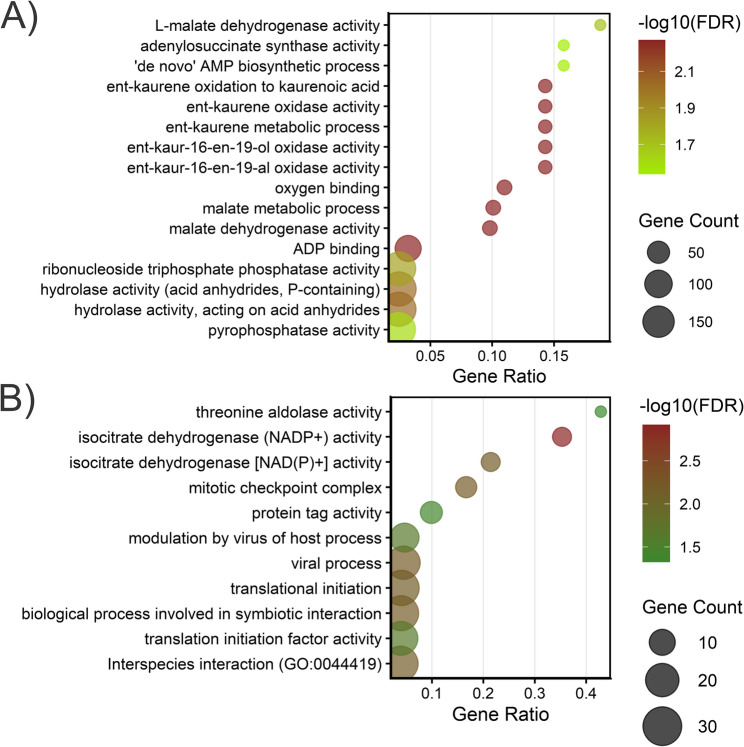 Figure 8
Figure 8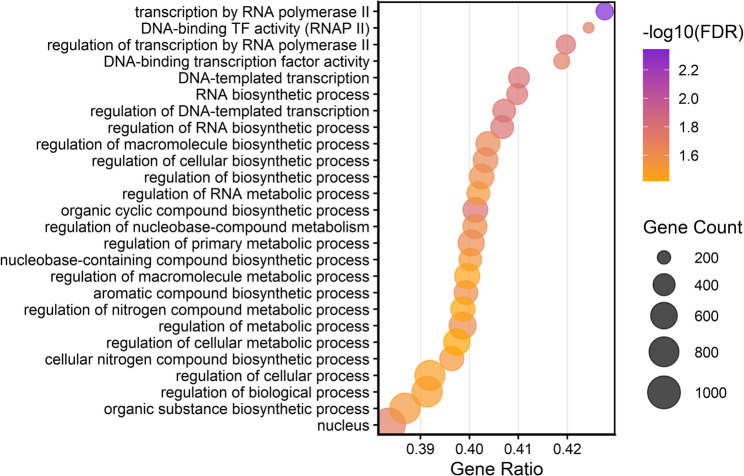 Figure 9
Figure 9- —Albert-Ludwigs-Universität Freiburg im Breisgau (1016)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant and Fungal Interactions Research · Plant-Microbe Interactions and Immunity · Plant Pathogens and Fungal Diseases
Background
In recent years, our understanding of the molecular mechanisms governing plant responses to pathogenic challenges has expanded significantly. Long non-coding RNAs (lncRNAs; >200 nt) have emerged as key regulatory molecules that fine-tune gene expression at transcriptional and post-transcriptional levels [1]. LncRNAs can be categorized by genomic location into long intergenic ncRNAs (lincRNAs), long intronic ncRNAs (incRNAs) and natural antisense transcripts (NATs) [2]. These transcripts have diverse molecular mechanisms by interacting with different biomolecules such as DNA, RNA and proteins, for example as decoys, guides, enhancers or scaffolds [3, 4]. With the advent of next-generation sequencing and high-resolution transcriptome analyses, plant lncRNA discovery has rapidly advanced in recent years [5]. Several pathogen-responsive lncRNAs have been experimentally validated, including ELENA1 in Arabidopsis thaliana [6], lncRNAs induced by Fusarium oxysporum [7], and cotton lncNATs whose silencing increased resistance to Verticillium dahliae and Botrytis cinerea [8]. These studies support a role for lncRNAs in plant immunity, however, lncRNAs remain poorly characterized in non-model plants, particularly in conifer species.
Although transcriptomic studies of interactions in important pathosystems involving conifers have increased considerably over the past decade [9–20], very few have studied the non-coding part [21]. The development of resistant genotypes through breeding and/or genetic engineering requires a deep understanding of the molecular mechanisms underlying plant-pathogen interactions. Using resistant plant material for reforestation may be one of the most effective long-term strategies for mitigating pathogen infections in natural forests and plantations. This holds particularly true when dealing with pathogens that cannot be eradicated, as is the case with Fusarium circinatum, the causal agent of Pine Pitch Canker (PPC) [22, 23]. PPC is a destructive disease affecting pines (Pinus spp.) and Douglas-fir (Pseudotsuga menziesii) globally, causing serious problems in nurseries, pine plantations and natural forests [24]. In this context, various transcriptome analyses aimed at unraveling defence responses have yielded detailed insights into the molecular mechanisms driving disease progression in the Pinus-F. circinatum pathosystem. These studies have explored host responses, ranging from high susceptibility in pine species like Pinus greggii, P. patula and P. radiata, to moderate or lower susceptibility in P. maximinoi,* P. oocarpa*,* P. pinaster*, P. pinea and P. tecunumanii [11, 13, 17, 18, 20, 25, 26]. The importance of jasmonic acid (JA), ethylene (ET), and salicylic acid (SA) signaling pathways in host resistance to F. circinatum has been linked to both resistant and moderately susceptible species. The biosynthesis of secondary metabolites appears to play a significant role in effective defence against the pathogen, with the induction of phenylalanine ammonia-lyase (PAL) expression and higher expression of genes related to this pathway observed in resistant pine species [17, 20]. Additionally, the ineffective response observed in susceptible pine species can likely be attributed to impaired perception of fungal infection and a subsequent failure in downstream defence signaling, as genes encoding pattern recognition receptors (PRRs) and resistance (R) proteins were weakly expressed in these plants [17, 20, 27].
Despite growing evidence that F. circinatum infection reprograms host coding and non-coding transcriptomes, including pathogen-responsive lncRNAs linked to kinase signaling, phytohormone regulation, and cell-wall reinforcement [21], the roles of lncRNAs in conifers, and particularly in P. pinea, remain unclear. Dual RNA-seq is particularly valuable in this system because it enables simultaneous profiling of host defence and pathogen infection programs from the same infected tissues and time point. Here we use dual RNA-seq to identify and compare defence-related lncRNAs in resistant P. pinea and susceptible P. radiata, and to profile F. circinatum lncRNAs expressed during pathogenesis. We catalog the number and features of host and pathogen lncRNAs, predict cis targets, and integrate differential expression with co-expression networks to place lncRNAs within defence modules. Functional annotation of lncRNA targets highlights candidate pathways that may underpin pine resistance and fungal virulence. To our knowledge, this is the first report of P. pinea and F. circinatum lncRNAs, providing a resource and testable hypotheses for lncRNA-mediated regulation of resistance.
Methods
Data collection and reads quality check
Two transcriptome datasets generated from stem tissues of P. radiata and P. pinea infected with F. circinatum [20] were used to identify defence-related lncRNAs. As described in the original study, plant material consisted of one-year-old seedlings from provenance seed lots (Galicia for P. radiata and Meseta Norte for P. pinea). Each biological replicate corresponded to an individual seedling, and seedlings were randomly allocated to inoculated and mock-inoculated treatments. Seedlings from both species were inoculated with 10 µL of a spore suspension (10^6^ spores mL^− 1^) of the F. circinatum isolate Fc072v, originally isolated in northern Spain (Cantabria) and previously described as virulent in P. radiata [20, 28, 29].
Control seedlings were mock-inoculated with sterilised distilled water. Plants were maintained in a growth chamber at 21.5 °C under a 16/8 h light/dark photoperiod, watered following routine nursery practice, and no fertilizers or fungicides were applied. Seedlings were kept under these controlled conditions prior to inoculation to allow acclimation.
Sampling at 4 days post-inoculation (dpi) captured the early infection phase (within the first week post-inoculation), when F. circinatum biomass increases rapidly [30], and ensures sufficient pathogen biomass for dual RNA-seq analysis. The dataset for each pine species included 4 inoculated and 4 control libraries of strand-specific RNA-seq that were sequenced on an Illumina NovaSeq 6000 platform, and is deposited in Zenodo (doi: 10.5281/zenodo.17591267).
