Why 11β-HSD1 inhibitors show variable efficacy in Alzheimer's therapy: an APOE4-dependent HSD11B1 mechanism
RaiHua Lai, FengShiun Shie, RenHua Chung, Paul WeiChe Hsu, YiChung Chen, KaHei Lam, JyhLyh Juang

TL;DR
This study explains why 11β-HSD1 inhibitors have mixed results in Alzheimer's treatment, showing that their effectiveness depends on whether a person carries the APOE4 gene variant.
Contribution
The study identifies an APOE4-dependent mechanism linking HSD11B1 activity to Alzheimer's pathology and cortisol metabolism.
Findings
APOE4 carriers show increased HSD11B1 expression in the entorhinal cortex, linked to higher cortisol and AD risk.
APOE4 upregulates HSD11B1 via C/EBPβ, increasing neuronal cortisol production.
A functional HSD11B1 variant is associated with elevated cortisol and accelerated brain atrophy in APOE4 carriers.
Abstract
Rationale: Clinical trials for Alzheimer's disease (AD) often yield inconsistent results despite promising preclinical findings. Inhibition of 11β-hydroxysteroid dehydrogenase type 1 (HSD11B1), a cortisone reductase, has demonstrated neuroprotective effects in preclinical models. However, clinical outcomes have varied. A potential explanation is the limited representation of apolipoprotein E ε4 (APOE4) carriers in preclinical studies, despite evidence that APOE4 alters stress responses and glucocorticoid regulation. We hypothesized that APOE4 status modulates the efficacy of HSD11B1 inhibition by influencing cortisol metabolism and AD pathology. Methods: We conducted a genetic association study to test whether HSD11B1 variants are linked to plasma cortisol levels, brain atrophy, and AD risk, stratified by APOE4 status. Postmortem human brain tissues and wild-type mice were analyzed for…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
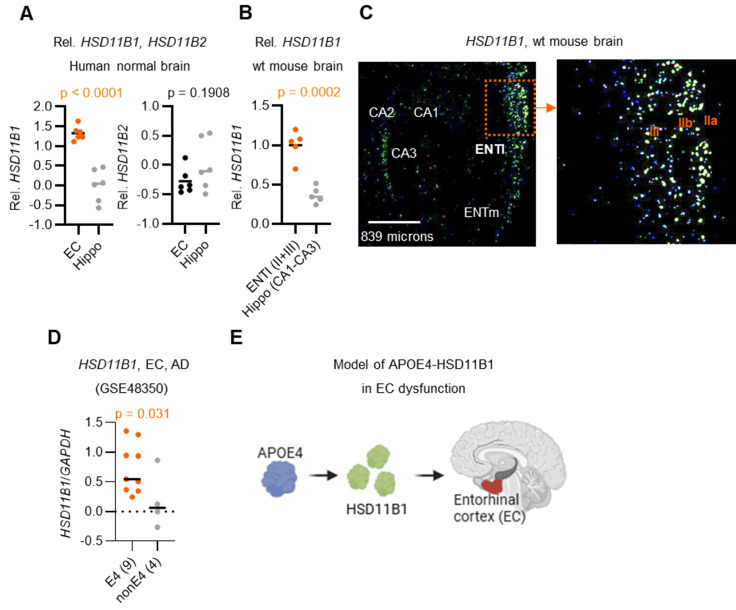 Figure 1
Figure 1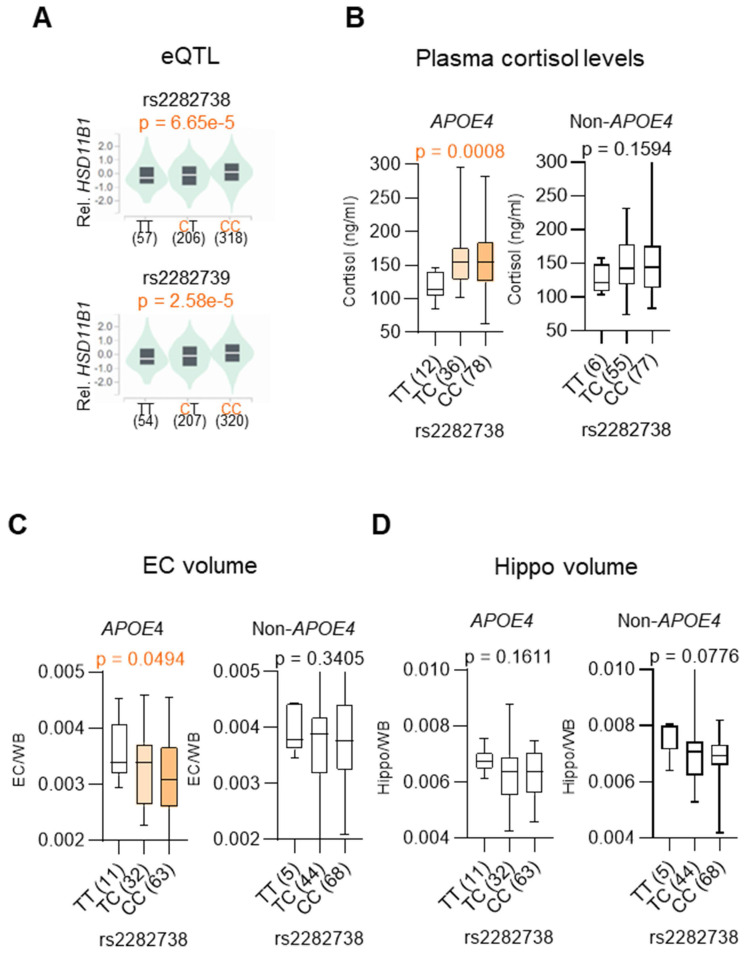 Figure 2
Figure 2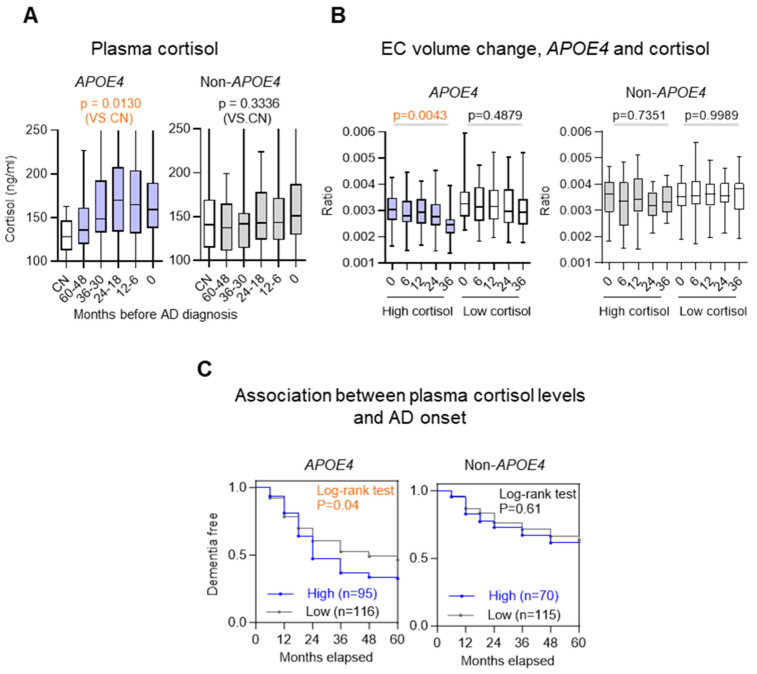 Figure 3
Figure 3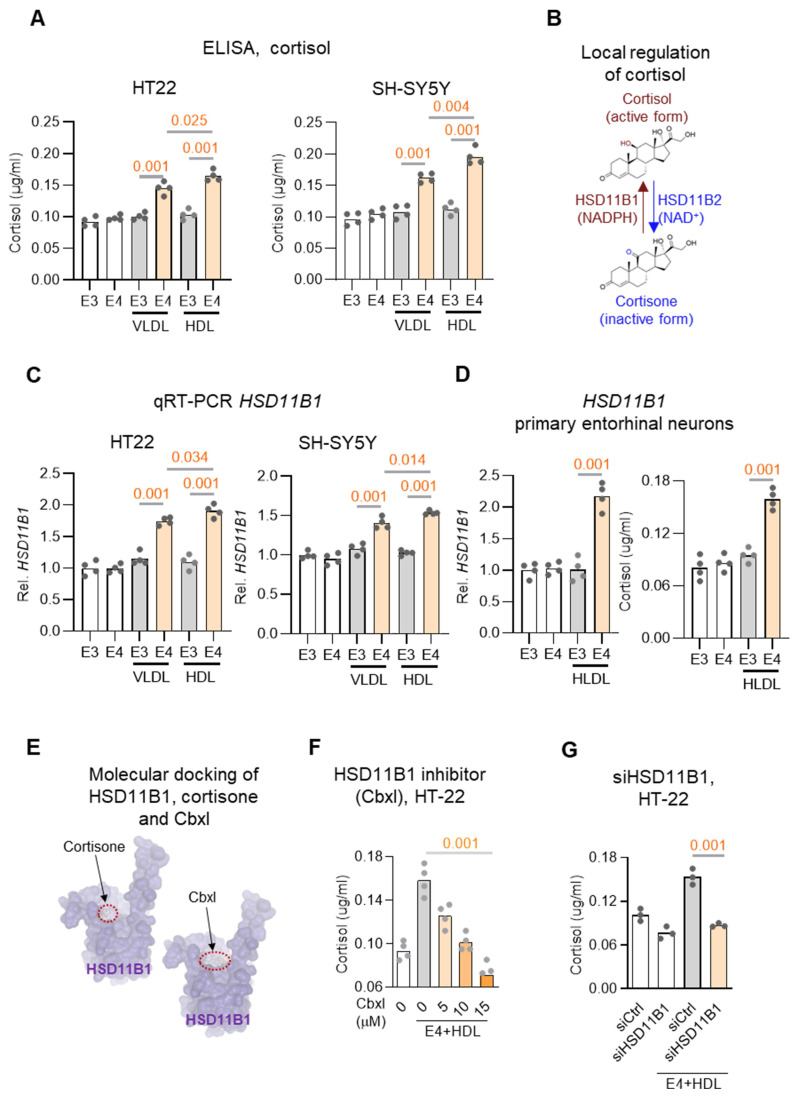 Figure 4
Figure 4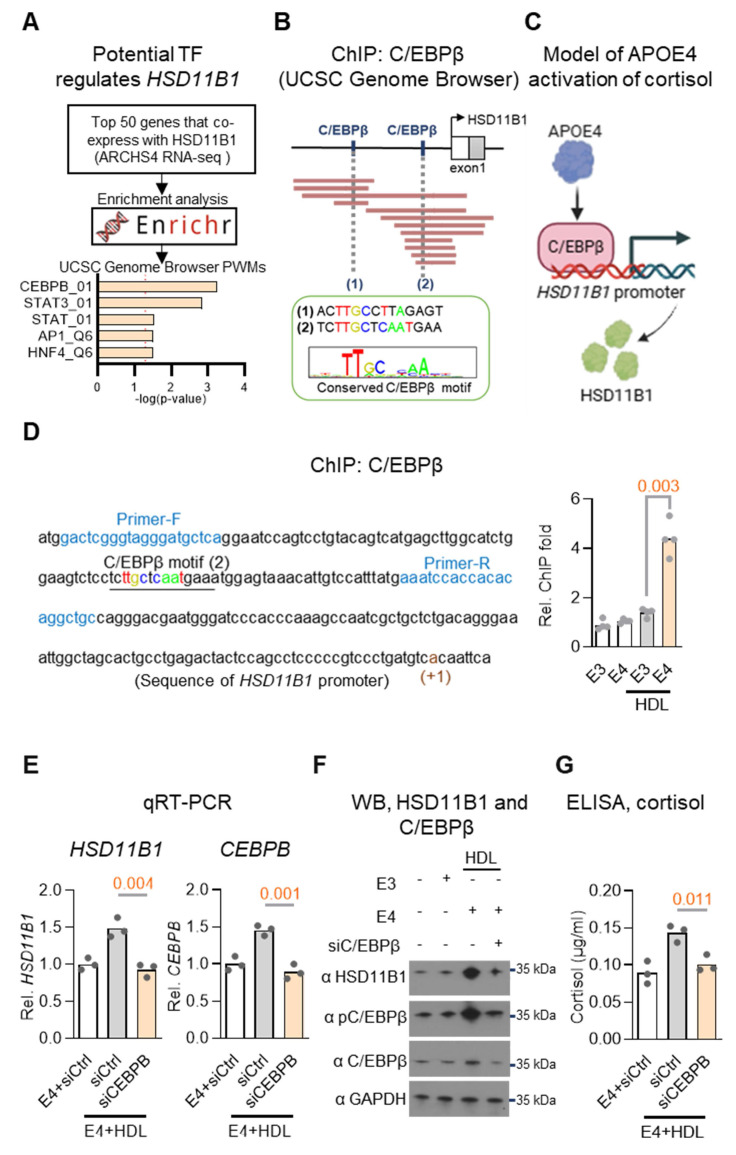 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHormonal Regulation and Hypertension · Stress Responses and Cortisol · Estrogen and related hormone effects
Introduction
Genetic variation substantially influences susceptibility to and progression of AD, among which the APOE4 allele has been consistently identified as the predominant risk factor for late-onset AD. Carriers of the APOE4 allele exhibit an increased multifaceted propensity for amyloid-β accumulation, heightened neuroinflammation, disrupted lipid metabolism, and impaired clearance of toxic proteins in the brain 1, 2. Despite the significant impact of APOE4 on AD pathogenesis, there remains a notable lack of understanding of APOE4-associated signaling mechanisms and therapeutic strategies specifically tailored to carriers of this genotype. Current clinical interventions largely adopt a one-size-fits-all approach, overlooking the distinct molecular and cellular pathways affected by this genotype. The absence of targeted therapies not only limits treatment efficacy in APOE4-positive individuals but may also contribute to the inconsistent outcomes observed in clinical trials. This highlights an urgent need to develop genotype-informed therapeutic approaches that address the unique biological mechanisms associated with APOE4 and improve outcomes for this high-risk population.
