Diastereodivergent and Enantioselective Organocatalytic Synthesis of Spiro-Fused Coumarins via [4+2] Cycloadditions
Raquel Hidalgo-León, José Trujillo-Sierra, José Miguel Sansano, Fernando P. Cossío, Abel de Cozar, María de Gracia Retamosa

TL;DR
Scientists developed a new method to create spiro-fused coumarins with high precision using organocatalysis and diastereodivergent control.
Contribution
A novel organocatalytic method for enantioselective and diastereodivergent synthesis of spiro-fused coumarins via [4+2] cycloadditions.
Findings
Trienamine-mediated Diels–Alder reactions with cyclic 2,5-dienones yield spiro-fused coumarin derivatives with high enantioselectivity.
Reversible diastereoselectivity is achieved using cinchonidine-based amine with p-methylbenzoic acid or triethylamine.
Electron-withdrawing groups at C3 of coumarins influence reactivity patterns and diastereodivergence.
Abstract
A highly enantioselective, organocatalytic, diastereodivergent synthesis of spiro-fused coumarin derivatives via trienamine-mediated Diels–Alder reactions with cyclic 2,5-dienones is reported. Using a cinchonidine-based primary amine and either p-methylbenzoic acid or triethylamine enables reversible diastereoselectivity, providing complementary enantioenriched diastereoisomers. The influence of electron-withdrawing groups at C3 of coumarins is delineated, revealing distinct reactivity patterns. Computational studies provide insight into the mechanism and additive-controlled diastereodivergence.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