Plant and pathogen transcript reconstruction
The bioinformatics workflow and versioned scripts are provided in the GitHub repository archived at Zenodo [31]. Fastq files were first assessed for quality control using FastQC v.0.11.9 [32]. The raw reads were trimmed for Illumina adaptor sequences and light 5′-end cropping using Trimmomatic v.0.38 [33]. Quality-filtered reads were then mapped to the Pinus taeda reference genome (Pita_v2.01; Treegenes database [34]) using HISAT2 v.2.0.0 [35] with parameters “--dta” and “--rna-strandness RF”. The SAM files from the pine mapping were processed with the SAMtools utility [36] for converting to binary alignment map (BAM) format and sorting by coordinates. The transcripts for each sample were reconstructed separately by StringTie v.2.1.4 [37] using the “-G" option with the annotation file of P. taeda (Pita.2_01.entap_annotations.tsv; Treegenes database [34]). This file was previously fixed with Gffread utility v.0.12.1 [38] for the correct readability by StringTie program. After transcripts assembly, 16 GTF files had been generated and subsequently merged based on the pine species using the StringTie “-merge” option. Transcripts with expression levels < 0.1 FPKM (Fragments Per Kilobase of exon per Million) mapped reads were excluded to reduce very lowly supported transcript models, which are more likely to represent spurious reconstructions [39]. Then, two non-redundant set of transcripts (P. radiata and P. pinea) with unique identifiers were generated and further compared with the P. taeda reference annotation GTF file (Pita_v2.01; Treegenes database [34]) using the software GffCompare v.0.12.1 [38]. Transcripts were classified in different class codes according to their nature/origin.
For the pathogen, the identical read alignment and file manipulation process was executed using solely the libraries from the inoculated samples and the sequenced F. circinatum Fc072v genome (accession number JAGGEA000000000). As there was no curated GTF file available for this pathogenic fungus, transcript reconstruction was carried out without the aid of an annotation file. Ultimately, the resulting eight GTF files were merged to create a non-redundant set of F. circinatum transcripts with unique identifiers, following the same procedure as with the pine files. The transcripts were then annotated using the program Eukaryotic Non-Model Transcriptome Annotation Pipeline (EnTAP) v.0.9.2 [40], which predicts open reading frames (ORFs), conduct a similarity search using DIAMOND v.1.9.2 [41] and assigns protein domains (Pfam), Gene Ontology (GO) terms and KEGG pathways using EggNOG v.1.0.3 [42]. A new GTF file was generated using the annotated transcripts to facilitate comparison with the previous non-redundant set of F. circinatum transcripts, categorizing them into different class codes. The annotated transcripts were then discarded from the pipeline (79.3%), and the remaining (20.7%) were retained for lncRNA identification.
LncRNA identification
Based on all the pine assembled transcripts, the already known transcripts marked with the class code “=” were excluded from each pine transcript set before conducting the potential long non-coding RNAs identification. For the unknown transcripts, stringent conditions were applied using the filter module of the FEELnc v.0.2 tool [43] by removing transcripts < 200 bp and single-exon models to reduce low-confidence short-read assemblies and prioritize a high-confidence lncRNA set. After that, the sequences of the resulting pine transcripts were extracted with Gffread v.0.12.1 [38] and further screened for their respective coding potential using six different computational approaches: [1] Coding Potential Calculator (CPC2 v.1.0.1) [2], Coding-Non-Coding Index (CNCI) [3], Coding-Potential Assessment Tool (CPAT) [4], PLEK [5], FEELnc codpot module and [6] EnTAP v.0.9.2. CPC2 is a tool for assessing protein-coding potential using sequence alignment and biological characteristics, categorizing transcripts as non-coding when the score is < 0 [44]. CNCI analysis distinguishes transcripts by evaluating adjacent nucleotide triplets, not relying on known annotations, where a transcript is considered as non-coding RNA when the score is < 0 [45]. We used a custom CPAT model trained on F. circinatum (hexamer/logit), classifying transcripts as non-coding at coding probability < 0.38 [46]. PLEK applies a support vector machine (SVM) algorithm based on an improved k-mer scheme to differentiate lncRNAs from mRNAs [47]. The codpot module of the FEELnc tool for assessing the coding potential of the transcripts was used with the shuffling mode to calculate a coding potential score (CPS) using a random forest algorithm trained by extracting features of ORF coverage, codon usage and nucleotide frequency. The specificity threshold was set at 0.95 to minimize false positives in a non-model, repeat-rich conifer–fungus system. This stringent setting prioritizes precision over recall so that only the most confidently non-coding transcripts are retained. Finally, EnTAP was employed to detect potential annotations among the transcript sequences. In this case, the similarity search was conducted using the NCBI non-redundant protein database (release-201) with E-value < 10^− 5^ as cut-off for identification, excluding genomic databases containing non-coding sequences. For each pine species, candidates for lncRNA transcripts that were consistently identified as non-coding across all methods were accepted as putative lncRNAs.
The lncRNA set for each pine species was further classified into the different GffCompare categories according to the locations relative to the nearest protein-coding genes, including lncRNAs without any overlap with other protein-coding genes are classified as intergenic lncRNAs (lincRNAs; class code ‘u’), natural antisense lncRNAs overlapping exons of a protein-coding transcript on the opposite strand (lncNAT; class code ‘x’), and intronic transcripts (class code ‘i’; [48]).
Putative characteristics of identified LncRNA transcripts
To understand the differences between lncRNAs and mRNAs, the genomic features of the predicted lncRNAs were analysed focusing on the length, the number of exons and their length, the expression levels and the GC content. For the pine hosts, feature distributions were compared across transcript classes (mRNA, lincRNA, intronic lncRNA and lncNAT) using Kruskal-Wallis tests, followed by Benjamini-Hochberg-corrected pairwise Wilcoxon rank-sum tests. For F. circinatum, differences between lncRNA and mRNA transcripts were evaluated using Wilcoxon rank-sum tests with Benjamini-Hochberg correction across features.
The conservation of pine lncRNAs was assessed using two recently updated databases of known plant lncRNAs. CANTATAdb 2.0 [49] includes 239,631 predicted lncRNAs from 36 plant species, while GreeNC 2.0 database [50] encompasses 496,903 lncRNAs from 94 plant and algae species. All the pine transcripts designated as lncRNA were aligned against the known plant lncRNAs using the blastn algorithm (E-value < 10^− 5^) of the BLAST v.2.5.0 software suite. Likewise, the transcripts were also aligned to the Rfam (version 14.10 Nov 2023, 4170 families) and miRBase (version 22.1) non-coding RNA databases using the blastn algorithm (E-value < 10^− 5^) in order to detect housekeeping non-coding RNAs including transfer RNA (tRNAs), ribosomal RNA (rRNAs) and snoRNAs, and miRNA precursors [51, 52].
To explore potential conserved or divergent lncRNAs between both pine species during the fungal infection, we performed a cross-species sequence comparison of the identified lncRNAs. Sequences of lncRNAs from each species were aligned using the blastn algorithm (E-value < 10^− 5^) of the BLAST v.2.5.0 software suite.
Differential expression analyses between inoculated and control pine seedlings
StringTie was employed to estimate expression for all transcripts of the experiment-level transcriptomes [53]. The output files were reformatted using the “prepDE.py” script for further expression analysis [37]. DESeq2 v.1.40.2 [54] was used to identify differentially expressed lncRNA (DELncRNA) transcripts based on the matrix of the estimated counts. The pairwise comparison of inoculated and control plants for each pine species were evaluated using Wald tests. LncRNAs were considered as differentially expressed if the adjusted p-values for multiple testing, using Benjamini–Hochberg to estimate the false discovery rate (FDR) [55], was less than 0.05 and the |log_2_ (Fold Change)| ≥ 1. To conduct co-expression analysis and assess potential target genes, we identified differentially expressed genes (DEGs) in both species equally.
LncRNAs target gene prediction and functional enrichment
Based on the genome location of the lncRNAs relative to the neighbouring genes, putative cis targets were defined as protein-coding genes located within 10 kb upstream and 100 kb downstream of each lncRNA, following common practice in lncRNA identification pipelines [56]. These genes both in P. radiata and P. pinea were identified using the FEELnc classifier module [43] and annotated using the EnTAP pipeline [40] as described above but implemented with two additional datasets, the RefSeq complete protein database (release-201) and the UniProtKB/Swissprot database (release-2020_05). Because libraries were strand-specific and read alignment was performed in a strand-aware manner, the assembled transcripts retained strand orientation. Strand and overlap relationships relative to annotated genes (intergenic, antisense exonic overlap, intronic overlap, and divergent/convergent configurations) were derived from GffCompare class codes and FEELnc output, and were used to distinguish antisense-overlapping lncRNAs (lncNATs) from intergenic lncRNAs when interpreting proximity-based cis candidates. Functional enrichment analysis of the target genes associated with the DELncRNAs was performed. Utilizing all assembled transcripts of each pine species as background, GO enrichment analysis was conducted using GOSeq v.1.38.0 [57]. This method is based on the Wallenius non-central hyper-geometric distribution, enabling adjustment for transcript length bias. The GO terms with corrected p-values (FDR) lower than 0.05 were considered to be enriched in the group.