Accumulating evidence suggests that APOE4 modulates HPA-axis function, perturbs glucocorticoid receptor (GR) signaling, and confers greater stress sensitivity 3. Actually, cortisol levels in the body can be elevated through two primary mechanisms. The first mechanism entails activation of the HPA axis, leading to stress-induced cortisol secretion from the adrenal glands into the circulation. The second, more local pathway operates at the cellular level, where 11β-hydroxysteroid dehydrogenase type 1 (HSD11B1) regenerates active cortisol by reducing cortisone 4. HSD11B1 functions as a NADPH-dependent reductase primarily in metabolic and neural tissues, including the brain. In this context, HSD11B1 enables tissue-specific amplification of glucocorticoid signaling independent of systemic HPA activation, thereby playing a critical role in modulating local stress responses and potentially contributing to region-specific neuronal vulnerability. In addition to the role in glucocorticoid metabolism, emerging evidence reveal that HSD11B1 also modulates neuroinflammation and cognitive decline, making it an attractive target for AD therapy 5. However, current understanding of HSD11B1's role in local cortisol signaling in AD brain remains limited.
Emerging evidence suggests that the long-term elevation of glucocorticoids increases the risk and accelerate the progression of AD 6, 7. APOE4 carriers exhibit early pathological stress and dysregulated cellular events during MCI, which may be more amenable to therapeutic intervention than the widespread neurodegeneration seen in advanced AD. Although cortisol functions as a stress hormone in the context of AD, the mechanistic link between APOE4, cortisol activation, and the brain subregions affected in early AD remains incompletely understood.
In this study, we focus on the response of EC to dysregulated cortisol signaling in AD. The EC lies in the medial temporal lobe, immediately rostral to the hippocampus, acts as a key interface between the neocortex and hippocampus 8. APOE4-related changes in the EC are detectable even in youth, and EC atrophy is one of the earliest structural hallmarks of AD 9, 10, raising the question of whether APOE4 engages EC-specific pathways. Supporting a direct role for stress hormones, glucocorticoids rapidly alter microcircuit activity in layer II of the medial EC, reducing inhibitory synaptic events and enhancing α1-adrenergic responses in principal cells 11. Anatomical studies show high levels of GR mRNA and protein in the adult EC 12, and EC manipulation can influence hippocampal GR expression 13, highlighting tight functional coupling within this network. Together, these findings suggest that cortisol may contribute to AD progression via its effects on EC circuitry.
Notably, convergent preclinical work shows that lowering brain 11β-HSD1 activity reduces local glucocorticoid amplification and improves memory and neuropathology 14, 15. In contrast, randomized clinical trials in AD have been largely negative at primary endpoints; for example, ABT-384 did not improve cognition versus placebo 16, although biomarker enriched analyses suggest possible signals, such as with Xanamem in participants with elevated plasma pTau181 17. This gap between preclinical and clinical findings suggests that host modifiers of glucocorticoid tone may shape treatment response. Given that APOE4 carriers display altered stress and glucocorticoid regulation and greater stress related cognitive vulnerability 3, 18, 19, an approach that evaluates HSD11B1 inhibition with stratification by APOE4 status and guidance by biomarkers is biologically plausible and may improve therapeutic yield.
In this study, we aim to address the translational gap between the promising effects of 11β-HSD1 inhibition observed in preclinical models and the inconsistent outcomes seen in AD clinical trials. We hypothesize that APOE4 status modulates the therapeutic efficacy of HSD11B1 inhibition by altering cortisol metabolism and contributing to AD pathology. To test this hypothesis, we performed a multimodal integrative analysis stratified by APOE4 genotype, incorporating genetic association studies, neuroimaging data analysis, plasma cortisol measurements, EC atrophy assessment, and evaluation of MCI-to-AD progression. Our findings suggest that APOE4 genotype may influence individual responses to HSD11B1 inhibitor therapy and provide mechanistic insight into an APOE4-dependent pathway involving C/EBPβ-mediated HSD11B1 upregulation and local cortisol dysregulation during early AD progression.
Materials and Methods
Cell lines and transfection
Mouse hippocampal neuronal cell line HT-22 (Sigma-Aldrich, SCC129) and the APOE3 (E3/E3) genotype human neuroblastoma cell line SH-SY5Y (ATCC, CRL-2266) were cultured in Dulbecco's Modified Eagle Medium (Gibco) and Minimum Essential Medium (Gibc), respectively, at 37°C in a 5% CO₂ humidified atmosphere. APOE genotyping was performed using DNA sequencing. The primers employed for amplification and sequencing were as follows: APOE_F: 5'-GAC CAT GAA GGA GTT GAA GGC CTA C-3' and APOE_R: 5'-CTC GCG GGC CCC GGC CTG GTA-3' 20. RNA interference-mediated knockdown of C/EBPB or HSD11B1 was achieved by transiently transfecting SH-SY5Y cells with either siC/EBPB, siHSD11B1 or a control siRNA (Stealth siRNA; Invitrogen) was transfected using DharmaFECT (Dharmacon).
