First multicenter real-world analysis of switching to next-generation enzyme replacement therapies in late-onset Pompe disease
Daniel H. Mendelsohn, Angela Rosenbohm, Anne-Katrin Güttsches, Cornelia Kornblum, Karl Christian Knop, Tanja Fangerau, Nam Nguyen-Younossi, Guljan Shahyrova, Natalia Garcia-Angarita, Benedikt Schoser, Stephan Wenninger

TL;DR
This study examines the real-world outcomes of switching enzyme replacement therapies in late-onset Pompe disease, finding that transitions are generally feasible and maintain clinical stability.
Contribution
The study provides the first multicenter real-world evidence on switching next-generation enzyme replacement therapies in late-onset Pompe disease.
Findings
Transitions between enzyme replacement therapies were associated with stable respiratory function and minimal changes in activity levels.
No significant effects of time or switch type were observed, with baseline performance being the main predictor of outcomes.
Switching therapies was generally feasible and linked to clinical stability in patients with late-onset Pompe disease.
Abstract
Next-generation enzyme replacement therapies (ERTs) for late-onset Pompe disease (LOPD), including avalglucosidase alfa and cipaglucosidase alfa with miglustat, have been developed to improve muscle targeting and enzyme stability. Real-world evidence on therapy switching between ERT preparations remains limited. A prospective, observational, multicenter cohort study was conducted across German neuromuscular centers. Adults with genetically confirmed LOPD who transitioned to avalglucosidase alfa (Aval) or cipaglucosidase alfa with miglustat (Cipa/Mig) between August/2022 and September/2024 were included. Clinical data were extracted from medical records using a standardized case report form. Following EPOC recommendations, data captured comprised six-minute walk test (6MWT), ten-meter walk test (10MWT), Rasch-built Pompe-specific Activity scale (R-PAct), upright and supine forced vital…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
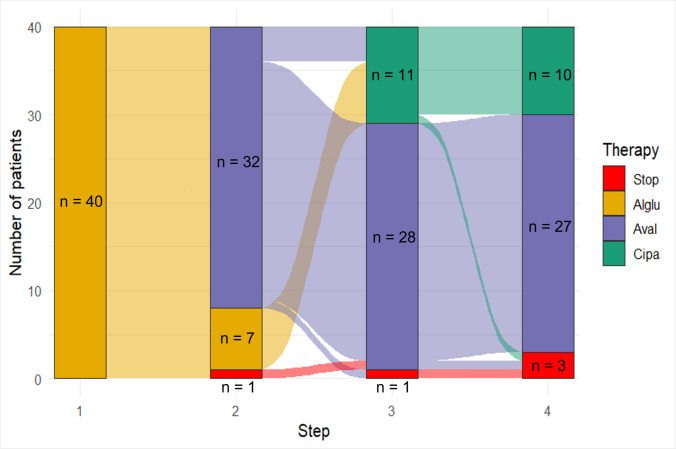 Figure 1
Figure 1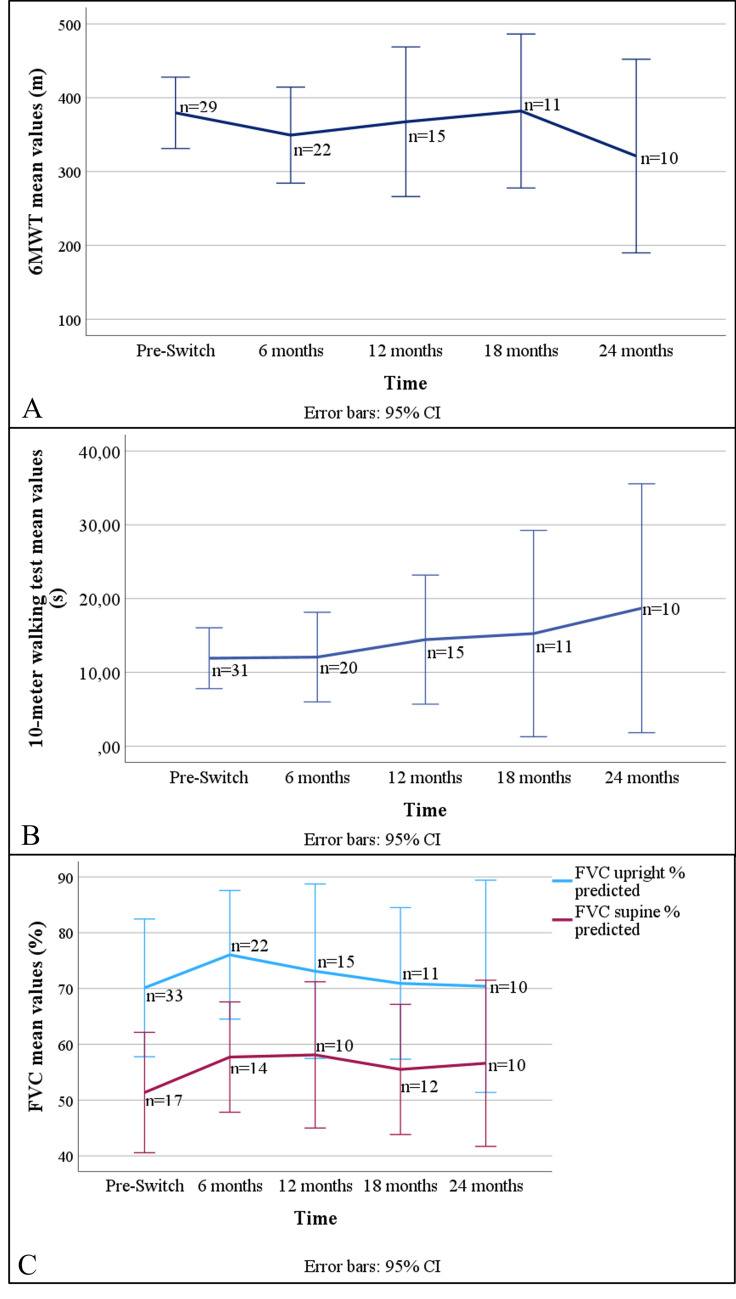 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLysosomal Storage Disorders Research · Glycogen Storage Diseases and Myoclonus · Whipple's Disease and Interleukins