Weighted gene co-expression network analysis (WGCNA) and identification of hub genes
In order to examine the co-expression patterns of protein coding genes and lncRNA genes under infection, read counts matrix for each organism was processed using the Weighted Gene Co-Expression Network Analysis (WGCNA) R package (version 1.73) [58]. This algorithm was used to construct a weighted correlation network and identify sets of highly correlated transcripts (modules) sharing similar expression patterns across samples. Transcripts with fewer than 10 reads and not detected in at least one condition were removed. For the remaining, variance-stabilizing transformation (VST) from DESeq2 was applied prior to network construction. The soft-thresholding power was selected using the scale-free topology criterion (R² ≥ 0.8) while also considering the corresponding slope and mean connectivity to avoid overly sparse networks. Based on this balance, the power was set to 6 for P. radiata (R² = 0.952; slope = − 1.88; mean.k = 2,684) and 12 for P. pinea (R² = 0.824; slope = − 1.39; mean.k = 852). A signed Topological Overlap Matrix (TOM) was used to quantify network interconnectedness, and modules were defined using the dynamic tree-cut algorithm with a minimum module size of 50 genes and merge cut height = 0.25. Other parameters were kept at default values. Module eigengenes were correlated with the infection trait to identify infection-responsive modules, and modules showing |r| ≥ 0.6 and p < 0.05 were considered significantly associated with infection. Within infection-associated modules, the intra-modular connectivity (kME) was examined to identify highly connected genes and lncRNAs (hub candidates). To gain functional insight, GO enrichment analysis of the protein-coding members of each significant module was performed using GOSeq as described above.
Results
High-throughput sequencing and transcripts assembly
High-throughput strand-specific RNA-seq of 16 libraries constructed from stem tissue of P. radiata and P. pinea inoculated with F. circinatum and mock-inoculated were analysed for lncRNA identification. Overall mapping rates from both pines and pathogen data sets are provided in Table S1. Sixteen high-depth transcriptomes were generated, with eight derived from each pine species. Within each pine species, four transcriptomes were reconstructed from seedlings inoculated with F. circinatum, while the remaining four were obtained from mock-inoculated seedlings. The number of assembled transcripts shared between treatments in each pine species is represented in Venn diagrams (Figure S1). After merging all the transcriptomes separately from each species, the unique transcriptome assembled for P. radiata was composed of 90,505 loci and 133,379 transcripts, with 43.1% GC content (Table S2). In the case of the P. pinea unique transcriptome, 107,395 transcripts were assembled located in 81,214 loci with 43.8% GC (Table S3). A total of 51,264 (38.44%) transcripts from P. radiata and 51,382 (47.84%) transcripts from P. pinea matched the P. taeda reference annotation (Pita_v2.01.gtf) and were excluded from lncRNA detection. These transcripts correspond to annotated protein-coding RNAs. The remaining 82,110 (P. radiata) and 56,013 (P. pinea) transcripts were processed through the lncRNA identification pipeline.
In the case of F. circinatum, the transcripts were assembled in a similar manner. Eight transcriptomes were generated, with four reconstructed from P. radiata seedlings and four from P. pinea seedlings, all of them inoculated with the pathogen. After merging all transcriptomes, the unique non-redundant transcriptome assembled for the fungus was composed of 10,486 loci and 11,706 transcripts, with 50.3% GC content (Table S4). The EnTAP program annotated 9,282 transcripts (79.3%), and the remaining 2,424 (20.7%) were utilized in the lncRNA identification pipeline (Table S5).
Genome-wide identification and characterization of pine LncRNAs
Unknown transcripts from each pine species were subjected to sequential filtering to obtain candidate lncRNAs. First, FEELnc excluded 54,597 (66.5%) and 38,964 (69.6%) transcripts in P. radiata and P. pinea, respectively, based on length and structure. We then assessed coding potential using six approaches (CPC2, CNCI, CPAT, PLEK, FEELnc and EnTAP). PLEK yielded the highest number of non-coding candidates, whereas EnTAP yielded the lowest, likely reflecting its primary role in annotation rather than coding-potential prediction. After removing candidates with coding potential, we retained 8,783 (P. radiata) and 5,255 (P. pinea) lncRNAs (Fig. 1). These lncRNAs were further categorized into different class codes based on their relationship with the closest reference transcripts. Table 1 summarizes the most representative class codes of assembled transcripts and predicted lncRNAs for each species. Percentages refer to the proportion within the total number of assembled transcripts and within the total number of predicted lncRNAs.
Fig. 1. Venn diagrams showing the numbers of candidate lncRNAs in (A) P. radiata and (B) P. pinea according to the Coding Potential Calculator (CPC2), Coding-Non-Coding Index (CNCI), Coding-Potential Assessment Tool (CPAT), PLEK, FEELnc codpot module and the Eukaryotic Non-Model Transcriptome Annotation Pipeline (EnTAP)
Table 1. Classification of P. radiata and P. pinea transcripts according to gffcompare class codesClass codeAfter assemblyLncRNAs predictedDescription^1^Pinus radiataTranscript no.Pinus pineaTranscript no.Pinus radiataTranscript no.Pinus pineaTranscript no. x 550 (0.41%)303 (0.28%)142 (1.62%)72 (1.37%)Natural antisense overlapping an exon of an annotated gene at the opposite strand (lncNATs) i 1,199 (0.90%)964 (0.90%)218 (2.50%)163 (3.10%)Fully contained in a known intron (intronic lncRNAs) y 559 (0.42%)331 (0.31%)160 (1.82%)129 (2.46%)Contains a reference gene within its intron u 49,678 (37.25%)34,635 (32.25%)8,262 (94.10%)4,889 (93.04%)Intergenic region (lincRNAs)^1^Brief explanation of the class codes
The results revealed that the majority of lncRNA transcripts were categorized as lincRNAs in both pine species, other classes being much less represented (Table 1). Differences in the characteristics of lncRNA transcripts compared to those of the protein-coding transcripts were identified in both species (Figs. 2 and 3) and were supported by non-parametric tests across transcript classes (Table S6). The average length of protein-coding transcripts of P. radiata (1,205 bp) and P. pinea (1,169 bp) was higher than that of lincRNAs (653 and 442 bp), lncNATs (632 and 417 bp) and intronic lncRNAs (557 and 411 bp). In general, lengths of P. radiata lncRNAs (614 bp) appeared to be longer than those of P. pinea (428 bp), while differences among lncRNA subclasses were generally smaller. The number of exons per transcript was significantly higher in the protein-coding transcripts than in the lncRNA transcripts in both species. The average exon length was 394 bp and 386 bp in protein-coding, and 272 bp and 192 bp in lncRNA transcripts of P. radiata and P. pinea, respectively. The GC content of pine lncRNAs (41%) was lower than that of protein-coding transcripts (45%) (Fig. 3A), whereas GC differences among lncRNA subclasses were modest, with lncNATs showing slightly higher values. Likewise, lncRNAs showed lower expression than mRNAs (2.6 vs. 5.0 FPKM in P. radiata; 5.4 vs. 9.4 FPKM in P. pinea), and overall expression differences among lncRNA subclasses were limited. Notably, P. radiata lncNATs differed from other lncRNA classes, with slightly higher GC content (42% vs. ~40.6%) and lower expression (1 FPKM vs. ~3.1 FPKM). The same tendency was observed in P. pinea lncNATs, although less pronounced.
Fig. 2 Characterization of pine lncRNA transcripts showed differences with the characteristics of protein-coding transcripts in P. radiata and P. pinea. (A) Transcript size distribution, (B) number of exons per transcript, and (C) exon size distributions.
Fig. 3. Differences in (A) GC content and (B) Fragments Per Kilobase of exon per Million (FPKM) distribution for lncRNAs and protein-coding RNAs in P. radiata and P. pinea.
We identified 82 lncRNAs in P. radiata and 19 in P. pinea as putative miRNA precursors (Tables S7-S8). These candidates belonged to 15 miRNA families, with MIR160 and MIR1314 most represented in P. radiata, and MIR160 and MIR3701 in P. pinea. In addition, Rfam annotated 69 transcripts in P. radiata and 35 in P. pinea (Table S9), spanning 13 conserved RNA families (rRNAs, tRNAs, histones, and snoRNAs). Notably, no histones or snoRNAs were detected among P. pinea transcripts.
