Significant but Temporary Efficacy of Statin for a Patient With Severe Autoimmune Pulmonary Alveolar Proteinosis: A Case Report
Fumihiko Makino, Kohei Shibayama, Yuki Muto, Rie Hayakawa, Kenta Izumi, Yohei Suzuki, Osamu Nagashima, Shinichi Sasaki, Kazuhisa Takahashi

TL;DR
A patient with severe autoimmune lung disease showed temporary improvement with statin therapy after standard lung lavage treatments.
Contribution
This case report demonstrates the temporary efficacy of oral statin therapy in a severe autoimmune pulmonary alveolar proteinosis patient.
Findings
The patient showed significant clinical improvement in oxygen saturation and pulmonary function after starting statin therapy.
Respiratory failure relapsed two years after initiating statin therapy, indicating temporary efficacy.
Statin therapy provided a promising but not long-term solution for severe autoimmune pulmonary alveolar proteinosis.
Abstract
Autoimmune pulmonary alveolar proteinosis (APAP) is a rare autoimmune lung disorder characterised by the presence of anti‐granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) antibodies. Whole‐lung lavage (WLL) therapy remains the standard treatment for severe cases. Recently, inhaled GM‐CSF therapy has been approved in Japan; however, the cost of the treatment remains a limiting factor. Several reports have suggested that oral statin therapy may be a promising therapeutic option for APAP. Herein, we report a case of severe APAP that underwent WLL therapy twice and achieved an excellent response and remarkable clinical resolution of respiratory failure after the initiation of oral statin therapy. Remarkable improvements in oxygen saturation, blood gas analysis, serum biomarker levels and pulmonary function test results were observed after statin administration. However, the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
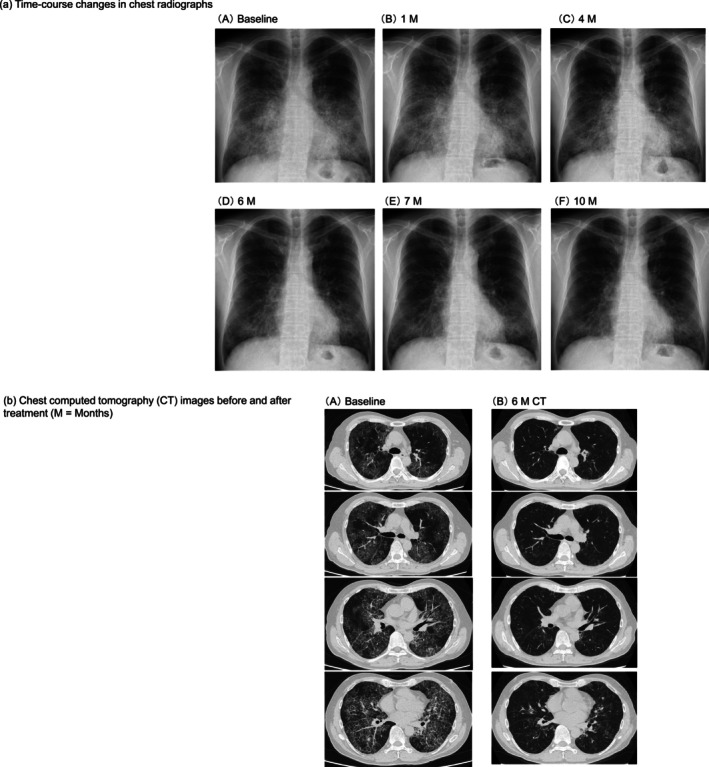 FIGURE 1
FIGURE 1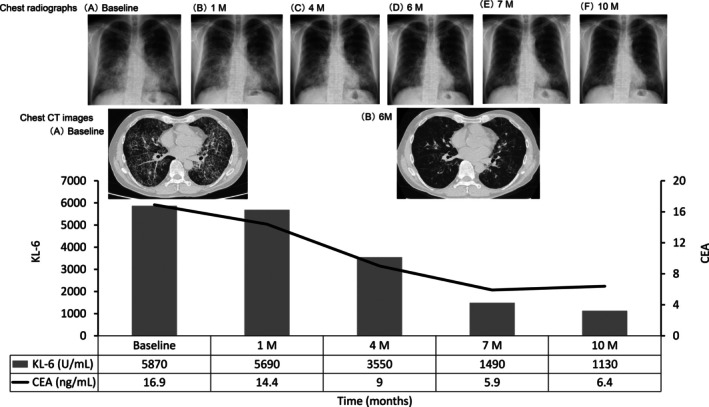 FIGURE 2
FIGURE 2| (a) Disease activity markers | |||||
|---|---|---|---|---|---|
| Biomarker | Baseline | 1‐month follow‐up | 4‐month follow‐up | 7‐month follow‐up | 10‐month follow‐up |
| KL‐6 (U/mL) | 5870 | 5690 | 3550 | 1490 | 1130 |
| SP‐D (ng/mL) | 330 | 244 | 238 | 125 | 111 |
| LDH (IU/L) | 260 | 231 | 183 | 178 | 166 |
| CEA (ng/mL) | 16.9 | 14.4 | 9.0 | 5.9 | 6.4 |
| Pulmonary function test | Baseline | 7‐month follow‐up |
|---|---|---|
| VC (L) | 3.05 | 3.63 |
| FEV1 (L) | 2.35 | 2.65 |
| FVC % pred (%) | 72.1 | 87.0 |
| FEV1% pred (%) | 68.3 | 78.3 |
| DLCO % pred (%) | 54.6 | 61.4 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInterstitial Lung Diseases and Idiopathic Pulmonary Fibrosis · Pulmonary Hypertension Research and Treatments · Neonatal Respiratory Health Research
Introduction
1
Pulmonary alveolar proteinosis (PAP) is a rare pulmonary disorder characterised by progressive respiratory failure due to excessive accumulation of surfactant proteins and lipids in the alveoli and peripheral airways. PAP is classified into three types based on the aetiology: autoimmune PAP (APAP), secondary PAP and congenital/hereditary PAP. Among these, APAP is the most common, accounting for approximately 92% of all cases. Additionally, APAP is caused by the presence of anti‐granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) antibodies, which disrupt the GM‐CSF signalling pathway. This impairment disturbs the maturation and functional maintenance of alveolar macrophages [1].
