Bacteriophages in gut metagenomes: from analysis to application
Natalia Zakharevich, Aleksandra Strokach, Egor Shitikov, Ksenia Klimina

TL;DR
This review discusses the role of bacteriophages in the gut, their impact on health, and how they can be studied and used for therapeutic purposes.
Contribution
The paper provides a comprehensive overview of current bioinformatic methods and therapeutic applications of gut bacteriophages.
Findings
Bacteriophages significantly influence gut microbiota structure and immune system interactions.
Advancements in metagenomics have improved understanding of phage diversity and 'viral dark matter'.
Phages show promise in treating cancer, inflammatory, and liver diseases as well as in diagnostics.
Abstract
Bacteriophages constitute a major component of the human gut virome, playing very important roles in shaping of the structure and function of the gut microbiota. Moreover, bacteriophages interact with the human immune system, thereby influencing various disease processes. Recent advancements in metagenomic sequencing and computational analysis have substantially expanded our understanding of gut phage diversity and the scale of the so-called ‘viral dark matter’. In this review, we summarize current bioinformatic approaches for identifying and annotating bacteriophage sequences in metagenomic data, discuss key challenges in taxonomic classification and host prediction of phages, as well as the limitations associated with the assembly and analysis of viral metagenome-assembled genomes (vMAGs). We also analyze the therapeutic potential of bacteriophages, including their application in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
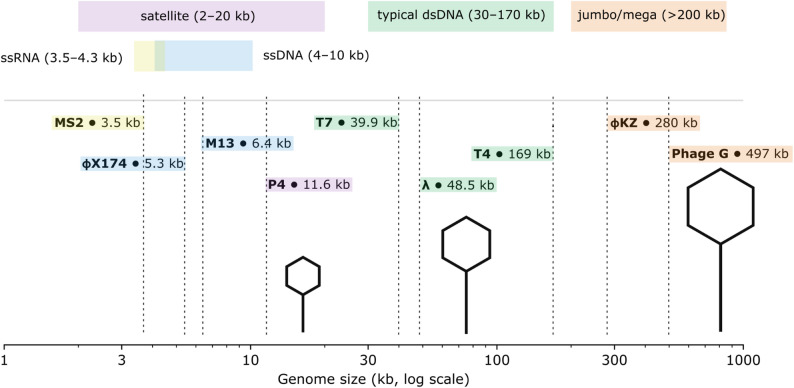 Figure 1
Figure 1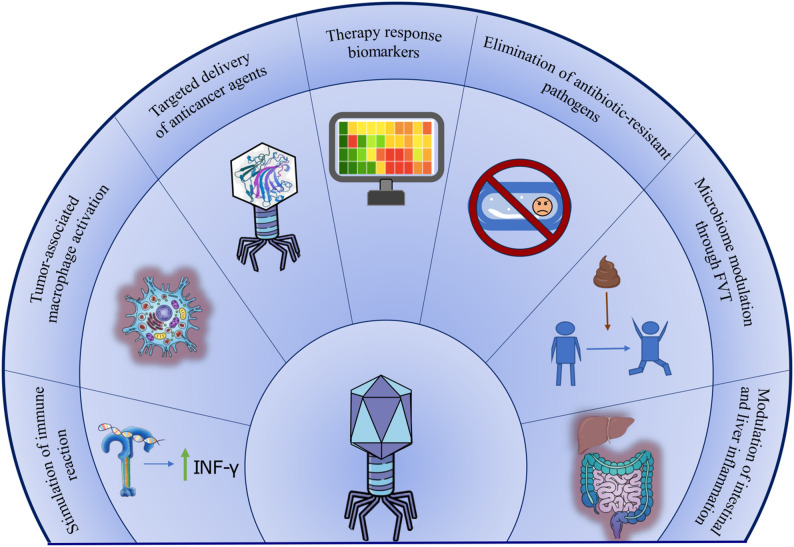 Figure 2
Figure 2- —https://doi.org/10.13039/501100006769Russian Science Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacteriophages and microbial interactions · Genomics and Phylogenetic Studies · Cancer Research and Treatments
Introduction
Viruses inhabiting the human body form a special and still insufficiently studied component of the microbiota – the virome. The human virome comprises of eukaryotic viruses, including pathogenic viruses that infect human cells, and viruses that infect bacteria, known as bacteriophages or phages. Plant viruses may also be present in the virome, likely originating from food sources [1, 2]. Viromes vary depending on the biological niche (such as skin, nasopharynx, gut, or urogenital tract), as well as in their impact on human health. In recent years, interest in the virome has grown significantly, driven by advances in metagenomic sequencing and bioinformatic analysis. However, a large proportion of viral sequences detected in human samples – from ~ 70% to ~ 95% – belong to the hidden expanse of viral diversity termed ‘viral dark matter’ [3–10]. Essentially, viral dark matter is the collection of viral sequences retrieved from sequencing data that remain entirely uncharacterized for now: they cannot be taxonomically assigned to established families, genera, or species; their putative hosts are undetermined; and their biological functions remain obscure [11–15].
The gut virome (GV) plays an important role in shaping the composition and function of the human gut microbiota (GM) in both health and in disease. Bacteriophages are predominant in the GV, accounting for up to 90% of its viral component. Phages influence GM diversity and contribute to horizontal gene transfer, thereby shaping and expanding the functional potential of the microbial community [3, 16, 17]. In addition to their direct impact on gut bacteria, there is growing evidence that phages can influence the human body, including the immune system. One such mechanism involves the release of pro-inflammatory molecules into the gut lumen when bacterial cells are lysed by phages. Additionally, evidence of phage transcytosis across gut epithelial cells suggests that phages may directly stimulate the human immune system [18–20]. It is also important to note that phages can significantly reduce the bacterial load and ameliorate disease by specifically targeting pathogenic bacteria in the infectious setting and/or dysbiosis, as has already been demonstrated in a number of animal models [21, 22]. Of particular interest is the ability of phages to enhance the immune response. For instance, it has been shown that a prophage of Enterococcus hirae can induce T-cells that cross-react with tumor antigens, thereby increasing tumor sensitivity to immunotherapy [23, 24]. These findings open the possibility of using specific phages to stimulate the immune system in order to improve the effectiveness of immunotherapy and increase the number of patients who respond to it.