To assess conservation at the sequence level, we searched for homologous lncRNAs in public datasets. Pine lncRNAs were aligned against 493,512 annotated lncRNAs from 94 plant species in GreeNC 2.0 (Table S10) and against the CANTATA 2.0 dataset (Table S11). Notably, these resources do not include gymnosperms. To avoid redundancy, we selected species so they did not appear in both databases. Overall, 1,991 (22.7%) lncRNAs of P. radiata and 2,057 (39.1%) lncRNAs of P. pinea exhibited homology with at least one hit with a known lncRNA from other plant species. Both pine species exhibited similar homology ratios with known plant lncRNAs (Fig. 4). The highest ratios, determined by the number of hits of pine lncRNAs relative to the total number of lncRNAs for each plant species, were consistently observed with Beta vulgaris (3.4% in P. radiata and 5.2% in P. pinea) and the woody plant Prunus dulcis (2.7% in P. radiata and 5.2% in P. pinea) in both pine species.
Fig. 4. Homology ratios of lncRNAs from *Pinus radiata *and P. pinea to known lncRNAs in the CANTATA 2.0 and GreeNC 2.0 databases. Bars represent the number of hits for each plant species relative to its total lncRNAs. Only ratios > 0.2 are shown.
Genome-wide identification and characterization of pathogen LncRNAs
The fungal transcripts that were not annotated using the EnTAP program (2,424) were subjected to sequential filtering steps to obtain the lncRNA transcripts. Based on transcript length and structure, the FEELnc filter module excluded 267 transcripts (11.01%) of F. circinatum. Next, the protein-coding potential of transcripts was predicted using five computational approaches (CPC2/CNCI/CPAT/PLEK/FEELnc). PLEK identified the highest number of non-coding transcripts (2,137 predicted lncRNAs), with the FEELnc tool being the most stringent, predicting 1,254 lncRNAs. A total of 1,020 transcripts were predicted as lncRNAs for the pathogen consistently across all approaches (Fig. 5). After categorizing the lncRNAs into different class codes, the majority of the lncRNAs were lincRNAs with 935 (91.7%) transcripts, followed by lncNAT with 75 (7.4%) transcripts, nine transcripts with the intron matching on the opposite strand, and one intronic transcript. In addition, one lncRNA was identified as miRNA precursor (MIR3250) using miRBase database (Table S7), and two others were annotated as rRNAs and tRNAs using Rfam database (Table S9).
Fig. 5. Venn diagrams showing the numbers of candidate lncRNAs in F. circinatum according to Coding Potential Calculator (CPC2), Coding-Non-Coding Index (CNCI), Coding-Potential Assessment Tool (CPAT), PLEK and FEELnc codpot module.
Overall, pathogen lncRNAs were considerably shorter (average = 388 bp, mode = 206 bp) than protein-coding transcripts (average = 1,519 bp, mode = 730 bp; Fig. 6A), and this difference was statistically supported (Table S6). Differences in the analysis of the exons were also observed. LncRNAs had fewer exons (median = 2) than protein-coding transcripts (median = 3; p < 0.001; Fig. 6B), with two-exon lncRNAs being the most frequent class. Consistent with transcript length, lncRNA exons were shorter (average = 181 bp, mode = 137 bp) than mRNA exons (average = 626 bp, mode = 430 bp; p < 0.001; Fig. 6C). The GC content of lncRNAs (48.5%) was slightly lower than that of protein-coding transcripts (50.6%; p < 0.001; Fig. 6D). Finally, lncRNAs showed lower expression (median log_10_(FPKM) = 0.26 vs. 1.20; p < 0.001; Fig. 6E).
Fig. 6. Characterization of lncRNA transcripts showed differences with the characteristics of protein-coding transcripts in F. circinatum. A Transcript size distribution, (B) number of exons per transcript, (C) exon size distributions, (D) GC content and (E) FPKM distribution for lncRNAs and protein-coding RNAs.
Differential expression analysis in pine-F. circinatum interaction
The expression changes of lncRNAs and protein-coding transcripts between F. circinatum-inoculated seedlings and controls were analyzed for the two different pine species separately. The PCAs revealed different patterns for each treatment (control or inoculated, Figure S2). A total of 37 lncRNA transcripts were identified as differentially expressed (adjusted p-value < 0.05, log_2_ (|Fold-change|) ≥ 1) in response to pathogen infection in P. radiata, with 25 up-regulated and 12 down-regulated (Fig. 7A). Among the differentially expressed lncRNAs (DELncRNAs), 35 were lincRNA transcripts, while the remaining consisted of one lncNAT and one lncRNA transcript containing a coding-protein in its intron (Table S12). One of the down-regulated DELncRNAs (PIRA.18271.1) was found to be homologous to a snoR64 sequence. In the case of P. pinea, 34 lncRNA transcripts were differentially expressed on infection, with 27 up-regulated and 7 down-regulated (Fig. 7B). These lncRNAs were categorized as lincRNAs [31], lncNATs [1] and intronic lncRNAs [2] (Table S13). The volcano plots displaying the top differentially expressed lncRNAs in both species are shown in Figure S3. On the other hand, 812 (618 up-regulated and 194 down-regulated) and 1,037 (830 up-regulated and 207 down-regulated) protein-coding transcripts showed significantly differential expression in P. radiata and P. pinea (inoculated vs. control), respectively (Table S14 and S15). DEGs were clustered in heat maps and represented in volcano plots in order to visualize the expression pattern of both conditions of the analyses (Figure S4 and S5).
Fig. 7. Hierarchical clustering plot of the differentially expressed lncRNAs of (A) P. radiata and (B) P. pinea in response to F. circinatum. The plot shows the scaled expression levels of the lncRNAs. Different columns represent different libraries, and different rows represent the differentially expressed lncRNAs. Purplish: relatively high expression; Blue: relatively low expression
PCA revealed different patterns for F. circinatum lncRNAs infecting each species (Fig. 8A). The differential expression analysis for the pathogen infecting P. pinea compared with infecting P. radiata revealed four up-regulated and one down-regulated lncRNAs (Fig. 8.B). It’s worth noting that three of the up-regulated lncRNAs were not expressed by the pathogen during the infection to P. radiata (FC072V.3858.1, FC072V.3349.1, FC072V.2859.2). Among the DELncRNAs, four were lincRNA transcripts, and one lncNAT (Table S16). The expression analysis of the protein-coding transcripts of F. circinatum revealed 216 DEG, with 145 up-regulated and 71 down-regulated genes in F. circinatum infecting P. pinea compared to infecting P. radiata (Table S17 and Figure S6).
Fig. 8. Visualization of differential expression analysis (DEA) of lncRNAs identified in F. circinatum. A Two-dimensional scatterplot of the principal component analysis (PCA) for pathogen lncRNAs, based on rlog-transformed counts. B Volcano plot illustrating the differentially expressed lncRNAs (DELncRNAs) in F. circinatum infecting P. radiata compared to infecting P. pinea. The x-axis represents log2 fold change, and the y-axis represents log10 p-value. Labeled transcripts represent the DELncRNA IDs (adjusted p-value < 0.05 and |log2 fold change| ≥ 1).
Analysis of lncRNAs cis-interacting genes
To predict the role of cis-acting lncRNAs in response to F. circinatum in both pine species, protein-coding transcripts located within a 10 kb window upstream and 100 kb downstream were investigated. A total of 2,577 and 1,892 lncRNA–mRNA interaction pairs were recorded in P. radiata and P. pinea, respectively, by the FEELnc classifier module. However, one lncRNA could have more than one target gene, and a target gene could be the target of one or more lncRNAs. In P. radiata, a number of 1,776 candidate cis target genes were observed for 2,267 lncRNAs (Table S18), of which 2,027 had a single candidate target gene and 240 lncRNAs had multiple interactions. The maximum number of target genes for a single lncRNA was five, which was reached by ten lncRNAs (Table S19). Putative cis targets were enriched for nucleotide/energy metabolism and gibberellin (GA) biosynthesis, suggesting an involvement in these pathways (Fig. 9A). In P. pinea, 1,422 candidate cis-target genes were identified for 1,654 lncRNAs (Table S20), of which 1,480 were associated with a single target gene, while 174 lncRNAs showed multiple interactions. The lncRNA PIPI.27358.5 was predicted to have potential cis interactions with eight distinct protein-coding transcripts, while five other lncRNAs exhibited five plausible interactions each with different genes (Table S21). Functional enrichments of the potential targets pointed to TCA/redox, and biotic interface/translation control (Fig. 9B).