Primary entorhinal cortical neuron cultures
Primary entorhinal cortical neuron cultures were prepared from postnatal day 1 C57BL/6J mouse pups of either sex, as previously described 21, 22. Brains were removed and placed in Hank's balanced salt solution. The entorhinal cortex was carefully microdissected under a stereomicroscope. Dissected tissues were pooled and enzymatically dissociated using a papain-based enzyme solution, followed by incubation with a trypsin inhibitor solution. After gentle trituration, a single-cell suspension was obtained and cells were seeded at a density of 5 × 10⁴ cells per well onto 48-well plates with pre-coated poly-D-lysine and laminin. The cells were maintained at 37 °C in a humidified incubator with 5% CO₂ in serum-free Neurobasal medium supplemented with 2% B27, 1% L-glutamine (2 mM), and 1% penicillin/streptomycin. Twenty-four hours after seeding, one-third of the medium was replaced with fresh medium, followed by medium changes every 3-4 days thereafter. Neuronal enrichment of the cultures was confirmed by immunolabeling with the neuronal marker MAP-2. Experiments were conducted at 6-10 days in vitro (DIV). The animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the National Health Research Institutes (NHRI) under protocol number NHRI-IACUC-103136-A and NHRI-IACUC-114027.
Cortisol ELISA assay
Culture cells were treated with VLDL (25 μg/mL; Sigma-Aldrich, LP1) and HDL (25 μg/mL; Sigma-Aldrich, LP3) in the presence of human APOE3 (10 μg/mL, Sigma-Aldrich, SRP4696) or APOE4 (10 μg/mL, Sigma-Aldrich, A3234) for 3 days, following modifications from previous literature 23. Following this treatment, cortisone (0.4 μg/mL; Sigma-Aldrich, C2755) was added to the culture medium, either alone or in combination with carbenoxolone disodium (5-15 μM; MedChemExpress, HY-B1367), for an additional 24 hours. The culture medium was then collected, and cortisol levels were measured using the Cortisol ELISA Kit (Elabscience, Cat# E-EL-0157) according to the manufacturer's instructions.
Quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR)
qRT-PCR was performed with modifications based on previously published methods 22. The primers used were as follows: Human GAPDH-F: 5'-CCT GCC AAA TAT GAT GAC ATC AAG-3; Human GAPDH-R: 5'-ACC CTG TTG CTG TAG CCA AA-3'; Human HSD11B1-F: 5'-GTT ACG TGG TCC TGA CTG TAG C-3'; Human HSD11B1-R: 5'- GCA GCA ACC ATT GGA TAA GCC AC-3'; Human HSD11B2-F: 5'- CAA ACC AGG AGA CAT TAG CCG C-3'; Human HSD11B2-R: 5'- TCA CCT CCA TGC AGC TAC GGA A-3'; Mouse HSD11B1-F: 5'- CTC TGC TCA CTA CAT TGC TGG C-3'; Mouse HSD11B1-R: 5'- GAG ACA GCG AGG TCT GAG TGA T-3'; Mouse HSD11B2-F: 5'- CAG AGG ACA TCA GCC GTG TTC T-3'; Mouse HSD11B2-R: 5'- GAA AGT CGC CAC TGG AGA CAG T-3'; Mouse GAPDH-F: 5ʹ-CAT CAC TGC CAC CCA GAA GAC TG-3ʹ; Mouse GAPDH-R 5ʹ -ATG CCA GTG AGC TTC CCG TTC AG-3ʹ.
Identification of potential transcriptional regulators of HSD11B1
To identify potential transcriptional regulators of HSD11B1, we first identified genes co-expressed with HSD11B1, we analyzed ARCHS4 RNA-seq data from the Enrichr database (https://maayanlab.cloud/Enrichr/), which integrates publicly available transcriptomic datasets. The top 50 co-expressed genes were selected for further analysis. Next, we performed enrichment analysis using the Enrichr transcription factor (TF) database and UCSC Genome Browser Position Weight Matrices (PWMs) to identify enriched TF binding motifs in its regulatory regions.