Besides to modulating the host immune system, phages can also influence brain function and memory [25, 26]. For example, it was discovered that bacteriophages from the Demerecviridae and Drexlerviridae families, class Caudoviricetes (the original publication uses the now-obsolete order name Caudovirales and family name Siphoviridae), have been associated with increased memory capabilities through upregulation of the expression of genes involved in neuroplasticity, neuronal development and memory formation [26]. Furthermore, Wu et al. reported a potential role for gut phages in the development of depression, suggesting that phage imbalance may serve as both a distinguishing feature of and a diagnostic marker for the disorder [25].
One of the most striking discoveries of recent years is that the human GV exhibits not only exceptional diversity but also a high degree of individual specificity and temporal stability [2]. Although bacteriophages constitute the majority of viral particles in the gut, their composition is unique to each individual and remains remarkably stable over the course of months. Thus, the phage composition may serve as an individual ‘viral fingerprint’, potentially useful for diagnosing and monitoring changes in the GM. Although much of the genomic diversity of gut phages is still hidden from us in the ‘viral dark matter’, but its contours are gradually becoming clear: exemplified by the two most common fecal phage clades, crAssphage and Gubaphage, identified only in 2014 and 2021 [9, 13], respectively, which together highlight the extent of the gut’s still unexplored viral diversity.
In this review, we consider current bioinformatic approaches to GV analysis using metagenomic data, and discuss the therapeutic potential of bacteriophages, particularly in the context of cancer treatment. Considering that the composition of the phage community appears to be linked to clinical outcomes and reflects the functional state of the GM and the patient’s immune system, we also discuss the potential of phages as biomarkers for predicting response to immunotherapy.
Identification of bacteriophages in metagenomic data
From culturing to bioinformatics
Historically, the identification and characterization of bacteriophages – like bacteria – relied on isolation and culturing techniques [27–31]. However, a significant proportion of bacteria and their viruses remain uncultured under laboratory conditions [32]. The development of next-generation sequencing has facilitated the acquisition of metagenomic data, enabling the direct analysis of all genetic material in a sample, regardless of the possibility of culturing.
In metagenomic research, one can either sequence the entire community and subsequently separate viral sequences using bioinformatic methods, or perform physical separation of the viral and bacterial fractions prior to library preparation to obtain a metavirome. The latter approach risks losing a significant proportion of phages due to their association with the cellular fraction. This occurs because phages may integrate into bacterial genomes as prophages, attach to the cell surface, or be involved in ongoing lysogenic infections. Purification methods can also remove certain groups of phages: for instance, chloroform treatment has been shown to inactivate lipid-containing bacteriophages, filamentous phages, and a number of other tail phages [33, 34]. Whole-community metagenomic sequencing not only enables the identification of viral sequences, but also provides the opportunity to study phage-host interactions. This is particularly relevant for analyzing the GV because current data indicate that temperate phages predominate in its composition [35, 36].
Recent decades have seen significant progress in both isolation, and culturing protocols and bioinformatic methods for virome analysis. To describe and understand bacteriophage population dynamics, their interactions with GM, and ultimately, their impact on human health, global phage diversity is currently being studied mainly in silico, using culture-independent tools that analyze the whole gut community.
Computational strategies for identifying viral sequences in metagenomic data
Detection bacteriophages in metagenomic data is a significant challenge. Identification and taxonomic classification of phage sequences are complicated by the absence of a universal marker gene – analog of the 16 S rRNA gene in prokaryotes. Furthermore, potential misclassification of viral sequences as bacterial due to their ability to integrate into the host genome. Mosaicism is a distinctive feature of bacteriophage genomes and results from the dominant role that horizontal gene transfer plays in shaping the architecture of these genomes. Mosaicism is certainly not limited to phage genomes, as bacteria also acquire DNA through horizontal gene transfer, but its scale and impact on bacteriophage evolution are much greater. This leads to two difficulties. First, this leads to assembly problems: traditional methods designed for more stable bacterial genomes often cannot correctly assemble the mosaic sequences of phages, resulting in fragmented or chimeric assemblies. Second, high mosaicism also hinders direct genomic alignment and the construction of correct phylogenetic trees. At a minimum, a balance between sensitivity and speed is necessary to address these challenges. High sensitivity is essential for successfully overcoming the high diversity and rapid evolution of phages, as well as the typically low and uneven coverage of viral sequences in metagenome. Speed is required for processing large data [37–42].
In recent years, advancements in sequencing technologies and metagenomic methods, coupled with the emergence and application of machine and deep learning approaches, have led to the development of a wide range of tools for identifying phage sequences. These tools can be categorized into two main groups: (i) reference-dependent and (ii) reference-independent approaches [8, 43].
- (i)Reference-dependent approaches identify viral sequences by aligning them to reference databases. Also, we include reference-dependent k-mer classifiers that search for exact/almost exact matches of k-mers with the database (when installing them, the user must download the database) in this group. These methods demonstrate high accuracy when applied to whole-genome sequencing data and perform well with data obtained by long-read sequencing platforms such as Oxford Nanopore and Pacific Biosciences. However, their performance decreases when processing contigs derived from metagenomic data because they are not adapted to recognize incomplete or short/fragmented sequences [42, 44, 45]. Moreover, their accuracy is limited by the completeness and quality of reference databases, making it challenging to detect novel phages that diverge significantly in sequence from phages from known viral families [46–51]. Such approaches can also be called database-dependent.
- (ii)Reference-independent approaches represent an alternative that does not rely on reference databases. These methods can be divided into two subgroups. The first includes tools that analyze gene composition and structural features of the genome, such as GC content, codon usage, gene density, etc., to distinguish between viral and bacterial sequences. These features are either analyzed statistically or utilized as input for machine learning classifiers, as in VIBRANT tool [50]. The second subgroup focuses on analyzing frequency-based characteristics of nucleotide sequences (k-mers) that are specific to viral and bacterial genomes. These features are used for training machine and deep learning classifiers. Machine learning models employ a variety of algorithms, ranging from traditional approaches such as support vector machines, logistic regression, random forests, and naive Bayes to deep learning architectures, including neural networks [42, 44, 45, 48, 50–53].