Fig. 9. Gene Ontology (GO) enrichment of cis-associated target genes predicted for lncRNA–mRNA interaction pairs in Pinus radiata (A) and Pinus pinea (B). Only categories with FDR < 0.05. Gene count represents the number of input genes annotated to each GO term. The Gene ratio is Gene count divided by the total number of input genes retained in the enrichment test.
Eight DELncRNAs in P. radiata displayed potential cis interactions with eight distinct candidate target genes. Notably, one of these target genes, predicted to have a phosphofructokinase domain, exhibited up-regulation in a magnitude similar to its potential regulator (Table 2). In contrast, P. pinea had eleven DELncRNAs, each with the potential to interact with an equal number of candidate target genes, including one up-regulated in line with its potential DELncRNA regulator (Table 2). The functional prediction for these DELncRNAs was based on the functional annotation of their nearby target genes. Among the targeted genes in P. radiata, there were genes involved in molecular signalling encoding protein kinases, genes with domains commonly found in proteins associated with pathogen resistance (TIR, NB-ARC), one gene related to cell-wall reinforcement and lignification (4-coumarate-CoA ligase), and one plant-specific transcription factor (NAC) involved in the regulation of stress responses. In P. pinea, DELncRNAs potentially targeted genes involved in cell redox homeostasis, including a gene encoding for a thioredoxin reductase (NTRA), hydrolysis (transcripts with PAP2 domains) and cellular stress (transcript with DnaJ domain), among others. One of these genes predicted to be cis-regulated was also associated with brassinosteroid responses, a plant hormone that plays key roles in plant adaptation to biotic stresses.
Table 2. Candidate target genes predicted to interact with DELncRNA transcriptsLncRNALog2FC^a^Target geneLog2FC^a^Direction and type^b^LocationDescription of targeted gene Pinus radiata PIRA.53532.16.23 ↓PITA_11394Sense, intergenic (125)Same strand, upstreamPhosphatidylinositol phosphate kinase (PIP5K9)PIRA.15849.37.84 ↓PITA_42244Sense, intergenic (13,546)Same strand, upstreamTranscript with toll/interleukin-1 receptor homology (TIR) domainPIRA.61112.17.37 ↑PITA_48846Antisense, intergenic (865)Divergent, upstreamTranscript with NB-ARC domainPIRA.41278.12.66 ↑PITA_19394Antisense, genicOverlapping, intronicNAC transcription factorPIRA.21207.27.17 ↓PITA_10260Sense, intergenic (46,688)Same strand, downstreamCalcium/calmodulin dependent protein kinase IPIRA.26018.13.78 ↓PITA_29813Antisense, intergenic (80,313)Convergent, downstreamProbable galactinol-sucrose galactosyltransferase 6 isoform X1PIRA.24844.16.47 ↑PITA_43179Sense, intergenic (5,950)Same strand, downstream4-coumarate-CoA ligase, partial (4CL3)PIRA.39930.18.48 ↑PITA_333937.87 ↑Sense, genicContaining, intronicTranscript with phosphofructokinase domain Pinus pinea PIPI.37419.16.60 ↓PITA_20432Sense, intergenic (90,655)Same strand, downstreamRIX1PIPI.46862.15.76 ↓PITA_14476Antisense, genicContaining, intronicTranscript with phosphofructokinase domainPIPI.23778.14.56 ↑PITA_41443Sense, intergenic (5,563)Same strand, upstreamUnknownPIPI.47263.53.16 ↓PITA_48741Sense, intergenic (555)Same strand, upstreamTranscript with PAP2 domainPIPI.49311.13.50 ↑PITA_22808Antisense, genicNested, intronicCarboxyl-terminal-processing peptidase 1, chloroplastic, partialPIPI.19443.27.50 ↑PITA_02979Sense, genicNested, intronicTranscript with phosphofructokinase domainPIPI.38312.16.41 ↑PITA_11059Sense, intergenic (9,141)Same strand, upstreamTranscript with DnaJ domainPIPI.17776.13.70 ↑PITA_09873Antisense, intergenic (21,628)Divergent, upstreamThioredoxin reductase NTRAPIPI.11444.33.57 ↑PITA_23988Sense, intergenic (402)Same strand, downstreamEXORDIUM-like proteinPIPI.38808.26.53 ↓PITA_08782Sense, intergenic (2,603)Same strand, downstreamTranscript with phosphofructokinase domainPIPI.39715.16.34 ↑PITA_366789.71 ↑Sense, intergenic (200)Same strand, downstreamTranscript with PAP2 domain Fusarium circinatum FC072V.9741.13.16 ↓FC072V.9742.1Sense, intergenic (5069)Same strand, downstreamERG24FC072V.9739.1Sense, intergenic (9910)Same strand, upstreamIntegral membrane proteinFC072V.9743.1Sense, intergenic (6863)Same strand, downstreamHomogentisate 1,2-dioxygenaseFC072V.9744.1Sense, intergenic (9576)Same strand, downstreamHypothetical protein FGLOB1_6208FC072V.4978.22.40 ↑FC072V.4980.1Sense, intergenic (1555)Same strand, downstreamFusaric acid biosynthesis 9 (FUB9)FC072V.4976.2Sense, intergenic (7495)Same strand, upstreamCutinase transcription factor 1 beta (CTF1-BETA)FC072V.4977.1Antisense, intergenic (5028)Convergent, downstreamMajor facilitator superfamily transporterFC072V.4981.1Sense, intergenic (4049)Same strand, downstreamLinear gramicidin synthase subunit DFC072V.4976.1Sense, intergenic (7495)Same strand, upstreamCutinase transcription factor 1 betaFC072V.4982.1Antisense, intergenic (8116)Divergent, upstreamO-acetylhomoserine (thiol)-lyaseFC072V.4981.2Sense, intergenic (4157)Same strand, downstreamLinear gramicidin synthase subunit DFC072V.3858.16.27 ↑FC072V.3856.1Sense, intergenic (2726)Same strand, upstreamHypothetical protein FVER53263_00133FC072V.3855.1Sense, intergenic (3573)Same strand, upstreamRelated to hsp70 proteinFC072V.3854.1Sense, intergenic (6770)Same strand, upstreamRhamnogalacturonan acetylesteraseFC072V.3349.18.44 ↑FC072V.3348.1Antisense, genicOverlapping, exonicHypothetical protein FBULB1_308FC072V.3351.2Sense, intergenic (5515)Same strand, downstreamBeta-glucosidase G (BGLG)FC072V.3350.1Sense, intergenic (615)Same strand, downstreamRelated to transcription factor PLM2FC072V.3351.1Sense, intergenic (5317)Same strand, downstreamBeta-glucosidase G (BGLG)FC072V.3346.1Sense, intergenic (9233)Same strand, upstreamTranscript with ring finger domainFC072V.2859.25.68 ↑FC072V.2860.1Antisense, intergenic (216)Convergent, downstreamMED7-member of RNA polymerase II transcriptional regulation mediator complexFC072V.2853.1Antisense, intergenic (9383)Divergent, upstreamProteasome subunit alpha type-4FC072V.2856.1Sense, intergenic (5406)Same strand, downstreamCTI6 Cyc8-Tup1 interactingFC072V.2857.1Sense, intergenic (1012)Same strand, downstreamAdenylosuccinate synthetase (ADSS)FC072V.2861.1Antisense, intergenic (2679)Convergent, downstreamF-box domain-containing protein^a^The symbol ↑ refers to up-regulated expression and ↓ refers to down-regulation expression of lncRNAs and genes^b^Numbers in parenthesis indicate the genomic distance between the lncRNA and its potential target gene
Similarly, the cis-acting activity of lncRNAs of F. circinatum was based on their location with respect to protein-coding transcripts. The number of lncRNA–mRNA interaction pairs predicted in F. circinatum was 4,584 (Table S22). A number of 3,298 candidate cis target genes were observed for 984 lncRNAs, of which 920 (87.3%) had multiple interactions. The majority of lncRNAs were predicted to interact with three distinct protein-coding transcripts. The lncRNA FC072V.10163.1 had the highest number of target genes, reaching thirteen (Table S23). Functional enrichment analysis of these candidate targets revealed a strong bias toward transcriptional regulation and biosynthetic processes (Fig. 10). Five DELncRNAs showed potential cis interactions with 24 candidate target genes (Table 2), none of which belonged to the set of differentially expressed genes. Specifically, the down-regulated DELncRNA in the pathogen infecting P. pinea compared with P. radiata (FC072V.9741.1), potentially cis-regulates genes associated with ergosterol biosynthesis and phenylalanine degradation. On the other hand, potential target genes of up-regulated DELncRNAs in F. circinatum infecting P. pinea, were implicated in protein degradation, including genes with F-box and RING finger domains, mycotoxin biosynthesis (fusaric acid), and transcriptional regulation, such as CTF1-BETA and MED7, the latter being involved in the regulated transcription of nearly all RNA polymerase II-dependent genes. It’s worth noting that two potential target genes with hydrolysis activity and only expressed during the infection to P. pinea, are associated with the degradation of plant cell walls, including rhamnogalacturonan and cellulose. The target genes regulating the DELncRNAs were not enriched for any GO term.
