cAMRah: a scalable and portable workflow for harmonized antimicrobial resistance gene prediction from bacterial genomes
Daniella L Matute, Thomas H Clarke, Andrew R LaPointe, Indresh Singh, Derrick E Fouts

TL;DR
cAMRah is a flexible workflow that combines multiple tools to accurately predict antimicrobial resistance genes in bacterial genomes.
Contribution
cAMRah introduces a harmonized, consensus-based approach for AMR gene prediction using multiple tools and databases.
Findings
cAMRah integrates six AMR-finder tools and databases for comprehensive AMR gene prediction.
The workflow harmonizes results into consistent gene symbols and ontologies.
cAMRah is portable and can run on any system with Docker.
Abstract
cAMRah is a curated workflow designed to predict antimicrobial resistance (AMR) genes in microbial genomes, either in the cloud or on any personal computer running Docker containers. Numerous AMR gene-finding packages exist, each utilizing different algorithms and prediction methods. cAMRah adopts a consensus-based approach to AMR prediction, recognizing that no single tool can identify all AMR genes. It integrates and runs six AMR-finder tools and databases (with plans for future expansion), scores the AMR predictions, maps all results to CDS coordinates and harmonizes the annotation, resulting in consistent gene symbols and ontologies. Source code, demo data and detailed documentation are freely available at https://github.com/JCVenterInstitute/CAMRA.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
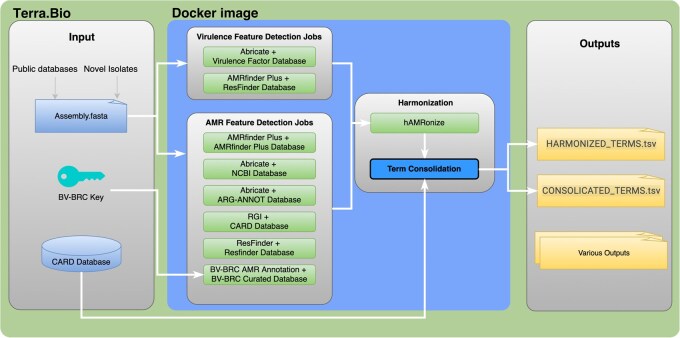 Figure 1
Figure 1- —National Institute of Allergy and Infectious Diseases, National Institutes of Health
- —Department of Health and Human Services10.13039/100000016
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntibiotic Resistance in Bacteria · Genomics and Phylogenetic Studies · Machine Learning in Materials Science
1 Introduction
Antimicrobial resistance (AMR) is a global threat. A 2024 study evaluating the global AMR burden during 2021 estimated 1.14 million deaths attributable to AMR, and 4.71 million deaths associated with AMR occurrence. By 2050 they estimate 1.91 million annual deaths globally and 8.22 million associatively (GBD 2021 Antimicrobial Resistance Collaborators 2024). Antibiotic resistance in the US alone is responsible for 23 000 deaths each year with an economic impact estimated at 35 billion in healthcare costs and societal loss of productivity, respectively (Antibiotic Resistance Threats in the United States 2013).
To aid in understanding the genetic basis of resistance and global spread, high throughput genomic sequencing has significantly bolstered epidemiological investigations and surveillance efforts (Baker et al. 2023, Duan et al. 2025). Due to increased availability of high-quality bacterial genomes, many AMR gene (AMG) prediction software tools and pipelines have been developed, with over 18 that are commonly used open-sourced command line tools (Mendes et al. 2024). Despite all the advancements in AMG detection software, no single tool can accurately predict every AMG. Further confounding efforts to compare multiple AMG detection tools and combine multiple predictions is that each tool uses different algorithms and databases as well as various non-standardized output file formats (Kordova et al. 2025). Efforts have been made to “harmonize” the results of multiple AMG detection tools (Mendes et al. 2024), but fail to map predictions to the same genetic loci.
With all the various AMG detection tools requiring a multitude of software and library dependencies, specific hardware platforms and computing knowledge, it is challenging for biologists and healthcare professionals to run large batches of genomes. With this in mind, AMRColab was created, which is a Google-based Jupyter Notebook for running AMRFinderPlus, ResFinder, a harmonization module and geographical visualization module (Lam et al. 2024); however, it only runs two AMG prediction tools, suffers from the same harmonization limitations and is not portable.
Here we introduce cAMRah, a portable, reproducible workflow that runs and combines the output of six AMG detection tools, including the k-mer-based novel AMG predictor from the BV-BRC to generate a conservative and comprehensive representation of the AMR features of a bacterial genome. The aims were inspired by (Baichoo et al. 2018, GLASS Whole-Genome Sequencing for Surveillance of Antimicrobial Resistance 2020) and are as follows: (i) make a reproducible workflow, (ii) make it portable, (iii) it should scale to the capacity available to the researcher to suit heterogeneous computing environments, (iv) harmonize outputs of AMR tools at various levels to permit ease of analysis, including CARD ontology mapping, and (v) assist in the data analysis bottleneck generated by sequencing data. The workflow leverages Docker containers and WDL workflow language for platform-independent computing and a novel AMG term harmonization heuristic to map AMG predictions to specific genetic loci.