It should be noted that deep learning techniques are successfully employed nowadays to solve a variety of problems in computational biology, and that deep learning is one of the advanced machine learning algorithms. Examples of tools utilizing deep learning methods are DeepVirFinder (https://github.com/jessieren/DeepVirFinder) and Seeker (https://github.com/gussow/seeker) [45, 53]. DeepVirFinder trained on a large number of sequences and applies convolutional neural networks (CNNs) to search viral genomic signatures, which are then used to construct a predictive model capable of determining whether a given sequence is of viral origin. Employing long short-term memory (LSTM) models and a type of recurrent neural network (RNN), Seeker enables rapid phage detection in metagenomic datasets by differentiating phage sequences from bacterial sequences, even when phage sequences exhibit minimal similarity with known phage families. This tool can be said to be less biased: it doesn’t rely on predefined sequence features. Instead, it is trained to read the entire DNA sequence and evaluate the likelihood of their belonging to phage genomes.
Experience shows that reference-dependent tools exhibit a low level of false positives. At the same time, reference-independent (in particular machine and deep learning tools) approaches demonstrate greater sensitivity to phages with lower representation [8, 45]. Furthermore, reference-independent tools can work with shorter contigs compared to reference-dependent tools, as they do not require, for example, the presence of several genes to classify sequences. Most importantly, reference-independent tools are capable of identifying novel phage sequences – part of the so-called ‘viral dark matter’ – unlike reference database dependent methods. However, it is worth noting that machine learning tools still depend to some extent on reference databases, as these are used to train the classifiers. In addition, long-read sequencing (which was already mentioned above (i)) represents a promising complement to short-read sequencing. For instance, it can be used to reconstruct megaphage genomes and obtain information about methylation patterns – which can aid in host prediction, as well as to study population structure at the level of an individual virions. In some cases, long-reads may span complete or nearly complete viral genomes [54].
In recent years, hybrid approaches that combine elements of reference-dependent and -independent methods have been actively developed. For example, VIBRANT (https://github.com/AnantharamanLab/VIBRANT) – hybridizes neural network machine learning and protein signatures, and geNomad (https://github.com/apcamargo/genomad) – combines machine learning with protein profile databases to improve prediction accuracy [50, 55].
The choice of tool for analyzing metagenomic data significantly influences the predicted viral community composition. Different computational approaches often yield divergent results; for instance, comparisons of predicted viral sequences show that the overlap between any two tools rarely exceeds 40% [8]. This highlights the need for a multi-tool strategy for predicting viral genomes in a metagenomic context, where the combination of different algorithms can provide a more reliable consensus. In Table 1, we compiled information on phage detection tools in metagenomic data published over the past decade, and also tried to briefly describe their strengths and limitations [44–53]. In addition, we would like to provide a brief overview of existing virus/bacteriophage databases – Supplementary Table 1 provides their description, content and some notes.
Table 1. An overview of available published metagenomic phage detection tools over the past decadeTool/year/referenceBased on/groupStrengths and limitationsRepositoryVirSorter2015[46]viral sequences prediction using probabilistic models and using reference-guided, gene-content based rules (i) • One of the first automated pipelines for finding viral sequences in metagenomic data• Able to classify prophage sequences; also predicts lytic phages; assigns a confidence category (1–3) (the algorithm is good at finding prophages, but was less successful at detecting new free-living viruses)• Runs slowly on large metagenomic datasets compared to modern tools• VirSorter do not classify fragments shorter than 1000 nucleotides (nt);• Effect of contig length on performance: strongly affected by fragment length, with performance a marginal increase with increasing length; performed well for longer contigs• Decrease in precision due to low viral abundance;• Demonstrate low false positive rates and robustness to eukaryotic contamination (as all tools that use a homology approach) https://github.com/simroux/VirSorter MetaPhainder2016[47]viral sequences prediction using integrated analysis of BLASTn hits to a phage database (i) • Account for the mosaic genome: taking into account all hits (not the best hit) to the phage database is advantageous when dealing with mosaic phage genomes• Effect of contig length: fragment length largely unaffected on tool performance; demonstrate fairly consistent sensitivity and precision (interestingly, a homology-based tool exhibits a pattern similar to the sequence-based tools for this property)• Has a higher sensitivity, including to phages with less representation in reference databases; decrease in precision due to low viral abundance• Eukaryotic contamination: show much lower specificity, and frequently misclassifying eukaryotic fragments as viral, with an FPR around 0.5• CheckV evaluate: contain a low proportion of contigs with Medium Quality or higher, suggesting a potential number of FPs• Using BLASTn and calculating ANI requires significant computational resources, especially on large datasets - a slower approach https://github.com/vanessajurtz/MetaPhinder Marvel2017[48]viral sequences prediction using a random forest machine learning approach and three simple genomic features (i)+(ii) • The first tool capable to effectively separate metagenomic bins containing dsDNA phage sequences from those containing bacterial sequences (as opposed to treating contigs as isolated objects)• Uses a machine learning approach and three simple genomic features extracted from contig sequences: gene density, strand shifts, and fraction of significant hits against pVOGs database• Requires bins as input• Demonstrate good performance on the recall (sensitivity) measure• The tool’s performance is less tightly coupled to contig length: shows more variability in sensitivity for a given length, since the tool performs classification of contigs after binning, and contigs of various lengths may be present in the same bin• CheckV evaluate: does not predict contigs below medium quality https://github.com/LaboratorioBioinformatica/ MARVELviralVerify2020[49]viral sequences prediction using a Naive Bayesian classifier (i) • Can be used as a standalone tool to predict contigs of viral origin in any assembled metagenome, or as a separate module (MetaviralSPAdes, viralFlye)• Supports creation of a custom training database (from viral, chromosomal and plasmid contigs) and working with a custom HMM database• High precision but have length-dependent sensitivity: performance increasing with length• Robustness to low viral abundance in the community, especially on shorter contigs (about 500 nt)• Gene content dependence• Lack of independent comparisons: viralVerify has not participated in any current major benchmarks https://github.com/ablab/viralVerify VIBRANT2020[50]viral sequences prediction using hybrid neural networks of protein annotation signatures (i)+(ii) • Recovery of both free and integrated viral genomes from metagenomes• Hybridizes neural network machine learning and protein signatures• Software that integrates: virus identification, annotation, estimation of genome completeness