Fig. 10. Gene Ontology (GO) enrichment of predicted cis-associated target genes for lncRNA–mRNA interaction pairs in F. circinatum. Only GO terms with FDR < 0.05. Gene count represents the number of input genes annotated to each GO term. The Gene ratio is Gene count divided by the total number of input genes retained in the enrichment test.
Weighted gene co-expression network analysis associated with pine defence response
A WGCNA was constructed on a filtered transcript dataset consisting of 83,366 and 65,524 transcripts in P. radiata and P. pinea, respectively, after removing low count reads to study the potential roles of trans-regulating lncRNAs in pine defence. Soft-thresholding powers were set to 6 in P. radiata and 12 in P. pinea, representing a balance between scale-free topology approximation and network connectivity (Figure S7). For P. radiata, the analysis identified 72 modules containing similar patterns of expression. Five of these modules (ME1^PR^, ME3^PR^, ME4^PR^, ME14^PR^, ME27^PR^) harboured lncRNAs that were differentially expressed upon F. circinatum infection, however, none of these modules showed a significant correlation with the infection condition (Figure S8). These modules were enriched for different biological processes (Table S24; Figure S9). We then built a global co-expression network of these DELncRNAs and DEGs, revealing 688 transcripts interconnected by 10,897 edges across all common modules (Fig. 11). Hub analysis in the largest such module identified ME4^PR^ as having the greatest number of DELncRNAs [21]. This module was therefore selected for further study. Hub connectivity (kME > 0.95) within ME4^PR^ highlighted 11 and 505 highly connected DELncRNAs and DEGs, respectively. The functional enrichment of the coding genes in ME4^PR^ (3,425) returned 178 significantly over-represented GO terms (FDR < 0.05; Table S24). The top categories included general defence response, chitin binding, chalcone metabolic process, chalcone biosynthetic process, and naringenin-chalcone synthase activity (Fig. 11B). These results suggest that the ME4^PR^ lncRNAs may participate in trans-regulation of key defence pathways, especially those related to cell-wall modification and flavonoid biosynthesis, during the infection.
Fig. 11. Co-expression modules containing DELncRNAs in P. radiata. A Global network of DELncRNAs and DEGs in common modules. Red nodes represent DELncRNAs and blue nodes represent DEGs. B Top 20 enriched Gene Ontology (GO) biological processes associated with genes from module ME4PR, which harbored the highest number of DELncRNAs. Gene count represents the number of input genes annotated to each GO term. The Gene ratio is Gene count divided by the total number of input genes retained in the enrichment test
For P. pinea, the analysis identified 75 modules containing similar patterns of expression. Five modules were significantly correlated with the infection condition (Figure S10). In this case, DELncRNAs were detected in two of these infection-associated modules, ME2^PP^ showing a strong positive correlation (r = 0.885, p = 0.0035) and ME5^PP^ a strong negative correlation (r = − 0.897, p = 0.0025). We then built a global co-expression network of these DELncRNAs and DEGs, revealing 846 transcripts interconnected by 17,241 edges across all common modules (Figure S10). Within ME2^PP^ (8,110 transcripts), hub analysis (kME > 0.95) identified 1,421 hub genes, including 7 DELncRNAs and 735 DEGs. Functional enrichment of the coding genes in ME2^PP^ returned 554 significantly over-represented GO terms (FDR < 0.05; Table S25; Fig. 12A). Within ME5^PP^ (2,444 transcripts), hub analysis identified 121 hub genes (none of them DELncRNAs) and 84 DEGs. The enrichment analysis yielded 116 GO terms (FDR < 0.05; Table S25; Fig. 12B). Together, these results suggest that P. pinea DELncRNAs tend to be concentrated in infection-associated modules with opposite signs of association, with ME2^PP^ harboring most of the DELncRNAs and displaying broad enrichment for defence-related functions. In contrast, ME5^PP^ was enriched in photosynthesis- and chloroplast-related processes, suggesting a reprogramming of primary metabolism associated with its negative correlation to infection. Together, P. pinea displayed infection-responsive modules enriched for defence-relevant functions, placing DELncRNAs at well-defined network positions. By contrast, P. radiata defence-associated modules containing DELncRNAs showed weak or no robust association with infection, consistent with a diffuse transcriptional response.
Fig. 12. Top enriched Gene Ontology (GO) terms associated with genes from infection-associated modules (A) ME2PP (positive correlation with infection, r = 0.885) and (B) ME5PP (negative correlation, r = −0.897), harboring DELncRNAs in P. pinea. Gene count represents the number of input genes annotated to each GO term. The Gene ratio is Gene count divided by the total number of input genes retained in the enrichment test.
Weighted correlation network analysis associated with F. circinatum pathogenesis
For the pathogen dataset, the co-expression analysis was conducted on 6,787 transcripts after filtering for low expression. The soft-thresholding power was set to 12 (R² = 0.878; slope = − 1.51; mean.k = 85.5), and the adjacency function was used to construct the adjacency matrix (TOMType = signed, minModuleSize = 30, mergeCutHeight = 0.25; Figure S11A). Hierarchical clustering of samples based on variance-stabilized counts showed clear separation according to the host species infected by the pathogen (Figure S11B). A total of 31 co-expression modules were identified, of which three (ME1^FC^, ME16^FC^, and ME4^FC^) were significantly correlated with the host species (Fig. 13A). Modules ME1^FC^ and ME16^FC^ showed positive correlations, being more highly expressed in P. pinea, while ME4^FC^ showed a negative correlation and was therefore associated with P. radiata. DELncRNAs were detected in ME1^FC^ [4] and ME4^FC^ [1]. In ME1^FC^ (1,457 transcripts), hub analysis (kME > 0.95) identified 55 hub transcripts, including 3 DELncRNAs, alongside 141 DEGs. Functional enrichment of coding genes in this module yielded a single significant category related to the extracellular region (Table S26). In contrast, ME4^FC^ contained 441 transcripts, including 35 DEGs and 70 hub genes, but no DELncRNAs among the hubs. Enrichment analysis of ME4^FC^ highlighted 49 over-represented categories, strongly dominated by ribosome- and translation-related functions as well as RNA binding and processing (Table S26; Fig. 13B). Together, these results suggest that pathogen-derived DELncRNAs tend to localize within species-associated modules.
Fig. 13. Co-expression modules associated with F. circinatum pathogenesis infecting resistant or susceptible pine species. A Barplot of the top 10 modules ranked by absolute correlation with host species. Asterisks indicate modules with significant correlations (|r| > 0.6, p < 0.05). B Top 20 significantly enriched Gene Ontology (GO) terms for coding genes in module ME4, which was more highly expressed in P. radiata. Gene count represents the number of input genes annotated to each GO term. The Gene ratio is Gene count divided by the total number of input genes retained in the enrichment test
Cross-species comparison
Sequence similarity was also assessed between the identified lncRNAs of both species. The analysis revealed that 3,567 lncRNAs were shared between the pine species (percentage identity > 75%; Table S27), representing 40.6% of P. radiata lncRNAs and 67.9% of P. pinea lncRNAs. Among them, only one common differentially expressed lncRNA was shared between the species (PIRA.54452.3/PIPI.43598.2), highlighting the low level of conservation among lncRNAs involved in the defence response to F. circinatum and/or potential species-specific regulatory mechanisms. Notably, the expression of this putative homologous lncRNA was similarly upregulated in both species, with a fold change of 11.8 in P. radiata and 9.1 in P. pinea. In the co-expression networks, PIRA.54452.3 belonged to ME4^PR^, where it was identified as a hub (kME: 0.957) and the module showed strong enrichment for chalcone/flavonoid biosynthesis, chitin response, and jasmonate signaling. Its homolog PIPI.43598.2 was placed in ME2^PP^, which was also enriched for these defence-related processes, but it did not reach hub status, suggesting conserved functional associations but divergent network centrality.
We identified several lncRNAs that were differentially expressed in P. pinea under pathogen infection, while their homologs in P. radiata were not (Table S28). These lncRNAs are therefore potentially associated with resistance mechanisms. Conversely, P. radiata DELncRNAs whose homologs in P. pinea were not involved in the defence response may represent susceptibility-associated regulators. Some of these lncRNAs were predicted to regulate protein-coding genes (Table S28). For instance, a susceptibility-associated lncRNA was predicted to cis-regulate a glycosyl hydrolase (PITA_29813), whereas a resistance-associated lncRNA was predicted to influence a thioredoxin reductase (NTRA).