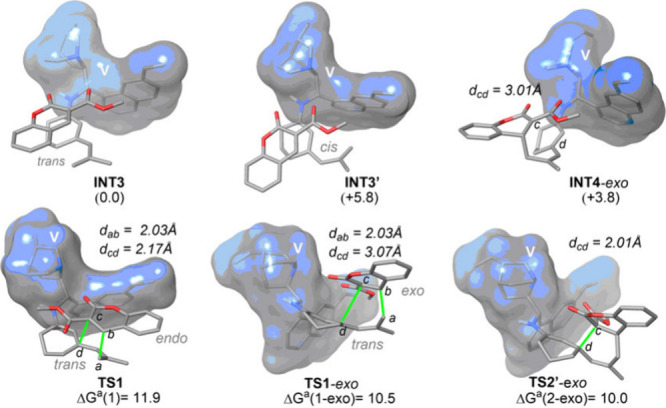 Figure 1
Figure 1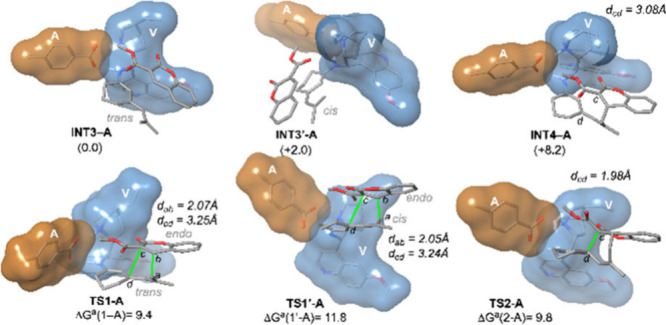 Figure 2
Figure 2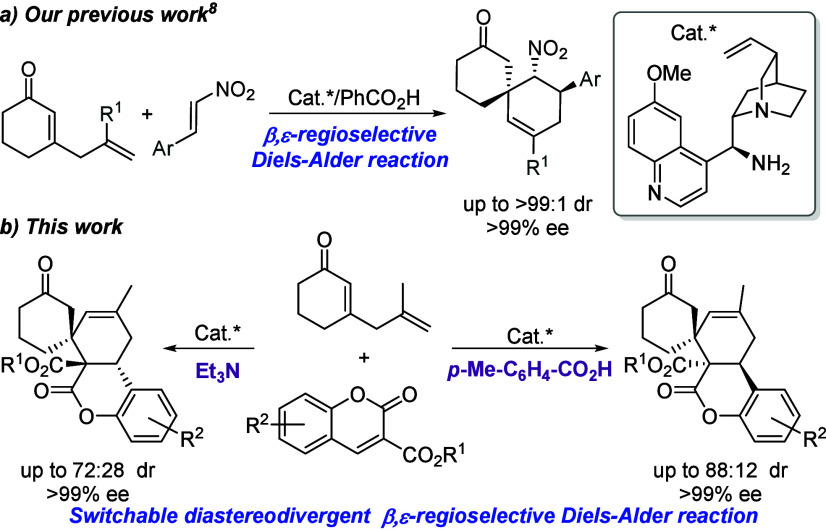 Figure 3
Figure 3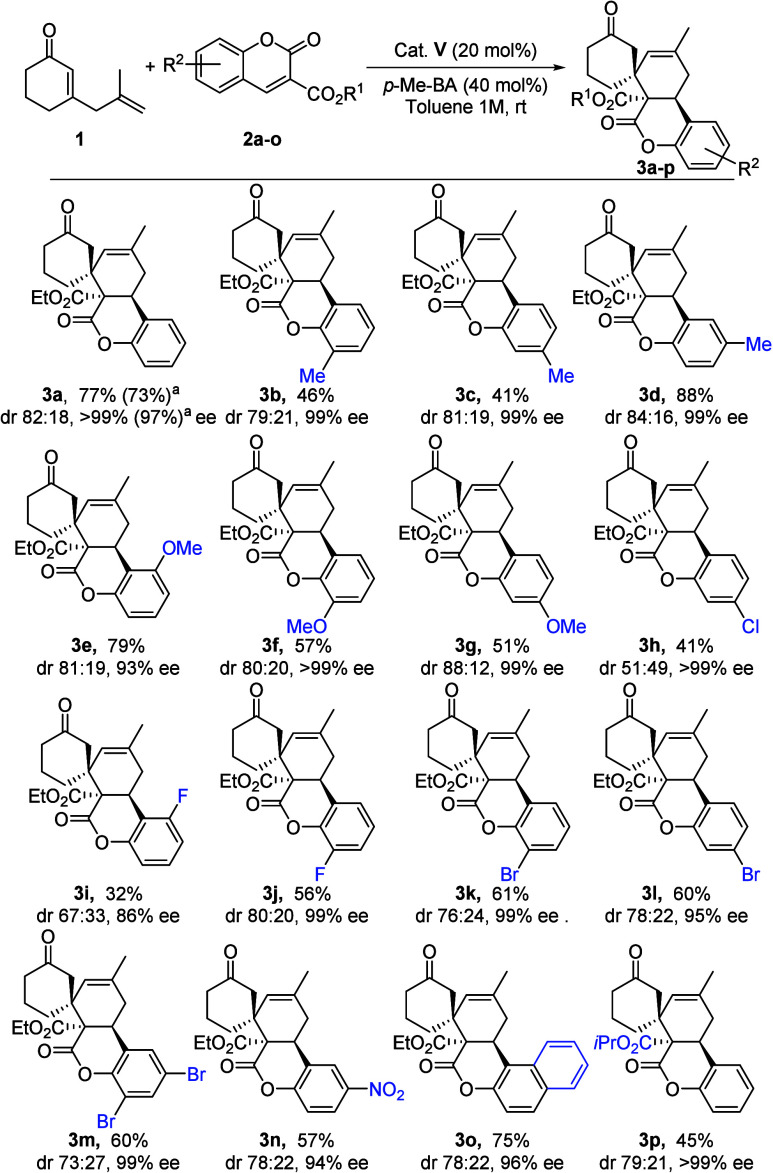 Figure 4
Figure 4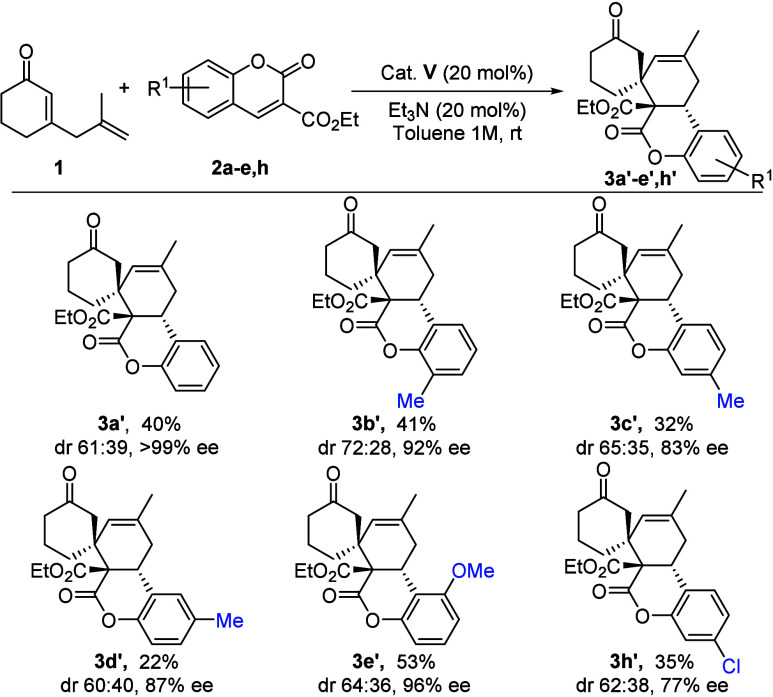 Figure 5
Figure 5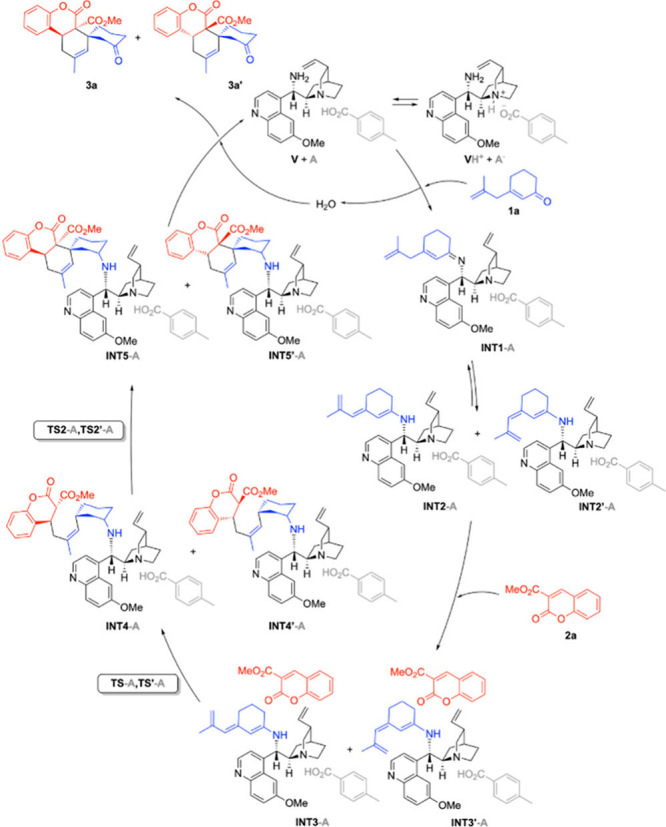 Figure 6
Figure 6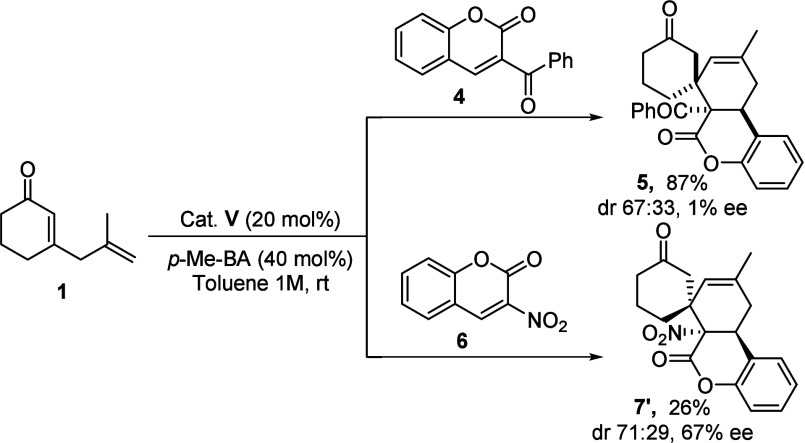 Figure 7
Figure 7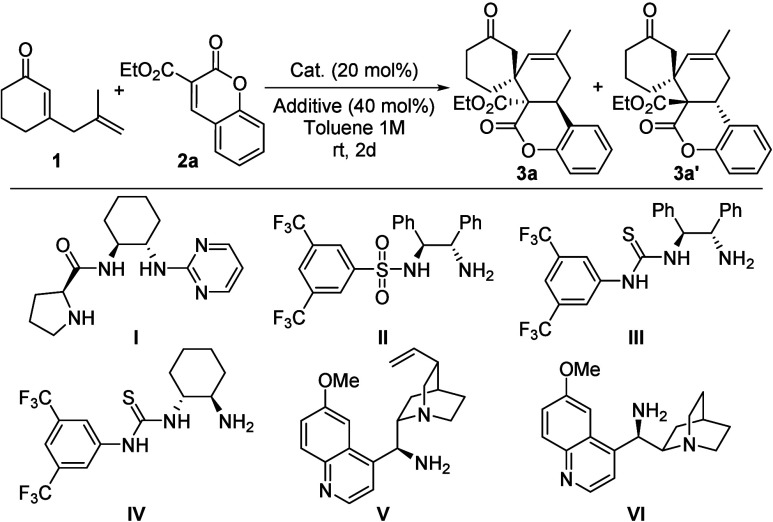 Figure 8
Figure 8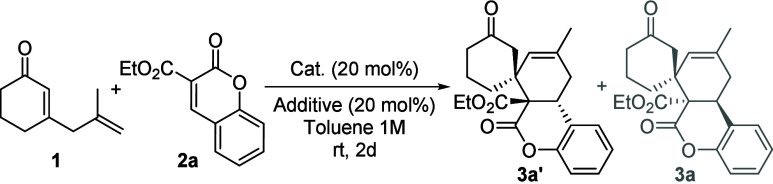 Figure 9
Figure 9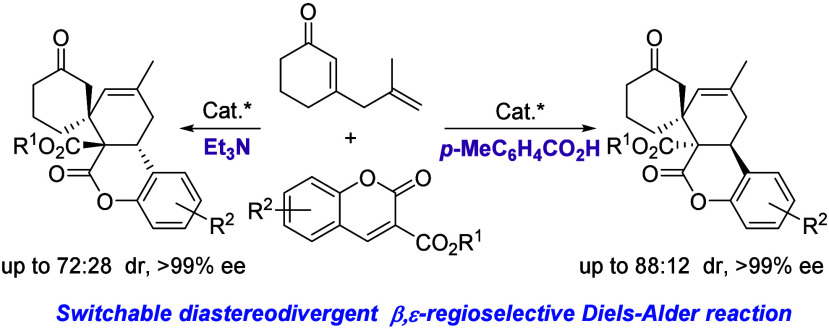 Figure 10
Figure 10 Figure 11
Figure 11- —Universidad de Alicante10.13039/100009092
- —Universidad de Alicante10.13039/100009092
- —Universidad de Alicante10.13039/100009092
- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —Ministerio de Ciencia, Innovaci?n y Universidades10.13039/100014440
- —Eusko Jaurlaritza10.13039/501100003086
- —Generalitat Valenciana10.13039/501100003359
- —Generalitat Valenciana10.13039/501100003359
- —Generalitat Valenciana10.13039/501100003359
- —European Regional Development Fund10.13039/501100008530
- —European Regional Development Fund10.13039/501100008530
- —European Regional Development Fund10.13039/501100008530
- —European Regional Development Fund10.13039/501100008530