and distinguishing between lytic and lysogenic viruses, and is also capable of assembling useful annotation data and categorizing the metabolic pathways of viral AMGs• Utilizes metrics of non-reference-based protein similarity annotation databases in conjunction with a unique “v-score” metric - give a quantifiable value to viral hallmark genes instead of categorizing them in a binary fashion• Prediction on short sequences: does not consider scaffolds shorter than 1000 bp or those that encode less than four predicted open reading frames• Reduced sensitivity on short contigs (< 3 kb): exhibits improved performance with scaffolds at least 3 kb in length• Eukaryotic DNA: VIBRANT effectively filtered out eukaryotic contamination by assigning them low v-scores, successfully recovering few false-positive viral sequences• May miss highly novel or divergent viruses https://github.com/AnantharamanLab/VIBRANT VirSorter22021[51]viral sequences prediction using random forest classifiers using an hmmsearch of predicted genes (i)+(ii) • Memory usage stays nearly constant with increasing data size, and scales nearly linearly with threads used• Written with snakemake: total CPU time is higher than other tools, but internal processes are highly parallelizable; this increased CPU time is mostly due to the annotation step (> 90% of CPU time)• Can be scaled to handle large-scale (> 100,000 sequences) datasets• Through modular framework: has a uniquely able to reliably detect different types of non-Caudoviricetes viruses; also, modular framework to enable modification and addition of new classifiers as our knowledge of viral sequence space increases• Users can omit the NCLDV, Lavidaviridae, and RNA virus classifiers for analysing bacterial and archaeal viruses to drop error rate• Approach is currently not optimal for short (< 3 kb) contigs• Plasmid sequences cannot be entirely distinguished from viruses: will be required the incorporation a dedicated tool for plasmid detection in VirSorter2 modular framework https://github.com/jiarong/VirSorter2 VirFinder2017[44]viral sequences prediction using machine learning based tool that uses k-mer frequencies (ii) • k-mer frequency based, machine learning method - entirely avoids gene-based similarity searches;• have good performance, especially for shorter contigs (i.e., 1000 bp)• Independent of the input contig set composition: generates static prediction scores for each contig regardless of the other tested at the same time contigs;• Statistical framework: generates prediction scores, p values and q values showing the probability that sequence is a virus; this is useful for assessing and controlling the false positive rate• Eukaryotic DNA: not trained on eukaryotic DNA; may falsely identify eukaryotic sequences as phages• Additional filtering needed: users must filter out potential eukaryotic contamination before analysis• Limits of k-mer patterns: sensitive to the diversity of known viruses represented in the training sequence database; if viruses have some signal of k-mer patterns that are specific and not shared “universally” - the performance will be lower https://github.com/jessieren/VirFinder PPR-Meta2019[52]3-class classifier for viral sequences prediction using novel neural network architecture –Bi-path Convolutional Neural Network (BiPathCNN) (ii) • The first tool that can simultaneously identify phage and plasmid fragments• Innovation: the design of BiPathCNN, which Uses a more detailed method of characterizing DNA sequences - consider coding or non-coding region to improve the performance• Sequences length: BiPathCNN directly extracts sequence features from the raw data represented and may be less sensitive to sequence length• The recognition rate for prophages lower than that for phage contigs• Non-target organisms: trained only on prokaryote chromosomes, plasmids, and phages; other organisms not including in the training set - may affect the analysis results• Chimeric sequences: PPR-Meta cannot detailed judgments chimeras (e.g., prophages or chromosome chimeras) https://github.com/zhenchengfang/PPR-Meta Seeker2020[53]viral sequences prediction using Long Short-Term Memory (LSTM) models (ii) • Few parameters: uses relatively few parameters (152–212), reducing risk of memorization and overfitting• Short sequences: performs well on short sequences, important for metagenomic data• Low computational cost: requires minimal resources, runtime scales linearly with input length• Major advantages speed and maintains a high level of performance on viral sequences with little similarity to training data - and thus is well suited to discover new groups of phages. Classification errors: may misclassify some phage and bacterial genomes (does not perfectly distinguish phages from bacterial genomes)• Eukaryotic DNA: not trained on eukaryotic DNA; may falsely identify eukaryotic sequences as phages• Additional filtering needed: users must filter out potential eukaryotic contamination before analysis• Prophage detection: cannot reliably detect prophages in bacterial genomes - this feature is not supported https://github.com/gussow/seeker DeepVirFinder2020[45]viral sequences prediction using convolutional neural networks (ConvNets) (ii) • Prediction accuracy: extending the training data with additional viral sequences from environmental samples improves the prediction accuracy for under-represented viral groups• Prediction on short sequences: the tool is able to accurately classify even individual reads as viral - this can potentially simplify the complexity in assembly of viral genomes, improve assembly accuracy and reduce computing resources• Automatically feature engineering: unlike pre-defined feature-based machine learning models (k-mer frequencies logistic regression, gene-based random forest), DeepVirFinder automatically learned using a large number of training examples - this leads to higher accuracy due to the inclusion of hidden patterns• Eukaryotic DNA: not trained on eukaryotic DNA; may falsely identify eukaryotic sequences as phages• Additional filtering needed: users must filter out potential eukaryotic contamination before analysis https://github.com/jessieren/DeepVirFinder geNomad2023[55]viruses and plasmids sequences prediction using a framework that combines information from gene content and a deep neural network (i)+(ii) • High classification performance for viruses and plasmids (outperforms other tools in benchmarks), also provirus detection with high precision• Useful for metagenomic/metatranscriptomic assemblies• Integrated and Automated Workflow• The end-to-end command is user-friendly, as it runs the entire workflow• An active project: well-documented, and issues are addressed by the developers• High computational requirements, especially for memory (RAM)• Prediction on short sequences: perform poorly on contigs < 10 kb, due to their reliance on complete protein information• Conservativeness: tend to generate fewer false positives, but more true positives may be missed• Misclassification issues: tendency to incorrectly identify prokaryotic chromosomal sequences as plasmids https://github.com/apcamargo/genomad (i) reference-dependent approaches (ii) reference-independent approaches
The resulting potential viral sequences can be evaluated using the CheckV tool (https://bitbucket.org/berkeleylab/checkv/src/master/), which is designed to assess the quality and completeness of viral genomes assembled from metagenomic data. Studies have shown that excluding contigs classified as ‘low-quality’ or ‘not-determined’ by CheckV assessment increases the consensus between independent phage prediction tools, highlighting a resulting set of high-confidence viral sequences. An additional advantage of the CheckV tool is that its database can be easily updated to include newly published viral genomes from the publicly available repositories [56, 57].