We focused on representative DELncRNAs and their cross-species homologs that potentially regulate the expression of the same coding gene, showing functionally conserved nodes (Table 3). DELncRNAs/homologs pairs belonged to co-expression modules (ME2^PP^ and ME4^PR^) strongly enriched for classic defence and specialized-metabolism programs (JA/SA signaling, chitin recognition/catabolism, phenylpropanoid/flavonoid and terpenoid biosynthesis, oxidative-stress and cell-wall remodeling terms). By contrast, P. radiata ME3^PR^, which contains the thioredoxin reductase (PITA_09873) targeted by the P. pinea DELncRNA PIPI.17776.1, is dominated by translational machinery and mitochondrial respiration functions. These patterns show that lncRNAs converge on conserved defence targets (e.g., PITA_29813, PITA_09873) but act through different co-expression modules.
Table 3DELncRNAs and their homologs in pine species that share the same predicted coding gene targetsDELncRNAWGCNA moduleHomologWGCNA modulePredicted targetWGCNA modulePredicted target annotationPIRA.26018.1ME4^PR^PIPI.19356.1ME1^PP^PITA_29813n.d.Galactinol–sucrose galactosyltransferase; Sip1 / raffinose pathwayPIPI.11444.3ME2^PP^PIRA.16474.4n.d.PITA_23988n.d.EXORDIUM-like protein; growth/abiotic stress, possible wall remodelingPIPI.17776.1ME5^PP^PIRA.24069.1ME3^PR^PITA_09873ME3^PR^ (hub gene, kME = 0.952)Thioredoxin reductase; redox/ROS
Discussion
The importance of the non-coding genome in plant biology has become evident over recent decades [59]. In model and crop species, lncRNAs have been shown to act across immunity layers, from perception and signalling to transcriptional and post-transcriptional control. They may operate in cis (affecting neighbouring loci), in trans (through RNA–protein or RNA–RNA interactions), and via ceRNA circuits with miRNAs [60, 61]. Despite this progress, non-model gymnosperms remain underexplored. Addressing this gap, we profile lncRNAs during early infection in P. pinea (resistant) and P. radiata (susceptible) challenged by F. circinatum, and concurrently survey pathogen-encoded lncRNAs during the pathogenicity. To our knowledge, this is the first identification and characterization of lncRNAs from P. pinea and F. circinatum within the PPC pathosystem, providing insights into the regulatory mechanisms engaged at the onset of disease.
Across published woody plant datasets, reported lncRNA catalog sizes vary with species, tissue breadth, and pipeline choices. The pine catalogs (8,783 lncRNAs in P. radiata and 5,255 in P. pinea) fall within the expected range for single-tissue experiments using a conservative multi-tool pipeline. For example, 3,689 lncRNAs were identified in Paulownia tomentosa under phytoplasma infection [62], 9,355 in Populus × euramericana infected by Melampsora [63], 6,417 in Eucalyptus urophylla under cold [64], and 3,094 in Hevea brasiliensis infected by Colletotrichum gloeosporioides [65]. In a comparable pathosystem, P. radiata infected by F. circinatum yielded 13,312 lncRNAs using a less conservative filtering pipeline [21]. Methodological non-standardization and the low primary-sequence conservation of lncRNAs (which limits homology-based discovery) make the results more variable [48]. It is also worth noting that, because host reads were aligned to the P. taeda reference genome, interspecific divergence may have influenced mapping efficiency and transcript reconstruction. Indeed, host mapping rates were consistently higher for P. radiata (~ 78–81%) than for P. pinea (~ 46–49%), which may have reduced sensitivity for transcript detection in the latter species.
The lncRNA catalogs generated here showed characteristic features consistently reported in plants and in other organisms [66–69]. These sequences were shorter in length, had fewer and shorter exons, with a dominance of intergenic lncRNAs (∼94.1% in P. radiata and 93.0% in P. pinea) and a smaller fraction of antisense and intronic classes. LncRNAs in A. thaliana generally show expression levels ~ 30–60-fold lower than mRNAs [70], and accordingly the lncRNAs we report here were expressed at relatively low levels in comparison with protein-coding genes. The GC content of the assembled transcripts of both pine species (45%) was similar to that of the transcriptome of other Pinus spp. such as P. tecunumanii (44%) [25]. Separately, the GC content in pine lncRNAs (41%) was lower than in protein-coding RNAs, a recurrent feature of plant lncRNAs attributed to distinct evolutionary pressures on ORFs [71]. Interestingly, lncNATs showed features that differed from the other lncRNA categories. This likely reflects their overlap with cognate protein-coding loci (in nearly 60% of loci), whereby biogenesis and expression are often coupled to the sense transcription unit, leading to distinct sequence/structure biases and dynamics [72]. For instance, at the FLC locus, the antisense lncRNA COOLAIR showed mutually exclusive sense–antisense transcription and contributed to cold-induced shutdown of FLC during vernalization [73]. Overall, the class proportions, genomic features, and expression patterns we observe are consistent with the architecture of lncRNAs reported across taxa, supporting the robustness of our identification pipeline.
LncRNAs are widely reported to contribute to the positive or negative regulation of gene expression [4]. One of the conserved mechanisms of action of the lncRNAs is their function as decoys by sequestering RNA-binding proteins (RBP), miRNAs or chromatin-modifying complexes [74]. It has been shown that some lncRNAs act as targets or target mimics of miRNAs, and that they regulate Triticum aestivum resistance to powdery mildew and stripe rust through miRNA-mediated pathways [75]. A subset of our pine lncRNAs overlaps hairpin-forming regions consistent with putative miRNA precursors, including MIR160, MIR1314, and MIR3701. Similar observations, where stress-responsive lncRNA loci either encode miRNA-like hairpins or participate in lncRNA–miRNA–mRNA (ceRNA) circuits, have been reported in woody systems under pathogen pressure [76, 77]. While we do not infer mature miRNA biogenesis without experimental validation, the co-occurrence of hairpin-bearing lncRNAs with defence-enriched coding neighborhoods supports post-transcriptional crosstalk in immunity [78]. Notably, we detected more lncRNA candidates with miRNA-like hairpins in P. radiata than in P. pinea (82 vs. 19). Family usage also differed (MIR160/MIR1314 in P. radiata vs. MIR160/MIR3701 in P. pinea), pointing to species-specific intersections with small-RNA pathways early after infection. MIR1314 appears conifer-restricted and has been linked to stress-related transcription factors in Araucaria angustifolia, suggesting roles at the hormone/abiotic–biotic interface [79], whereas MIR3701, more abundant in the resistant P. pinea, is a gymnosperm family connected to NBS-LRR/defence networks [80]. These patterns align with the earlier pathogen recognition reported for resistant pines in this pathosystem [20, 81].
Predicted lncRNA–mRNA pairs in our datasets are consistent with canonical roles of lncRNAs in chromatin modification, transcriptional regulation and posttranscriptional regulation [59]. Fusarium circinatum interaction targets were strongly enriched for transcriptional programs, including transcription by RNA polymerase II, regulation of transcription, RNA biosynthetic process or DNA-binding transcription factor activity. This pattern is consistent with lncRNAs interfacing with transcription factors and chromatin machinery to modulate gene expression. In P. pinea, the enrichment for translation initiation/translation factor activity terms is compatible with lncRNA effects on RNA-binding proteins (RBPs)-rich translation complexes. On the other hand, ADP/oxygen binding, malate metabolism/dehydrogenase activities, and ent-kaurene oxidation (GA biosynthesis) were enriched in lncRNA–mRNA pairs in P. radiata, regulatory roles also reported for lncRNAs in other plant species [82–84]. However, as noted above, the use of P. taeda as reference may affect inferred genomic distances and should be considered when interpreting proximity-based cis candidates.