- —European Regional Development Fund10.13039/501100008530
- —Agencia Estatal de Investigaci?n10.13039/501100011033
- —Agencia Estatal de Investigaci?n10.13039/501100011033
- —Agencia Estatal de Investigaci?n10.13039/501100011033
- —Agencia Estatal de Investigaci?n10.13039/501100011033
- —Agencia Estatal de Investigaci?n10.13039/501100011033
- —Medalchemy S. L.NA
- —Spanish Ministerio de Econom?a, Industria y CompetitividadNA
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCyclopropane Reaction Mechanisms · Asymmetric Synthesis and Catalysis · Radical Photochemical Reactions
Coumarins constitute a significant class of oxygenated heterocycles commonly found in numerous natural products and biologically active compounds. Various coumarin derivatives demonstrate a broad spectrum of bioactivities, including anticancer, anti-inflammatory, antibacterial, antineurodegenerative, and antitubercular properties.? Due to their versatile application in pharmaceuticals, modifications to the coumarin scaffold could significantly improve their pharmacological properties, leading to new advances in drug discovery. Numerous synthetic methodologies for the development of various coumarins and their derivatives have been reported.? Coumarins bearing a suitable electron-withdrawing group at position C3 have been employed by several research groups as starting material to build more complex coumarin derivatives via 1,4-conjugated addition,? [2+n]? and [3+n]? annulation reactions, cyclopropanations,? and cycloadditions.? However, spiro-fused coumarins have rarely been synthesized, and only a few examples have been described. ?,?,? On the other hand, our research group has recently developed an enantioselective synthesis of spirocyclic compounds by Diels–Alder and aldol/cyclization reactions via trienamine employing δ-substituted 2,5-dienones (Schemea).? In this context, continuing with the elaboration of complex chiral organic entities and considering that coumarins are found in several molecules with biological activity, we envisioned that the use of coumarins with a suitable electron-withdrawing group at position C3 would increase the reactivity of the coumarin double bond, allowing the formation of sterically hindered spiro-fused coumarins via a Diels–Alder reaction (Schemeb).
Based on the above considerations, the reaction of 2,5-dienone 1 and ethyl 3-coumarincarboxylate 2a was evaluated. The reaction was carried out using different organocatalysts with BA as additive in toluene at room temperature (Table 1, entries 1–6). The proline derivative I afforded the desired spiro-fused coumarins in good conversion as a mixture of diastereoisomers, with moderate enantiomeric excesses for both diastereoisomers (Table 1, entry 1). In contrast, the bifunctional amine-sulfonamide II and amine-thioureas