Prophages and the range of possible genome sizes for phages
Prophages are present in most bacterial taxa and play a crucial role in the evolution of prokaryotes [58–61]. They are often major sources of genetic diversity between closely related bacterial strains or pathovariants [62]. Prophages integration can significantly alter the phenotype of the bacterial host, facilitating the acquisition of new functions, including virulence, stress resistance, tolerance to phages and antimicrobials. Notably, many bacterial exotoxins involved in disease pathogenesis are encoded by prophages [63].
Prophages may contain so-called ‘accessory’ or ‘moron’ loci that are not required for the phage life cycle but influence host bacterial fitness, through a process known as lysogenic conversion. These loci are often highly expressed during lysogeny, while most phage-encoded genes are normally repressed at this stage. It has been hypothesized that accessory loci of prophages may possess unique transcriptional signatures distinct from lytic phage genes. However, identifying these genes solely on the basis of DNA sequence is very difficult [64, 65]. To clarify their functional role, approaches that include transcriptomic analysis are required.
In the context of studying the gut phageome, prophages represent a pivotal subject of analysis due to their predominance in the GV. However, annotating prophages in bacterial genomes and metagenomic data is associated with several methodological challenges. Over the past two decades, numerous bioinformatic tools have been developed to predict prophage regions in bacterial genomes [66]. Among these, VIBRANT [50], geNomad [55], DBSCAN-SWA [67], Phigaro [68], and CheckV [56] have demonstrated high prediction accuracy. Despite these advancements, many algorithms still face the difficulty of distinguishing between the bacterial chromosome and integrated prophage sequence, resulting in inaccurate predictions [10, 66]. Machine learning methods for prophage prediction are currently under active development, but their main limitation lies in the lack of manually annotated prophage regions in bacterial genomes. Such reference datasets are needed to test and train prophage region prediction algorithms and to improve the accuracy of prophage annotation. Several research groups are working toward the creation of such databases, which are expected to significantly improve the capabilities of modern prediction tools [66, 69–71].
Cryptic prophages – defined as prophages that cannot lyse their hosts and produce active phages – present an additional phage annotation challenge. These residual viral sequences can persist in the bacterial genome for many generations without functioning as true phages. This raises a classification dilemma: should these sequences be regarded as part of the bacterial genome, or as prophage because of their origin? Since temperate phages predominate in the GV, such classification difficulties pose a relevant problem [65, 72, 73]. It is also important to emphasize here that, contrary to the perception of them as a passive genetic cargo, cryptic prophages are capable of influencing the phenotype of the host bacterium. As a relatively permanent reservoir of genes, they provide the bacterium with numerous advantages for survival in competitive and unfavorable conditions. The influence of cryptic prophage genes on host cell physiology includes, for example, the acquisition of antibiotic resistance and tolerance, enhanced growth in rich and minimal media, as well as a strengthened host response to various environmental stresses, including oxidative and osmotic stress. Thus, these phage fossils significantly contribute to the metabolic potential of the host bacterium [74, 75].
When predicting viral sequences in metagenomic data, it is important to consider the range of possible phage genome sizes (Fig. 1). Metagenomic studies of various ecological niches have led to the discovery of the largest bacteriophages to date – the so-called megaphages – whose genomes can reach 500 kb or more [76–78]. The current record for the largest phage genome is a 735 kb megaphage from Lac Pavin, a freshwater meromictic crater lake in France (discovered through metagenomes sequenced from diverse ecosystems) [76]. Interestingly, some of megaphages use an alternative coding strategy. This poses a serious challenge for tools based on open reading frame homology searches with known viral genes, as recoded sequences are not recognized by standard annotation methods [79]. It should be noted here that recoding is also widespread among crAss-like phages and has been identified in a range of other phages [79–81]. On the other hand, emerging evidence suggests the existence of extremely small bacteriophages, with genome sizes well below the 10 kb threshold often used in metagenomic filtering [9, 10, 37, 56, 79]. Analysis of phage capsid structure by cryogenic electron microscopy (cryo-EM) revealed that icosahedral capsids can encapsulate DNA molecules of much smaller size. In addition, satellite phages – lacking the full set of genes necessary for independent replication and virion assembly – have been identified. These phages depend on co-infecting helper phages and may possess genomes as small as 5 kb (Fig. 1) [79].
Fig. 1. Comparative sizes of bacteriophage genomes. The following phages are shown in the figure: MS2 – Emesvirus zinderi (GenBank: V00642); phiX174 – Escherichia phage phiX174 (Sinsheimervirus phiX174, GenBank: J02482); M13 – Escherichia phage M13 (Inovirus M13, GenBank: V00604); P4 – Enterobacteria phage P4 (satellite phage P4, GenBank: NC_001609); T7 – Escherichia phage T7 (Teseptimavirus T7, GenBank: V01146); lambda – Escherichia phage Lambda (Lambdavirus lambda, GenBank: J02459); T4 – Escherichia phage T4 (Tequatrovirus T4, GenBank: AF158101.6); phiKZ – Pseudomonas phage phiKZ (Phikzvirus phiKZ, GenBank: AF399011); phage G – Bacillus phage G (Donellivirus gee, GenBank: JN638751)
Thus, the potential existence of both exceptionally large and unusually small phage genomes must be taken into account when analyzing metagenomic data. The use of methods focused solely on the traditional size of bacteriophages may result in the loss of a significant portion of viral diversity. This is especially important when developing new tools for identifying viral sequences, as genome size thresholds can significantly influence prediction outcomes.