The pathophysiology of APAP involves the impaired clearance of surfactant in the alveoli as a result of reduced GM‐CSF‐dependent cholesterol mechanism in alveolar macrophages [1, 2, 3].
Whole‐lung lavage (WLL) therapy is the standard treatment for severe cases of PAP and is highly effective [1]. However, the treatment modality is invasive and can only be performed in limited facilities. Recently, inhaled GM‐CSF was approved in Japan as a viable option for APAP. However, the high cost of APAP remains a significant concern.
Evidence on statin efficacy in PAP is limited and remains controversial [4, 5]. In this report, we present a case of APAP that recovered from respiratory failure following statin therapy and explore the potential role of statins in PAP management.
Case Report
2
The patient, a 60‐year‐old man, was diagnosed with APAP at a prior hospital 2 years ago. At the initial visit, he presented with progressive dyspnoea on exertion (DOE). Chest radiography during a routine health checkup revealed ground‐glass opacities (GGO) in both lungs. Computed tomography demonstrated a crazy‐paving pattern, while bronchoscopy revealed white, milk‐like bronchoalveolar fluid (BALF). Blood tests revealed anti‐GM‐CSF antibodies at a level of 46.9 μg/mL, confirming the diagnosis of APAP. His condition gradually worsened. He underwent four partial lung lavages 16 months ago, followed by one unilateral lung lavage 6 months ago. The patient was transferred to our hospital due to relocation.
At the initial visit to our hospital, he presented with chronic respiratory failure, an oxygen saturation of 89% at room temperature and dyspnoea. Home oxygen therapy was not initiated, as the patient declined treatment. Blood tests revealed abnormalities in lipid metabolism (high low‐density lipoprotein [LDL] cholesterol, 204 mg/dL); therefore, statin pitavastatin calcium therapy (1 mg/day) was initiated.
One month after initiating statin therapy, not only did LDL cholesterol levels improve, but lung injury biomarkers (Krebs von den Lungen‐6, surfactant protein D, lactate dehydrogenase and carcinoembryonic antigen) also decreased. Additionally, a trend toward improvement in DOE was observed. In response, we increased the statin dose from 1 to 2 mg to further reduce lung injury caused by protein deposition in macrophages. Following statin dose escalation, the GGO areas on chest radiographs and CT scans demonstrated further improvement (Figures 1 and 2). One year after initiating statin therapy, the GGO on CT had nearly completely resolved. Oxygen saturation increased from 89% to 97% at room temperature, with the blood gas analysis confirming recovery from chronic respiratory failure (Table 1b). The elevated biomarkers returned to normal within 10 months (Table 1a), and pulmonary function tests demonstrated improvements in mixed ventilation dysfunction and diffusion impairment (Table 2). Ultimately, statin therapy resulted in the remission of PAP in the patient.
Sequential radiological imaging findings (M = Months). (a) Time‐course changes in chest radiographs at the following time‐points: (A) baseline, (B) 1 M, (C) 4 M, (D) 6 M, (E) 7 M and (F) 10 M. (b) Chest computed tomography (CT) images before and after treatment (M = Months). (A) Baseline and (B) 6 M CT.
Time‐course changes in biomarkers and radiological images (M = Months). Chest radiographs at the following time‐points: (A) baseline, (B) 1 M, (C) 4 M, (D) 6 M, (E) 7 M and (F) 10 M.
However, the therapeutic efficacy of statin therapy appears to be transient. Approximately 2 years after beginning statin therapy, APAP aggravated again, leading to respiratory failure. Lung injury biomarkers increased again, with the following values: KL‐64600 U/mL, SP‐D 444 ng/mL, LDH 252 IU/L and CEA 25 ng/mL. Hence, the patient was transferred to another hospital for re‐administration of WLL.