III and IV gave rise to the desired adducts with high conversions and enantiomeric excesses but low diasterediastereoselectivities (Table 1, entries 2–4).Moreover, whereas quinine derivative V provided spirocyclic adduct 3a with high diastereoselectivity, quinidine derivative VI led to the formation of a mixture of spirocycles 3a and 3a′ with almost no diastereoselectivity (Table, entries 5 and 6). Further optimization was carried out with the best organocatalyst V using different amounts of the catalyst and reagents, and a variety of solvents (see the Supporting Information for details). Although these changes had a minimal impact on the reaction, the choice of additive emerged as a critical factor influencing the reaction outcome (Table, entries 7–10). While benzoic acid derivatives led to spirocyclic adducts with similar diastereomeric ratios (Table, entry 5 vs entries 7 and 8), the use of phenol and triethylamine resulted in lower diastereoselectivities (Table, entries 9 and 10, respectively). Notably, when both triethylamine and phenol were used as additives (Table, entry 10), 3a′ was obtained as the major diastereoisomer with a large enantiomeric excess. Finally, employing p-methylbenzoic acid (p-Me-BA) as the additive, the reaction was scaled up to 0.2 mmol, affording the desired cycloadduct 3a with a high diastereomeric ratio and excellent enantiomeric excess (Table, entry 11).