To further validate potential TF regulation, chromatin immunoprecipitation sequencing (ChIP-seq) data from the ReMap database within the UCSC Genome Browser (https://genome.ucsc.edu/) was analyzed. This analysis focused on the presence of CCAAT/enhancer-binding protein beta (C/EBPβ) motifs in the HSD11B1 promoter region. The identified binding sites were visualized to confirm the potential regulatory role of C/EBPβ in HSD11B1 transcription.
Chromatin immunoprecipitation (ChIP) assay
Culture cells were treated with HDL (25 μg/mL) or recombinant human ApoE3 or APOE4 proteins (10 μg/mL) for 3 days and then ChIP assays were performed as described 24. Briefly, 2 × 10⁷ cells were cross-linked with 1% formaldehyde at 37°C, followed by quenching and washing with cold phosphate-buffered saline (PBS). A portion of the cells was reserved for input chromatin DNA analysis. Cells were lysed in 200 μL ChIP lysis buffer and incubated on ice. Chromatin was sonicated using a Bioruptor plus at low intensity for 10 pulses of 30 seconds each. The sonicated lysate was diluted to 2 mL with ChIP dilution buffer and precleared with 80 μL of protein A Plus agarose beads at 4°C for 1 hour. The precleared lysate was incubated overnight at 4°C with an anti-C/EBPβ antibody (4 μg), with an IgG-antibody control as a negative control. Immunocomplexes were collected using 60 μL of protein A Plus agarose beads and sequentially washed with low-salt buffer, and TE buffer. Complexes were eluted with 1% SDS and 0.1 M NaHCO₃, followed by cross-link reversal at 65°C and proteinase K digestion. DNA was extracted using phenol/chloroform, ethanol-precipitated, and resuspended in 40 μL distilled water. Finally, 1 μL of ChIP DNA was used for qPCR amplification on a QuantStudio 1 Real-Time PCR System with specific primers for HSD11B1: 5'- GAC TCG GGT AGG GAT GCT CA-3' and 5'- GCA GCC TGT GTG GTG GAT TT-3'.
Antibody
The antibodies used for Western blotting were as follows: anti-GAPDH, GeneTex, Cat# GTX100118; anti-HSD11B1, ABClonal, Cat# A1619; anti-APOE4, Cell Signaling Technology, Cat# 8941; anti-C/EBPβ, ABclonal, A0711; anti-pC/EBPβ (T235), Cell Signaling Technology, #3084.
Brain HSD11B1 and HSD11B2 expression data analysis
Brain expression data for HSD11B1 and HSD11B2 were obtained from the Allen Human Brain Atlas microarray database (https://human.brain-map.org/microarray/), using probes A_23_P63209 and A_23_P14986, respectively. The dataset comprises genome-wide microarray expression profiles from six neurologically normal adult donors (aged 24-57 years; five males and one female), covering multiple anatomically defined brain regions. To represent the entorhinal cortex (EC), samples from the lateral bank of the parahippocampal gyrus and the bank of the collateral sulcus were selected, as these regions are largely occupied by the entorhinal area 25. The hippocampus was defined to include the dentate gyrus and the CA1-CA4 subfields. Based on these anatomical definitions, HSD11B1 expression levels in the EC and hippocampus were calculated separately for each individual donor. In addition, in situ hybridization (ISH) data for HSD11B1 were retrieved from the Allen Mouse Brain Atlas (https://portal.brain-map.org/) and analyzed using experimental datasets 79762381 and 73520987.
Gene expression data for HSD11B1 in the EC were obtained from the GSE48350 dataset on NCBI, which includes microarray expression data from human AD brains. To ensure age-matching, a total of 13 AD subjects aged 85-94 years were selected for analysis, including 9 APOE4 carriers and 4 non-carriers. For comparison, the expression levels of HSD11B1 and CEBPB were analyzed between the two groups to assess the differences in expression in the EC tissues, with GAPDH used as an internal control.
HSD11B1 SNP analysis using ADNI and ROSMAP datasets
SNP data for HSD11B1 were obtained from the ADNI_1_GWAS_Plink file within the ADNI database and from the Religious Orders Study and Memory and Aging Project (ROSMAP) cohort. Baseline demographic characteristics of the study participants are summarized in Table S1. A total of 556 participants from the ADNI cohort and 1272 participants from the ROSMAP cohort were included in the analysis. Participants were first stratified by APOE4 status, and subsequently categorized into cognitively normal (CN) and late MCI/AD groups. Genotypic and statistical analyses were conducted using PLINK software, employing logistic regression models with adjustments for age and sex.
The association between the HSD11B1 SNP rs2282738 and cortisol levels, EC, and hippocampal (hippo) volumes in MCI patients was evaluated. A range of variables evaluating disease progression and cognitive function were obtained from the “ADNIMERGE.csv” file provided by ADNI. These included the time from MCI to AD diagnosis, APOE4 genotype information, and MRI brain volume data for the EC, hippocampus (Hippo), fusiform gyrus, whole brain (W) and blood cortisol level. Statistical significance was assessed using the Brown-Forsythe ANOVA test, with a threshold of p < 0.05. Ethical approval for all study protocols was granted by the Research Ethics Committee at the National Health Research Institutes (protocol no. EC1001103).