Discussion
3
In this case, the patient demonstrated significant improvement in type I respiratory failure within 6–12 months after initiating statin therapy; however, the efficacy of therapy was not sustained. Unfortunately, the worsening of autoimmune PAP could not be avoided 2 years after the initiation of statin therapy. For hyperlipidaemia, statin therapy was initiated along with lifestyle counselling for dietary fat restriction. This resulted in notable improvement of the APAP. Paradoxically, this improvement may have caused the patient to let their guard down. Although medication adherence remained intact, the patient completely discontinued fat restriction and overindulged in food and drink as a rebound effect. This may have ultimately led to marked worsening of the APAP.
ATP‐binding cassette transporters ABCA1 and ABCG1 mediate cholesterol efflux from alveolar macrophages [1, 2, 3, 5]. In PAP, the messenger ribonucleic acid (mRNA) transcription levels of ABCA1 and ABCG1 are reduced [1, 2, 3, 5]. This results in impaired GM‐CSF‐dependent cholesterol clearance by alveolar macrophages, leading to foamy body formation accompanied by excessive free and esterified cholesterol. Cormac et al. reported that statins increase the mRNA levels of ABCA1 and ABCG1 by inhibiting 3‐hydroxy‐3‐methylglutaryl‐CoA reductase, thereby increasing the expression of sterol regulatory element‐binding protein‐2 in alveolar macrophages and promoting the clearance of pulmonary surfactants [1, 5].
Previous reports on autoimmune PAP have confirmed that the efficacy of statins lasts for approximately 1 year. Reports from China have also demonstrated that statins are effective in patients with APAP without dyslipidaemia; however, their efficacy is short‐lived. This may be because the presence of anti‐GM‐CSF antibodies induces the continuous formation of foamy macrophages through an autoimmune reaction, which may outweigh the benefits of statin therapy.
As the production of foamy macrophages induced by autoantibodies cannot be inhibited, disease progression is presumed to occur once intracellular lipid accumulation exceeds the improvement in cholesterol metabolism in alveolar macrophages achieved by statin therapy. Therefore, GM‐CSF supplementation is considered the only treatment with the potential to provide long‐term therapeutic benefits.
Traditionally, the standard treatment for severe PAP has been WLL; however, inhaled GM‐CSF has recently been approved in Japan as a new option for APAP. Compared to conventional WLL, inhaled GM‐CSF therapy is less invasive and can be administered at home; however, its high cost is a concern. Statins could serve as an effective treatment option to bridge the gap between WLL and inhaled GM‐CSF for severe APAP.
Author Contributions
Fumihiko Makino was responsible for patient management and drafting the initial manuscript. Kohei Shibayama, Yuki Muto, Rie Hayakawa, Kenta Izumi, Yohei Suzuki and Osamu Nagashima reviewed the clinical course and laboratory findings, interpreted the radiological images and critically revised the manuscript. Shinichi Sasaki and Kazuhisa Takahashi supervised the case study and approved the final version of the manuscript. All the authors contributed equally to the discussion of the results and approved the final version of the manuscript.
Funding
The authors have nothing to report.
Consent
The authors declare that written informed consent was obtained for the publication of this manuscript and accompanying images using the consent form provided by the journal.
Conflicts of Interest
K.T. is an Editorial Board member of Respirology Case Reports and a co‐author of this article. He was excluded from all editorial decision‐making related to the acceptance of this article for publication. The other authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1C. Mc Carthy , B. C. Carey , and B. C. Trapnell , “Autoimmune Pulmonary Alveolar Proteinosis,” American Journal of Respiratory and Critical Care Medicine 205 (2022): 1016–1035.35227171 10.1164/rccm.202112-2742 SOPMC 9851473 · doi ↗ · pubmed ↗
- 2A. Sallese , T. Suzuki , C. Mc Carthy , et al., “Targeting Cholesterol Homeostasis in Lung Diseases,” Scientific Reports 7 (2017): 1–14.28860566 10.1038/s 41598-017-10879-w PMC 5579240 · doi ↗ · pubmed ↗
- 3J. Perez‐Gil and T. E. Weaver , “Pulmonary Surfactant Pathophysiology: Current Models and Open Questions,” Physiology 25 (2010): 132–141.20551227 10.1152/physiol.00006.2010 · doi ↗ · pubmed ↗
- 4S. Shi , R. Wang , L. Chang , et al., “Long‐Term Follow‐Up and Successful Treatment of Pulmonary Alveolar Proteinosis Without Hypercholesterolemia With Statin Therapy: A Case Report,” Journal of International Medical Research 49 (2021): 1–9.10.1177/03000605211010046 PMC 811394233926277 · doi ↗ · pubmed ↗
- 5C. Mc Carthy , E. Lee , J. P. Bridges , et al., “Statins as a Novel Pharmacotherapy of Pulmonary Alveolar Proteinosis,” Nature Communications 9 (2018): 1–9.10.1038/s 41467-018-05491-z PMC 608144830087322 · doi ↗ · pubmed ↗