Barriers to reliable phage genome assembly in metagenomics
At first glance, the obvious solution to the problem of identifying viral sequences in metagenomic data might seem to be assembling viral genomes directly from metagenomes, an approach known as viral metagenome-assembled genomes (vMAGs). This strategy relies on metagenomic binning, which clusters DNA sequences obtained from metagenomic sequencing into groups (bins) based on sequence similarity. In theory, such binning can enable the reconstruction of viral genomes. However, this approach faces a number of serious limitations that currently preclude its use as a reliable method.
First, viral sequences are often characterized by low and non-uniform coverage, which makes correctly assembling genomes difficult. Metagenomic binning tools are mainly focused on bacterial genomes, for which coverage is usually more stable and specific to different taxonomic groups. In contrast, for viruses, particularly temperate phages and rare virotypes – low coverage leads to fragmented assemblies and low binning accuracy [54, 65, 82].
Second, bacteriophage genomes exhibit a mosaic structure due to extensive horizontal gene transfer. Viral sequences, assembled in vMAGs, are often a concatenation of genome fragments from multiple unrelated phages, and represent chimeric assemblies. As a result, there are low-quality vMAGs that cannot be reliably classified [41].
Finally, genomes assembly from metagenomes produces many short contigs that are either discarded or incorrectly binned. This is particularly critical for viral genomes, because they often contain terminal repeat regions that are usually present in the assembly as short fragments. The loss of these regions can result in the omission of important portions of phage genomes [41].
Thus, at the current stage of development of computational methods and algorithms, viral binning tools cannot reliably reconstruct a complete, non-chimeric, and high-quality phage genome from metagenomic data.
Difficulties in taxonomic classification and host prediction
In addition to the techniques used for phage identification, the approaches and methods for their classification, including taxonomic systematization, remain a challenging task. The taxonomic classification of bacteriophages is more challenging than that of bacteria: bacteria possess universal marker genes (e.g., 16 S rRNA) which reflect their evolutionary relationships. By contrast, viruses lack any universally conserved genes on which a phylogeny could be constructed. Historically, phage classification was based on their morphology. The first taxonomic scheme was proposed by David Bradley in 1967 and focused on the best-studied phages of the time – tailed bacteriophages [83]. Later, in 1971, David Baltimore proposed classifying viruses based on the type of nucleic acid in virions, reflecting differences in viral replication strategies [84]. For a long time, the Baltimore scheme together with phage morphology, remained the basis for the bacteriophage classification.
With the advancement of molecular biology and sequencing technologies, it became evident that the genomic diversity of bacteriophages is much broader than previously recognized, particularly among tailed phages. It was discovered that there are significantly more uncultivated phages than cultured ones in natural ecosystems, leading to a significant expansion of bacteriophage taxonomy [85–88]. The resulting exponential growth in the number of new phage taxa has necessitated revising traditional classification systems.
In March 2022, the International Committee on Taxonomy of Viruses (ICTV, https://ictv.global/) approved a new classification system for tailed phages, abolishing the order Caudovirales and uniting all tailed phages into the class Caudoviricetes. New orders were introduced to replace the previous taxon and better reflect the origin and evolutionary relationships among of the various phage groups. Unfortunately, most of the viruses identified to date remain unclassified: among the ~ 55 500 records in the NCBI Virus portal (https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/, December 2025, host filter: Bacteria, taxid:2), ~ 25 000 (46%) do not have taxonomic annotation [88, 89].
Modern taxonomic classification of bacteriophages relies on a range of bioinformatic approaches, such as genome structure and organization analysis, proteomic analysis, clustering based on average nucleotide identity, and phylogenetic reconstruction. Accurate classification is complicated by the frequent genetic exchanges among phages, which can involve both individual genes and groups of genes. It is also important to consider, especially for phages with double-stranded DNA (dsDNA), extensive sharing of genes and/or gene cassettes – which require strict alignment criteria in their classification to avoid false-positive associations and artificial links between unrelated taxa. Another challenge of bioinformatic classification approaches is the rapid evolution of viral proteins.
With the development of metagenomics, it has begun to play an important role in the revelation of new phage taxa, allowing the identification of genomes of uncultured viruses. Parallel to this metagenomic studies have shown that a significant proportion of bacteriophages, particularly those infecting non-pathogenic bacteria, remain unclassified despite the existence of current classification systems.
In addition to the above, there are also the following limitations:
-
viral genome databases do not adequately represent the true diversity of viruses present in nature, because, for example there is a problem of overrepresentation of certain phage groups in reference databases. So, most studies of viromes show that they are dominated by tailed phages from the Caudoviricetes class, which is probably a consequence of the bias in databases towards the well-studied viruses. The problem of overrepresentation of the Caudoviricetes class in databases reduces the sensitivity of the tools to other groups of bacteriophages. Among the available tools, DeepVirFinder demonstrates the highest sensitivity in detecting a broad spectrum of phages, including understudied groups [3, 8, 45];
-
The viral taxonomy, as defined by the International Committee on Taxonomy of Viruses (ICTV), is constantly changing. Despite these challenges, recent years have seen the emergence of specialized tools for classifying viruses assembled from metagenomes, most of which rely on sequence similarity approaches, including BLAST-based or HMM-based searches [85]. These computational methods form the foundation for current efforts to systematically catalog viral diversity. A broader overview of classification approaches, algorithms, and software solutions is provided in two comprehensive reviews, discussing the state of the field and its development [85, 90].
Determining the phage host is one of the most challenging tasks in virology, especially for unculturable bacteriophages. Without reliable experimental methods to establish phage-host associations, researchers must rely on bioinformatic predictions. These predictions are usually based on molecular features indicating co-evolution and/or an arms race between phages and their hosts, identity with the genomes of reference viruses or hosts, as well as matches with CRISPR spacers encoded by the host and on sequence composition analysis [91].
Despite the development a number of tools to predict potential hosts of uncultured phages developed over the past decade (Table 2), the ability to associate newly identified phages with their hosts remains limited [92–101].