2 Methods
2.1 Workflow description language (WDL) and docker integration
Workflow description language (WDL) is a standard for describing computational workflows, allowing parallel job execution and dependency resolution by means of a Directed Acyclic Graph (DAG). Key benefits of WDL adoption are increased automation, scalability, reproducibility and open-community support. These features provide bioinformaticians reproducibility and scalability capacity as highlighted by Baker et al. (2023), Baykal et al. (2024), and Afolayan et al. (2021) , who emphasize the application of workflow systems to overcome research barriers.
WDL workflows function by running commands within Docker containers, which envelops all necessary software, dependencies, and resources for a specific job. Docker containers provide a consistent environment for tool execution, while supporting portability and reliability. WDL manages the location of input and output files, and the computational resources allocated for processing. WDL was chosen due to its global adoption by researchers and its native support by the Terra.Bio cloud analysis and collaboration platform.
2.2 Cloud enabled
Terra.Bio is a secure, cloud-native platform for biomedical research enabling data access, pipeline execution, analysis and collaboration (https://app.terra.bio/). eLwazi Open Data Science Platform has established its own instance (https://elwazi.terra.bio/) to provide African scientists access to high-throughput, reproducible analysis capabilities while maintaining strict access and authorization control, enabling cross-consortium project collaboration. By leveraging Terra’s existing infrastructure in the African research community, we aim to contribute to the Data Science building effort of the continent.
Locally-run WDLs are useful for debugging, testing, or when data privacy is of particular concern. This platform flexibility enables scalability and democratic access to the pipeline, promoting reproducibility across environments of various-sized projects and skill levels.
2.3 cAMRah workflow
Whole Genome sequencing (WGS) is an essential input for antimicrobial surveillance and clinical application. The CAMRA (Combating Antibiotic-Resistant Bacteria in Africa) project required an AMR workflow compatible with eLwazi’s Terra instance and scalable across various user settings. We developed cAMRah, a single-command WDL-based pipeline that generates the outputs of multiple AMR detection tools, harmonizes output file types and data structure, and consolidates ontologies to harmonize AMR gene allele naming. The workflow includes six AMR analysis tools and databases combinations, including:
AMRFinderPlus v3.12.8 with the AMRFinderPlus database 2024-01-31.1 (Feldgarden et al. 2021),Resfinder v4.5.0 with the Resfinder database v2.3.1 (Bortolaia et al. 2020),RGI v6.0.3 with the latest CARD database (Bortolaia et al. 2020, Alcock et al. 2023),Abricate v1.0.1 (Seemann 2015) with the NCBI database (Feldgarden et al. 2019),Abricate v1.0.1 (Seemann 2015) with the ARG-ANNOT database (Gupta et al. 2014), andthe BV-BRC AMR detection tool with their manually curated data collection produced from literature, data resources, direct submissions and AMR phenotypic predictions (VanOeffelen et al. 2021).
While there are at least 18 commonly-used AMR-finding programs that utilize different search algorithms, parameters, input and output formats, we chose four of the eight top-performing and well-supported programs that have been recently compared (Kordova et al. 2025). Because of its k-mer-based AMR predictive power, the BV-BRC AMR detection algorithm was also included (VanOeffelen et al. 2021). Using a federated workflow design, we have included the BV-BRC AMR prediction tool using the BV-BRC command-line interface tool as in Fig. 1. Users will be required to create a free account with BV-BRC to benefit from their publicly available resource.
cAMRah workflow.
Because each AMR annotation tool produces different output file formats, harmonization of these files is performed with hAMRonize, an open source tool that eases the downstream bioinformatic analysis of results (Mendes et al. 2024). Although hAMRonize parses 17 tools, it fails to harmonize the multitude of AMR annotations to single gene loci (Fig. 1a, available as supplementary data at Bioinformatics Advances online). It also does not support AMRFinderPlus version v3.12.8 or the BV-BRC AMR prediction tool; therefore, we contributed to their code by providing these advancements, and our changes were accepted by the hAMRonize Team.
2.4 Installation and usage instructions
Step-by-step installation and usage instructions, including the final results, can be found on our project wiki located at https://github.com/JCVenterInstitute/CAMRA/wiki/AMR-Analysis.
2.5 Required input and example dataset
The cAMRah workflow requires an assembly fasta file (gzip or not) and the following strings: sample_name (string), genus (string), species (string), BVBRC_username (string), and BVBRC_password (string). The required strings are entered on the submission page when running on Terra or via a JSON file when running locally using Cromwell. An example assembly fasta file and JSON file are provided as File 1, available as supplementary data at Bioinformatics Advances online, and on the GitHub wiki at 2.ii.a.a and 2.ii.a.b. Other optional inputs can be declared; however, they are not necessary to run the fundamental analysis but provide the opportunity to customize the workflow. If no optional inputs or parameters are provided, cAMRah will run using parameters of the AMR prediction tools as noted in Fig. 2, available as supplementary data at Bioinformatics Advances online.