Having determined the best reaction conditions, we explored the general nature of this process. The results of a series of experiments are summarized in Scheme. First, a wide variety of aryl-substituted ethyl 3-coumarincarboxylates 2b–n bearing electron-donating and -withdrawing groups were tested. The reaction proceeded smoothly with coumarins containing electron-donating groups, affording cycloadducts 3b–g in moderate to high yields with high diastereoselectivities and excellent enantiomeric excesses. Coumarins bearing electron-withdrawing groups on the aromatic ring were also tolerated. Whereas electron-withdrawing substituents at positions 1 and 3 afforded cycloadducts 3h and 3i in moderate yields and diastereoselectivities with excellent enantiomeric excesses, substituents at positions 2 and 4 afforded cycloadducts 3j–n in good yields and diastereoselectivities with excellent enantiomeric excesses. More sterically hindered coumarin 2o proved to be amenable for this reaction, and expected product 3o could be obtained in 75% yield and 96% ee. On the other hand, isopropyl 3-coumarincarboxylate 2p was also tolerated, leading to the desired spirocycle 3p in moderate yield, high diastereomeric ratio, and excellent enantiomeric excess. To demonstrate the synthetic value of this methodology, the model reaction was performed at a 1 mmol scale for the synthesis of 3a (73%, 82:18 dr, 97% ee) (see the Supporting Information). Adduct 3a was crystallized, and its absolute 6aR,7S,10aS configuration was unequivocally confirmed by XRD analysis, assuming the same absolute configuration for other products 3.
As previously mentioned, the use of triethylamine as an additive resulted in 3a′ being the major diastereoisomer. To further investigate the diastereodivergence of the process, alternative reaction conditions were evaluated using different bases as additives (Table and the Supporting Information). Initially, the reaction was evaluated using quinine V and quinidine VI derivatives as catalysts and triethylamine as the additive, obtaining in both cases the desired spirocycle 3a′ as the major diastereoisomer after reaction for 2 days (Table, entries 1 and 2, respectively). Although quinidine derivative VI produced a higher diastereomeric ratio compared to that of quinine derivative V, its lower conversion after 2 days did not allow the determination of enantioselectivity. Notably, quinine derivative V afforded a large enantiomeric excess of 3a′ (Table, entry 1). Then, other organic and inorganic bases were evaluated (Table, entries 3–5). Similar results in terms of conversion and diastereoselectivity were obtained with DIPEA and Na_2_CO_3_ as with Et_3_N, but in both cases, a lower enantioselectivity was obtained for 3a′ (Table, entries 3 and 5 vs entry 1). DBU resulted in higher conversion and diastereoselectivity, but the Michael adduct was obtained as the major product (Table, entry 4). On the other hand, an increase in the amount of ketone 1 to 4 equiv allowed the conversion to increase to 73% after reaction for 2 days and >99% ee after reaction for 7 days, providing the desired spirocycle with good diastereomeric ratio and excellent enantiomeric excess (Table, entries 6 and 7, respectively). It is noteworthy that the reaction could be carried out without solvent with a slight decrease in enantioselectivity (Table, entry 8). Finally, the reaction was scaled to 0.2 mmol, affording the desired cycloadduct 3a′ with a good diastereomeric ratio and excellent enantiomeric excess (Table, entry 9).
Having determined the best reaction conditions to synthesize 3a′, we investigated the general nature of this process (Scheme). A selection of aryl-substituted ethyl 3-coumarincarboxylate 2a–e and 2h bearing electron-donating and -withdrawing groups was successfully evaluated, affording spirocyclic adducts 3a–e′ and 3h′, respectively, in moderate yields, good diastereoselectivities, and high to excellent enantioselectivities.
In order to understand the insights into the mechanism and its diastereodivergence depending on the additive, DFT calculations at the B3LYP-D3BJ(SCRF=PCM, toluene)/6-31G(δ) level of theory were performed (see the Supporting Information for further details). Our calculations indicate that, in an independent manner of the use of an acid additive, the reaction follows the catalytic cycle depicted in Scheme. Initially, 1a interacts with the catalyst to generate α,β-unsaturated imine intermediate INT1 that can evolve toward two different trienamines, namely INT2 and INT2′. These latter trienamines can react with π-deficient alkene 2a, giving rise to final spirocycles 3a and 3a′. Remarkably, in both scenarios, the reaction follows a stepwise mechanism in which the first C–C bond formation step (TS1) determines the geometry of the final spirocycle.