Table 2. An overview of phage host prediction tools(bioRxiv)/YearpublishedTool/referenceHost predictionapproachFeatures/notes2016 HostPhinder [92]predicts a phage’s host by k‑mers based genomic similarity to reference phages with known hosts• virus-dependent (virus-virus similarity)• alignment-free• independent of gene prediction and host reference database• sequence composition-dependent tools similarity to at least one reference phage genome is required2017 WIsH [93]predicts a phage’s host by comparing k-mer composition between a query phage sequence and host genomes, via trains Markov models on each host and ranks hosts• host-dependent (virus–host similarity)• alignment-free• independent of gene prediction and phage reference database sequence composition-dependent tools(2019)/2022 PHISDetector [94]predicts a phage’s host by integrative tool that detecting and integrating diverse in silico phage–host interaction signals (PHISs), and scoring their probability using machine learning models based on PHIS features• virus–host alignment-free similarity (based on k-mer frequencies)• virus–host alignment-based similarity• virus–host CRISPR-based similarity• integrates additional features from putative prophage regions• integrates additional features from protein-protein interactions• use machine-learning approaches provides fancy visualizations for users2020 VirHostMatcher-Net [95]predicts a phage’s host by network-based, Markov random field framework, using integrate multiple alignment-free and alignment-based features• virus-virus similarity• virus–host alignment-free similarity (based on k-mer frequencies)• virus–host alignment-based similarity• virus–host CRISPR-based similarity• use machine-learning approaches• improved accuracy and sensitivity by considering and integrating multiple signals integrative methods require a longer compute time since they need results from several individual prediction approaches(2020)/2022 vHULK [96]predicts a phage’s host by neural network-based tool based on protein-coding genes• alignment-dependent• independ of host reference database• predicted protein sequences are affiliated to the pVOGs database• use two deep neural networks• high accuracy for phages related to known references required sharing at least one marker gene with a known phage reference(2020)/2021 RaFAH [97]predicts a phage’s host by machine-learning approach - Random Forest classifier based on similarity in protein content• alignment-dependent/based• virus-dependent• based on protein content (using marker genes and HMM profiles)• for training database use host-dependent alignment-based methods• use machine-learning approaches only take mVCs as input2021 PredPHI [98]predicts a phage’s host by deep learning-based tool capable of predicting from sequence data• alignment-free• use machine-learning approaches• convolutional neural network (CNN)• host proteomes dependent• informative features based on properties of protein sequences:- amino acid residue frequency- chemical composition molecular weight2021 Bacteriophage-Host-
Prediction [99]predicts a phage’s host by machine-learning-based pipeline based on annotated receptor-binding protein (RBP) sequence data• use machine-learning approaches• uses 218 features:- nucleotide/codon frequencies, codon usage bias and GC-content- relative abundance of amino acids- physicochemical properties of the sequences (protein length, molecular weight, isoelectric point, aromaticity, and others)- protein secondary structure (α-helix and β-sheet frequencies)- features describing protein sequences (composition, transition, and Z-scale)• model predicts bacterial hosts only for phages infecting S. aureus,* K. pneumoniae*,* A. baumannii*,* P. aeruginosa*,* E. coli*,* S. enterica* and C. difficile2022 CHERRY [100]predicts a phage’s host by formulates the host prediction problem as a link prediction in a multimodal graph; apply a graph convolutional encoder and use a two-layer neural network decoder to calculate the probability virus–host pair• alignment-free• multimodal graph integrates different types of features(into the nodes and edges):- protein organization- CRISPR- sequence similarity- k-mer frequency(edges connect virus-host):- from labeled (training) and unlabeled (test) data(2022)/2023 iPHoP [101]predicts a phage’s host by integrate multiple approaches for host prediction and uses modular machine learning framework• host-based and phage-based• integrative methods• run 6 host prediction approaches:- Blast- CRISPR- VirHostMatcher- WIsH- PHP- RaFAH• provides reliable host predictions at the genus rank because it relies on a suite of different tools, remains relatively slow compared to other tools
The advent of long-read metagenomic sequencing has provided the opportunities to overcome some of these limitations. By producing contiguous reads that can span entire prophages and their flanking host sequences, this technology can empirically link an integrated prophage with the genome of its host within complex communities like the gut microbiome, overcoming the challenges of fragmented short-read assemblies [102, 103].
Comparing spacers from bacterial CRISPR-Cas systems with phage genomes shows that most spacers have no matches, confirming the existence of a significant pool of ‘viral dark matter’. An additional problem is that the host ranges of phages have been studied extremely unevenly. Phages that infect well-studied pathogenic bacteria, such as Escherichia coli or Klebsiella pneumoniae, are much better described than phages infecting commensal or difficult to culture bacteria. This creates a substantial data bias, reducing the accuracy of predictions for rare or ecologically important microorganisms [104]. Therefore, the most effective strategy for phage-host prediction is integration of different computational approaches. For example, the iPHoP tool, which integrates host-based and phage-based approaches for reliable host taxonomy prediction, demonstrates good results on metagenomic data and is widely used [101]. Furthermore, developing large-scale datasets containing experimentally validated phage-host pairs also plays a crucial role in improving prediction accuracy [91]. As data accumulate and algorithms continue to improve, substantial progress in predicting phage-host relationships can be expected.
Bacteriophages: promising therapeutic agents
Bacteriophages are emerging as promising therapeutic tools with broad applications, extending beyond their classical use against antibiotic-resistant bacteria. Recent studies have highlighted their potential to modulate host immune responses, influence tumor progression, alleviate inflammatory and liver diseases, and serve as diagnostic and prognostic biomarkers in oncology (Fig. 2) [105, 106].