2.6 AMR annotation term consolidation
Because hAMRonize does not perform consolidation of AMR annotation terms to specific gene loci, making the results difficult to interpret, cAMRah incorporates a term consolidation script that groups AMR hits from various tools that fall within the same loci/loci proximity (Fig. 1b, available as supplementary data at Bioinformatics Advances online). This was done using the following heuristic (Fig. 3, available as supplementary data at Bioinformatics Advances online). (i) All AMR hits are mapped to locus coordinates. (ii) The AMR hits are then perfectly matched to the CARD database based on either reference_accession, gene_symbol, gene_names or synonyms. The CARD database was chosen because it is highly curated, updated frequently and has a standardized AMR ontology to facilitate the consolidation of AMR annotation and naming conventions. Hits that did not have a CARD equivalent underwent blast-based searches. BLASTP, BLASTX, and BLASTN searches were performed against the CARD protein and nucleotide homolog and variants databases. The best quality blast alignment became the CARD match. (iii) If a query locus has only a single AMR tool hit, then its associated AMR tool annotation or CARD database match (if mapped), is reported as the consolidated term. (iv) If a query locus has multiple tool matches, then priority is given to hits with CARD exact mapping, (v) followed by AMR tool exact matches. (vi) For loci with multiple non-exact mappings, priority is given to the match with the CARD mapping with highest percent identity, (vii) followed by the AMR mappings. The pipeline returns the final output of the term consolidation WDL as (i) HARMONIZED_TERMS.tsv, which contains all the AMR hits with their CARD equivalents, and (ii) CONSOLIDATED_TERMS.tsv, which is a smaller file that contains the final decision of the identity of all the loci, ready for analysis. Users can judge the quality of the CONSOLIDATED_TERMS.tsv loci by observing the number of hits it received, the number of hits that congruently agreed on its identity and the BLAST percent identity given to the hit. The output files HARMONIZED_TERMS.tsv and CONSOLIDATED_TERMS.tsv from the cAMRah term consolidation as well as hamronize_amr_output.tsv from hARMonize are provided as File 2, available as supplementary data at Bioinformatics Advances online, and located on the GitHub wiki at 2.iii.a and 2.iii.b.
3 Results
cAMRah can run multiple AMG prediction tools on assembled bacterial genomes and harmonize to single genetic loci with standardized AMG ontologies assigned using a flexible, platform-independent and cloud-enabled workflow. This includes the ability to utilize the BV-BRC k-mer-based novel AMG prediction software, which can infer potential AMR phenotypes to hypothetical proteins. In addition to AMG-finding, this workflow can be adapted to predict virulence factors.
Supplementary Material
vbag017_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Afolayan AO, , Bernal JF, , Gayeta J Met al Overcoming data bottlenecks in genomic pathogen surveillance. Clin Infect Dis 2021;73:S 267–74. 10.1093/cid/ciab 78534850839 PMC 8634317 · doi ↗ · pubmed ↗
- 2Alcock BP , Huynh W, Chalil R et al CARD 2023: expanded curation, support for machine learning, and resistome prediction at the comprehensive antibiotic resistance database. Nucleic Acids Res 2023; 51: D 690–9.36263822 10.1093/nar/gkac 920PMC 9825576 · doi ↗ · pubmed ↗
- 3Antibiotic Resistance Threats in the United States. 2013. https://www.cdc.gov/drugresistance/threat-report-2013/index.html (18 June 2018, date last accessed).
- 4Baichoo S , Souilmi Y, Panji S et al Developing reproducible bioinformatics analysis workflows for heterogeneous computing environments to support African genomics. BMC Bioinformatics 2018;19:457.30486782 10.1186/s 12859-018-2446-1PMC 6264621 · doi ↗ · pubmed ↗
- 5Baker KS , Jauneikaite E, Hopkins KL et al; SEDRIC Genomics Surveillance Working Group. Genomics for public health and international surveillance of antimicrobial resistance. Lancet Microbe 2023; 4: e 1047–55.37977162 10.1016/S 2666-5247(23)00283-5 · doi ↗ · pubmed ↗
- 6Baykal PI, , Łabaj PP, , Markowetz F, et al Genomicreproducibility in the bioinformatics era. Genome Biol 2024;25:213.39123217 10.1186/s 13059-024-03343-2PMC 11312195 · doi ↗ · pubmed ↗
- 7Bortolaia V , Kaas RS, Ruppe E et al Res Finder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother 2020;75:3491–500.32780112 10.1093/jac/dkaa 345PMC 7662176 · doi ↗ · pubmed ↗
- 8Duan B , Zeng X, Peng J. Advances in genotypic antimicrobial resistance testing: a comprehensive review. Sci China Life Sci 2025;68:130–43.39300049 10.1007/s 11427-023-2570-4 · doi ↗ · pubmed ↗