Cortisol, APOE4, and brain changes in MCI progression
We analyzed data from the ADNI cohort to investigate how plasma cortisol levels and APOE4 status affect brain volume changes and progression from MCI to AD. MCI participants were stratified by APOE4 genotype and baseline plasma cortisol levels (high vs. low). EC and hippocampal volumes were assessed at baseline and follow-up (6-36 months) using MRI, normalized to total brain volume. Differences in EC volume over time were compared using ANOVA. Separately, we examined how baseline cortisol levels relate to the time to AD conversion using Kaplan-Meier survival analysis, with log-rank tests assessing group differences. MCI subjects who progressed to AD within 3 months were excluded. Analyses were performed to evaluate whether cortisol-related effects differ by APOE4 status.
Statistical analysis
Details on sample size, central tendency, and significance are provided in figure legends or tables. Group comparisons were conducted according to data type, using two- or one-tailed t tests for continuous variables and χ^2^ tests for categorical variables. One-way ANOVA was used for comparisons among multiple groups, with Brown-Forsythe ANOVA applied when variance was unequal or outliers were present. MCI-to-AD progression was analyzed using Kaplan-Meier curves and log-rank tests. A p-value < 0.05 was considered significant.
Results
Association of HSD11B1 polymorphisms with AD risk is APOE4-dependent
Although HSD11B1 SNPs have been proposed to affect AD risk, these associations have not been consistently replicated 5. To determine whether APOE4 modifies the genetic contribution of HSD11B1 to AD pathogenesis, we examined the association between HSD11B1 genetic variants and AD susceptibility, stratified by APOE4 carrier status in two large independent clinical cohorts. Analysis of SNP array data from the ADNI cohort revealed two variants, rs2282738 and rs2282739, that were significantly associated with elevated AD risk among APOE4 carriers, but not in non-carriers. Similarly, two different SNPs (rs7539852 and rs6672256) in the ROSMAP cohort were identified to be also significantly associated with increased AD risk in APOE4 carriers (Table 1). We conducted linkage disequilibrium analysis and found all four SNPs were actually located within a single LD block in the EUR population. Among them, rs2282738 was selected as the tag SNP, while rs2282739, rs7539852, and rs6672256 showed strong linkage disequilibrium with rs2282738 (R² = 0.977, 0.886, and 0.886; GRCh38). This suggests that rs2282738 can be used as the representative variant for this locus in subsequent functional analyses. In contrast, no significant associations were observed among APOE4 non-carriers in either cohort. These results support our hypothesis that the genetic influence of HSD11B1 on AD risk may be specific to individuals carrying the APOE4 allele.
Predominant HSD11B1 expression in the EC of APOE4-positive brains
Because HSD11B1 mediates local cortisol regeneration, and that brain cortisol levels are known to increase with age 26, it is important to investigate whether local cortisol is elevated in AD, particularly in the EC and hippocampus, regions among the earliest and most severely affected by AD pathology 9. To explore this, we analyzed HSD11B1 expression patterns using publicly available data from the Allen Brain Atlas (Figure 1A, 1B). Unexpectedly, our analysis revealed that HSD11B1 expression was significantly higher in the EC compared to the hippocampus in both normal humans (p < 0.0001) and mouse brains (p = 0.0002). In the EC, HSD11B1 expression was highest in the lateral subdivision and was mainly detected in layer II/III cells of the mouse brain that morphologically resembled pyramidal neurons (Figure 1C, right). In contrast, HSD11B1 expression in the hippocampus was substantially lower and was primarily confined to the CA3 subfield (Figure 1C, left; Figure S1A). This finding is consistent with prior immunohistochemical evidence indicating that AD-related pathology initially arises in layer II neurons of the EC 27-29. These findings suggest that the selective enrichment of HSD11B1 in the EC may drive localized cortisol activation under normal conditions. Furthermore, its preferential localization to layers II and III highlights a potential mechanism by which the EC becomes particularly vulnerable to atrophy during AD progression.
Building on these findings, we next examined HSD11B1 expression in AD patients to assess whether genetic risk factors might further influence local cortisol activation. Importantly, the individuals carrying the APOE4 allele exhibited significantly higher levels of HSD11B1 in the EC compared to non-APOE4 carriers (Figure 1D). Notably, this APOE4-associated increase was not observed in the hippocampus (Figure S1B). These results further support our hypothesis that APOE4 may enhance local cortisol activation selectively within the EC, thereby exacerbating regional vulnerability in AD. Based on these observations, we propose a model in which APOE4 promotes HSD11B1 expression in the EC, leading to excessive cortisol regeneration and contributing to EC dysfunction and neurodegeneration in AD brains (Figure 1E).