We detected differentially expressed lncRNAs (DELncRNAs) during early infection in both pine species, with similar counts at 4 dpi (P. radiata: 37; P. pinea: 34), despite larger differences in coding DEGs reported for this pathosystems [20]. This scale is comparable to other biotic-stress datasets generated from single tissues and early time points. For example, 53 DELncRNAs in P. × euramericana leaves during early Melampsora infection [63], ~ 100–120 infection-related lncRNAs reported for Paulownia under phytoplasma challenge [85], and 15 lncNATs responsive to F. oxysporum infection in A. thaliana [7]. This supports that only a small fraction of lncRNAs is rapidly mobilized at early stages of pathogen perception. Many DELncRNAs identified here lie in cis proximity to defence-related coding genes. A subset al.so shows coordinated expression changes with nearby genes, which is consistent with local regulatory effects. In P. radiata, several DELncRNAs were located near perception/signaling and lignification nodes. In P. pinea, DELncRNA-cis pairs were comparatively enriched near redox and protein-homeostasis components. Although the P. pinea cis targets are consistent with a rapid attempt to stabilize cellular status and contain oxidative/proteotoxic stress after infection, the P. radiata configuration needs closer examination, as impaired pathogen perception leading to weaker downstream defence has been reported for this species [20, 81]. This suggests that, in P. radiata, the lncRNAs may indicate compensatory or delayed sensing rather than effective early recognition, a possibility that requires experimental testing. Consistent with a possible cis relationship in the lignin pathway, we detected PIRA.24844.1 (fold change + 6.47) adjacent to PITA_43179 (4CL3), and in Zamora-Ballesteros et al., (2022) the same lncRNA was detected (lncRNAPiRa.33098.2: fold change + 6.8) likewise adjacent to PITA_43179. This cis-regulation was predicted to be sense, downstream in the same strand, ~ 5,950-6,072 nt apart. Notably, in our dataset PITA_43179 did not pass DEG thresholds (fold change: +5.8; FDR: 0.09), indicating that cis coupling does not always produce a detectable mRNA change at this stage.
To complement cis evidence, we placed lncRNAs within co-expression modules to explore potential trans-associated candidates based on shared expression patterns. Importantly, because module-level GO enrichment and connectivity-based hub prioritization can be influenced by module size, these results should be interpreted with caution as hypothesis-generating rather than definitive evidence. Our co-expression analysis placed pine DELncRNAs into species-specific network contexts. In P. radiata, DELncRNAs occurred in several modules, none of which showed a significant eigengene-infection association. However, the module containing the largest number of DELncRNAs (ME4^PR^) was functionally coherent for defence chemistry and wall-adjacent processes (e.g., defence response, chitin binding, chalcone/phenylpropanoid terms, and naringenin-chalcone synthase activity). This suggests that, although infection-wide modulation of those modules was not detected, DELncRNAs are candidates positioned within defence-relevant co-expression neighborhoods in P. radiata. In contrast, P. pinea showed infection-responsive modules containing DELncRNAs with opposite signs of association. ME2^PP^ harbored most DELncRNAs and was positively associated with infection, with enrichment in plant immunity-related categories such response to oxidoreductase activity, flavonoid biosynthetic process, immune response and secondary metabolite biosynthetic process. Terms that have been associated with resistance to C. gloeosporioides of walnut fruit bracts [67]. In addition, ME5^PP^ was negatively associated with infection and was enriched for photosynthesis/chloroplast terms, consistent with a downshift of primary metabolism during infection. Thus, in P. pinea, modules containing DELncRNAs appear to shift with infection, whereas in P. radiata those DELncRNAs reside in defence-like modules without a significant infection-wide change. This may indicate that, although P. radiata possesses the defensive machinery, it is not properly activated, consistent with previous observations [20, 81]. Additionally, convergence on similar coding targets (e.g., carbohydrate flux via the raffinose pathway and redox buffering) occurs in distinct modules across hosts, suggesting species-specific trans use of lncRNAs to reach shared defence processes. This is compatible with the notion that plant lncRNAs show limited primary-sequence conservation while preserving positional or functional roles across species [86].
Across plants, lncRNA primary sequences show little conservation that further declines with evolutionary distance [4, 48]. Consistent with this, pine lncRNAs showed little similarity to angiosperm databases, and few sequence matches were detected between P. pinea and P. radiata despite shared tissue, age, and pathosystems. Notably, P. pinea displayed a higher apparent homology (39.1%) than P. radiata (22.7%). Given the limited cross-lineage similarity even for coding transcriptomes (pine vs. angiosperms) contrasted with high within-conifer xylem conservation (~ 78–82%; E-value ≤ 10⁻⁵) [87], we cannot draw robust conclusions regarding their sequence conservation. Positional or functional features can be conserved despite sequence divergence. For instance, conserved antisense transcript positions at FLC/COOLAIR-like loci across six grass species [88] and analogous DNA methylation-reader roles for human lncRNA UHRF1 protein-associated transcript (UPAT) and plant lncRNA AUXIN-REGULATED PROMOTER LOOP (APOLO) [89], may still share secondary structures [90]. However, the lack of chromosome-level assemblies for our study species precluded rigorous tests of positional or structural conservation.
Beyond the global transcriptional-control signal in cis and trans, our pathogen-side results highlighted ergosterol biosynthesis and cell-wall–degrading activities as plausible lncRNA-connected axes relevant to virulence in the pine–F. circinatum pathosystem. Host plants can sequester pathogen sterols through PR-1–type resistance proteins, thereby inhibiting pathogen growth as a resistance mechanism [91]. In our dataset, the sole down-regulated lncRNA in the pathogen during infection of P. pinea (vs. P. radiata) was cis-positioned near sterol-biosynthetic loci. Consistently, genes in this pathway, including δ14-sterol reductase (ERG24), were expressed at lower levels when F. circinatum infected resistant hosts [18]. Our results extend this hypothesis by suggesting that sterol-pathway genes and nearby fungal lncRNAs (FC072V.9742.1) show host-dependent expression patterns that may be sensitive to host resistance. In parallel, during infection of P. pinea we observe up-regulation of lncRNAs potentially cis-regulating genes associated with the cell-wall degradation, consistent with a reinforced offensive program under a more restrictive host environment. This aligns with previous analysis showing early induction of secreted enzymes/CWDEs in P. pinea upon F. circinatum infection [20]. Taken together, these data support fungal lncRNAs as candidates potentially modulating virulence-associated programs under different host defenses.
In summary, this work contributes to expanding the molecular framework of the Pinus–Fusarium circinatum interaction by providing evidence for a new regulatory layer mediated by long non-coding RNAs. By jointly profiling host and pathogen transcriptomes, we report species-specific lncRNA responses and network architectures that may underlie resistance and virulence strategies. At the lncRNA level, the resistant P. pinea showed a more coordinated infection-associated regulatory landscape, with DELncRNAs enriched in infection-responsive programs that couple defence activation with down-regulation of photosynthesis/chloroplast functions. By contrast, in susceptible P. radiata, DELncRNAs were embedded in defence-related co-expression neighborhoods but without a strong infection-wide module shift, consistent with a diffuse or incompletely engaged regulatory response at 4 dpi. Beyond providing the first catalogues of P. pinea and F. circinatum lncRNAs, our study underscores the importance of integrating non-coding RNA regulation into forest pathosystem research. Importantly, our conclusions are based on computational predictions from this dataset. Hub lncRNAs/genes highlighted here should be viewed as priority candidates that will require independent validation to confirm their roles and improve confidence in their regulatory relevance. These findings open avenues for exploiting lncRNA-based mechanisms to enhance conifer resilience against emerging diseases.
Supplementary Information
Supplementary Material 1.
Supplementary Material 2.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Carrasco A, Wegrzyn JL, Durán R, Fernández M, Donoso A, Rodriguez V et al. Expression profiling in Pinus radiata infected with Fusarium circinatum. Tree Genet Genomes. 2017;13(2).
- 2Amaral J, Lamelas L, Valledor L, Castillejo MÁ, Alves A, Pinto G. Comparative proteomics of Pinus–Fusarium circinatum interactions reveal metabolic clues to biotic stress resistance. Physiol Plant. 2021;173(4):2142–54 .10.1111/ppl.1356334537969 · doi ↗ · pubmed ↗
- 3Zamora-Ballesteros C. Long non-coding RN As (lnc RN As) in the pathosystem Pinus spp.–Fusarium circinatum. Zenodo; 2025. Available from: https://zenodo.org/records/17591556
- 4Andrews S. Fast QC a quality control tool for high throughput sequence data. 2012. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- 5Wegrzyn JL, Lee JM, Tearse BR, Neale DB. Tree Genes: A forest tree genome database. Int J Plant Genomics. 2008;412875:1–7. 10.1155/2008/412875 PMC 251785218725987 · doi ↗ · pubmed ↗
- 6CCB. Transcript assembly and quantification for RNA-Seq. 2019. String Tie. Available from: http://ccb.jhu.edu/software/stringtie/index.shtml?t=manual
- 7Domínguez-Rosas E, Hernández-Oñate MÁ, Fernandez-Valverde SL, Tiznado-Hernández ME. Plant long non-coding rnas: identification and analysis to unveil their physiological functions. Front Plant Sci. 2023;14:1275399. 10.3389/fpls.2023.1275399 PMC 1064488638023843 · doi ↗ · pubmed ↗
- 8Wang L, Wang J, Chen H, Hu B. Genome-wide identification, characterization, and functional analysis of Lnc RN As in Hevea Brasiliensis. Front Plant Sci. 2022;13:1012576.10.3389/fpls.2022.1012576 PMC 958127736275565 · doi ↗ · pubmed ↗