In absence of an acid additive, our calculations indicate that unsaturated imine INT1 can generate trienamines INT2 and INT2′, in which the central double bond of the trienamine adopts a trans or cis configuration, where the former is ca. 4 kcal mol^–1^ more stable (see the Supporting Information). These trienamines can interact with electro-deficient alkene 2a to generate reactive complexes INT3 and INT3′. Analogously to the trienamines mentioned above, reactive complex INT3, related to the trans configuration of the central double bond of the trienamine moiety, is the most stable one (Figure). In line with that trend, the most stable transition structures are related to that trans configuration. Our calculations indicate that the diastereoselectivity arises from the endo or exo approach of 2a to trienamine INT3. In fact, transition structure TS1 -exo, related to the formation of 3a′, is 1.4 kcal mol^–1^ more stable than its endo analogue, TS1, related to 3a formation. We hypothesize that this effect can be ascribed to the free rotation of the N–C(sp^3^) bond of the quinine moiety that could easily adapt to both approaches, thus being capable of allocating both the bicyclic moiety of catalyst V and the methoxycarbonyl group of coumarin 2a. After the initial C–C bond formation, transient zwitterionic intermediate INT4-exo can form a ring closure transition structure (namely TS2-exo in Figure) to generate spirocyclic intermediate INT5′.
The computed energetic difference in the rate-determining step of the reaction is related to a theoretical 3a:3a′ ratio of 10:90, with 3a′ being the major diastereoisomer. Moreover, the theoretical TOF value, related to the effective “activation” energy of the catalytic cycle δG (see the Supporting Information), is 1.2 h^–1^.
With respect to the acid additive (Figure), the presence of p-methyl benzoic acid strongly affects the geometry of the stationary points and the energetic profile of the catalytic cycle. This additive engages in H-bonding interactions with the nitrogen atom of the quinuclidine moiety, increasing the rigidity of the system. Consequently, the energetic difference between INT2-A and INT2′-A is reduced to only 1.6 kcal mol^–1^ (contrary to the ca. 4 kcal mol^–1^ energetic difference obtained for the absence of an additive case). Interaction with 2a leads to the formation of reactive complexes INT3-A and INT3′-A, which are also close in energy (difference of ca. 2 kcal mol^–1^). Since both reactive complexes are energetically accessible, in this scenario transition structures associated with the two configurations of the trienamine moiety were explored. It is noteworthy that these H-bonding interactions provide a framework that enhances the reactivity of alkene 2a via HOMO increasing activation and determines the 2a approach. That dual activation is reflected in the lower computed Gibbs activation barrier as compared with the absence of the additive case (10.5 and 9.4 kcal mol^–1^ for TS1- exo and TS1-A, respectively).
In this case, least energetic transition structures TS1-A and TS1′-A correspond to the endo approach of 2a to trans and cis trienamines, respectively. In fact, all attempts to isolate transition structures related to 2a-exo were unsuccessful. Remarkably, in contrast to the previous case, the presence of the acid additive favors the formation of spirocycle 3a. Analogously, once transient zwitterionic intermediate INT4-A is formed, it can evolve toward spirocyclic cycloadduct INT5-A. However, in this case, the computed Gibbs activation barrier of this second step is slightly higher than that obtained for the initial C–C bond formation.
Since formation of the final cycloadducts is related to the existence of different energetically accessible reactive complexes, we have considered a Kurtin–Hammet kinetic scenario for the estimation of the theoretical diastereoselectivity (see the Supporting Information). Within that framework, the computed energetic difference would lead to a higher theoretical diastereoselectivity toward 3a formation than that observed for 3a′ in the absence of acid (3a:3a′ ratio of 97:3), in qualitative agreement with the experimental evidence. Additionally, the computed TOF is 0.1 h^–1^, thus indicating enhancement of the catalytic performance due to the presence of the acid additive.