Fig. 2. Therapeutic potential of bacteriophages
While phage therapy has traditionally targeted antibiotic-resistant pathogens, recent studies have begun exploring the potential of phages in oncology. Research into phage-based anticancer vaccines is underway, though none have yet received approval from the FDA or EMA [107]. Some studies suggest that phages can directly influence tumor cells. For example, phages T4 and HAP1 were shown to inhibit melanoma cell migration on fibronectin in vitro, indicating a potential role in limiting tumor dissemination [108]. Other studies have demonstrated that certain phages can activate tumor-associated macrophages, thereby promoting tumor cell destruction and stimulating innate immune responses [109]. Phages capable of modulating the immune reaction are also of growing interest. In germ-free mice, the administration of specific phages increased immune cell counts in the gut. For example, phages targeting Lactobacillus, Escherichia, and Bacteroides were shown to stimulate IFN-γ production via the TLR9 receptor, which detects viral DNA [21]. Moreover, engineered phages are being actively investigated for targeted delivery of anticancer agents and for immunostimulatory purposes. One study developed a modified T7 phage carrying a melanoma-targeting peptide and expressing the cytokine GM-CSF. Administration of this phage to mice resulted in 72% tumor growth suppression within 16 days and induced tumor infiltration by macrophages, dendritic cells, and CD8^+^ T-cells [110]. Another study engineered M13 phages expressing tumor-associated MAGE-A1 peptides, which elicited strong immune responses in mice, underscoring their potential in melanoma vaccine development [111]. Moreover, phage therapy has shown promise in treating hospital-acquired bacterial infections in cancer patients undergoing chemotherapy [112].
The development of inflammatory bowel diseases (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), is caused by the interaction of genetic, immunological and bacterial mechanisms. Numerous experimental models of enterocolitis support the immunological hypothesis of IBD pathogenesis, linking disease development to defects in both innate and adaptive immune responses. There is evidence of the influence of gut bacteriophages on the course of diseases associated with immune system disorders. Research indicates that patients with CD and UC exhibit reduced bacterial diversity in their GM, and the virome in these diseases differs from the norm and is characterized by a significant increase in the number of bacteriophages of the class Caudoviricetes (the original publication uses the now-obsolete order name Caudovirales) [113]. In addition, it is suggested that the virome plays an important role in the advancement of colorectal cancer (CRC). The increased alpha diversity of gut phages in CRC presumably leads to the depletion of the bacterial component of the GM due to an increase in the number of lytic infections, which in turn stimulates the development of CRC. An association with CRC has been shown for a number of viral families: in particular, enrichment of the Myoviridae,* Podoviridae*, and Siphoviridae families and depletion of Herelleviridae family (taxonomic names for virus families are given here according to the original cited works). The Herelleviridae family appears to promote the integrity of the gut barrier function, and its depletion in CRC potentially promotes tumor progression. Additionally, increased activity of fatty acid biosynthesis pathways has been identified in CRC patients, which has been previously shown to increase the risk of this disease [114, 115].
For liver disease, bacteriophages also have shown their therapeutic potential in pathologies, like alcoholic hepatitis. Transplanting bacteria from severe alcoholic hepatitis patients into humanized mice induced similar liver damage symptoms. It is known that alcoholic hepatitis has been linked to the presence of cytolysin-positive Enterococcus faecalis in the GM. Treatment with E. faecalis specific phages resulted in decreased levels of cytolysin in the liver, as well as decreased liver damage [116].
Another emerging therapeutic strategy is the fecal virome transplant (FVT), which involves the transfer of viral particles, including bacteriophages, and their associated metabolites from a healthy donor to a recipient. Unlike conventional fecal microbiota transplantation, this approach focuses specifically on the viral component of the GM. FVTs have already demonstrated clinical benefits in the treatment of recurrent Clostridium difficile infections in humans [117, 118], and preclinical studies suggest their potential to modulate obesity and metabolic alterations in mouse models [119]. Although still at an early stage of investigation, FVTs represent a promising avenue for harnessing the gut virome to restore microbial balance and influence host physiology. Other studies on FVT demonstrate its broad potential: in mice, FVT has been shown to modulate phenotypes (lean/obese) [120], improve metabolic status [119], and improve the proliferation of probiotic bacteria such as Akkermansia muciniphila [121]. Additionally, research in a yellow-feathered broilers using FVT has demonstrated that the virome and its metabolites, besides the microbiota, are also functional components contributing to the growth-promoting effect of fecal microbiota transplantation [122].
Importantly, an increasing number of publications suggest that intestinal bacteriophages may serve as biomarkers for various cancers, with potential diagnostic and prognostic applications. Predictive models based on gut bacteriophages have already demonstrated reproducibility and high predictive accuracy for colorectal cancer and colorectal adenoma [123, 124]. Furthermore, intestinal virus biomarkers have been shown to significantly distinguish patients with pancreatic cancer from healthy individuals, suggesting their potential utility in the early prognosis of the disease [125].
Together, these findings support the expanding therapeutic and diagnostic potential of gut phages, underscoring the need for further studies of the bacteriophage interaction mechanisms with both bacterial and host cells and its validation on large-scale clinical cohorts.
Conclusion and perspectives
The human body is constantly exposed to large numbers of bacteriophages – an estimated 31 billion phage particles passing into the body daily [3, 18]. However, the impact of the gut phage community (gut phageome) on human health remains poorly understood, primarily due to challenges in virome isolating and sequencing, assembly, and interpretation.
Virome research, particularly in the context of the gut, is still in its early stages, with bioinformatic tools and standards actively evolving. Although efforts between 2016 and 2018 attempted to establish methodological guidelines on the most effective ways to analyze viruses in large metagenomic datasets – however, these remain insufficient [126–130]. Metagenomic studies of the GV typically provide qualitative rather than quantitative insights, identifying dominant phage groups, their possible expression strategies, and potential interactions with the host. It bears emphasis that the choice of bioinformatic tools and their parameters can significantly influence the results obtained [36]. To address this, the most promising seem to be either emerging hybrid or complex approaches, which combine complementary methods, like reference-dependent and independent tools, for example [79, 131]. As emphasized by Benler and Koonin in their article, that given the diversity of currently available tools, an integrated approach could make complete mapping of the global phageome a realistic task [79].
Beyond basic research, the therapeutic potential of bacteriophages is becoming increasingly evident. In addition to their established application against antibiotic-resistant bacteria, phages show promise in the treatment of inflammatory diseases, liver disorders and even in the treatment of cancer. Recent studies suggest that phages can affect not only bacterial communities but also directly interact with human immune cells, potentially influencing antitumor immunity. Nonetheless, many questions remain unresolved – including understanding the mechanisms of phage interaction with eukaryotic cells, the long-term effects of the GV on human health, and developing reliable predictive models to personalize phage therapy.
Supplementary Information
Supplementary Material 1