Apart from 3-coumarincarboxylates 2, we also evaluated enantioselective β,ε-regioselective [4+2] cycloadditions with other coumarins bearing different electron-withdrawing groups at position C3 (Scheme). While 3-benzoylcoumarin 4 gave rise to the desired adduct in good yield and moderate diastereomeric ratio as a racemic mixture of major diastereoisomer 5, 3-nitrocoumarin 6 gave rise to spirocyclic adduct 7′ in good diastereomeric ratio and moderate yield and enantioselectivity. Notably, the major diastereoisomer obtained from 3-nitrocoumarin 6 shares the same relative configuration as the minor diastereoisomer observed in reactions with 3-coumarin carboxylates. Due to the moderate enantiomeric excess obtained for 7′, the absolute configuration has to be determined by electronic circular dichroism (ECD) (see the Supporting Information).
In conclusion, this work describes a synthetic diastereodivergent strategy to obtain chiral spiro-fused coumarin compounds with three stereogenic centers via trienamine catalysis in a Diels–Alder reaction. By using a cinchonidine-based primary amine as the catalyst and p-methylbenzoic acid or triethylamine as cocatalysts, the diastereodivergent construction of a wide variety of spiro-fused coumarins has been achieved. The addition of benzoic acid or triethylamine as a cocatalyst could effectively switch the diastereoselectivity. Additionally, the importance of an ester group at position C3 of the coumarins has been demonstrated by using other electron-withdrawing groups, which show different behaviors. Computational studies have provided insight into the mechanism and diastereodivergence of the reaction, depending on the additive used.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1a Fylaktakidou K. C.Hadjipavlou-Litina D. J.Litinas K. E.Nicolaides D. N.Natural and Synthetic Coumarin Derivatives with Antiinflammatory/Antioxidant Activities Curr. Pharm. Des.200410323813383310.2174/138161204338271015579073 · doi ↗ · pubmed ↗
- 2a Medina F. G.Marrero J. G.Macías-Alonso M.González M. C.Córdova-Guerrero I.Teissier García A. G.Osegueda-Robles S.Coumarin Heterocyclic Derivatives: Chemical Synthesis and Biological Activity Nat. Prod. Rep.201532101472150710.1039/C 4NP 00162 A 26151411 · doi ↗ · pubmed ↗
- 3a Kuang Y.Liu X.Chang L.Wang M.Lin L.Feng X.Catalytic Asymmetric Conjugate Allylation of Coumarins Org. Lett.201113163814381710.1021/ol 201312 y 21688863 · doi ↗ · pubmed ↗
- 4a Ivanov I. C.Raev L. D.Addition of Some Enamino Esters to 3-Substituted Coumarins Synth. Commun.198616151679169010.1080/00397918608056427 · doi ↗
- 5a Poronik Y. M.Gryko D. T.Pentacyclic Coumarin-Based Blue Emitters-The Case of Bifunctional Nucleophilic Behavior of Amidines Chem. Commun.201450445688569110.1039/C 4CC 01106 F 24652376 · doi ↗ · pubmed ↗
- 6a Tsuchida H.Tamura M.Hasegawa E.Cyclization and Ring-Expansion Processes Involving Samarium Diiodide Promoted Reductive Formation and Subsequent Oxidative Ring Opening of Cyclopropanol Derivatives J. Org. Chem.20097462467247510.1021/jo 802749 g 19216503 · doi ↗ · pubmed ↗
- 7a Jian T. Y.Chen X. Y.Sun L. H.Ye S.N-Heterocyclic Carbene-Catalyzed [4 + 2] Cycloaddition of Ketenes and 3-Aroylcoumarins: Highly Enantioselective Synthesis of Dihydrocoumarin-Fused Dihydropyranones Org. Biomol. Chem.201311115816310.1039/C 2OB 26804 C 23117299 · doi ↗ · pubmed ↗
- 8a Hidalgo-León R.Alberro N.Cossío F. P.Sansano J. M.Retamosa M. G.Remote Asymmetric Bisvinylogous [4+ 2] Cycloaddition Reaction to Synthesize Spirocyclic Frameworks Adv. Synth. Catal.20233653253325910.1002/adsc.202300811 · doi ↗