Association of HSD11B1 polymorphisms with local cortisol activation and early EC atrophy
Given our earlier finding that the association between HSD11B1 and AD risk is specific to individuals carrying the APOE4 allele, we then examined whether AD-associated HSD11B1 SNPs represent functional variants that influence local gene expression 30. To address this, eQTL analysis was performed using data from the GTEx Portal (https://gtexportal.org/). We assessed whether AD-associated SNPs (rs2282738 and rs2282739) were associated with HSD11B1 expression. The analysis revealed significant eQTL associations for two SNPs, indicating that these variants are functionally linked to HSD11B1 gene expression (rs2282738: p = 6.65 × 10⁻⁵; rs2282739: p = 2.58 × 10⁻⁵) (Figure 2A).
Building on these findings, we next examined whether HSD11B1 SNPs influence downstream cellular effectors in an APOE4-dependent manner. Notably, rs2282738 was significantly associated with plasma cortisol levels among APOE4 carriers (p = 0.0008), but not among non-carriers (p = 0.1594) (Figure 2B). Specifically, APOE4 carriers with the rs2282738 CC or CT genotype showed a 1.34-fold increase in plasma cortisol, compared to a modest 1.18-fold increase in individuals lacking the APOE4 allele. These results suggest that the AD risk-associated rs2282738 variant may modulate systemic cortisol levels in a genotype-specific manner, with APOE4 potentially acting as an enhancer of this effect.
Given that elevated cortisol is linked to neurodegeneration, we further examined whether rs2282738 is associated with AD-related early brain atrophy, focusing on the EC and hippocampus. Using structural MRI data, we analyzed the correlation between rs2282738 genotypes and regional brain volumes. We observed a significant association between rs2282738 and EC volume (p = 0.0494), with a trend across TT, TC, and CC genotypes (Figure 2C), whereas no significant association was detected with hippocampal volume (Figure 2D). Although a similar trend was observed in APOE4 hippocampal, the effect was more pronounced in EC: those with the rs2282738 CC genotype exhibited a 9% reduction in median EC volume compared with TT carriers, whereas a 5% decrease in hippocampal volume relative to TT carriers. These findings suggest that rs2282738 interacts with APOE4 to exacerbate EC atrophy, with a comparatively stronger effect in this region than in the hippocampus.
These findings support a model in which HSD11B1 polymorphisms contribute to AD risk by promoting cortisol dysregulation and selective vulnerability of the EC. Collectively, our genetic and imaging analyses reinforce the hypothesis that HSD11B-driven cortisol activation may underlie APOE4-specific susceptibility to neurodegeneration in AD.
Elevated plasma cortisol levels predict accelerated EC atrophy and MCI-to-AD progression
Building on the observation that HSD11B1 SNPs are linked to both elevated plasma cortisol levels and selective EC vulnerability, we next investigated whether plasma cortisol could serve as a prognostic biomarker for EC atrophy and disease progression in individuals with APOE4. In APOE4 carriers, higher plasma cortisol was observed across diagnostic stages spanning MCI to AD compared with CN (p = 0.0130 vs CN), a pattern not evident in non-carriers (Figure 3A). To further validate this relationship, we stratified MCI subjects into high- and low-cortisol groups based on baseline plasma cortisol levels and tracked EC volume changes over 36 months (Figure 3B). Among APOE4 carriers, individuals with high cortisol showed significantly greater EC atrophy than those with low cortisol (p = 0.0043 vs. p = 0.4879). In contrast, plasma cortisol levels showed no significant association with changes in hippocampal volume, irrespective of APOE4 status (Figure S2).
We next extending these findings to clinical outcomes. We assessed whether elevated cortisol levels predict conversion from MCI to AD. Among APOE4 carriers, individuals with high baseline cortisol progressed to AD significantly faster than those with lower cortisol levels (χ² = 4.192, p = 0.04; HR = 1.464), whereas no significant association was found in non-carriers (χ² = 0.266, p = 0.61; HR = 1.133) (Figure 3C). Our findings are supported by prior studies demonstrating that the APOE4 allele shows a relationship with increased EC atrophy in both AD and MCI 31, 32. Together, these results suggest that elevated cortisol has a pronounced and APOE4-specific impact on EC atrophy and accelerates disease progression, supporting its potential utility as a prognostic biomarker in at-risk individuals.
APOE4 protein enhances local cortisone-cortisol conversion by upregulating HSD11B1 in neuronal cells
To functionally validate the mechanistic model derived from our genetic, transcriptomic, biochemical, and neuroimaging analyses in two clinical cohorts, we next turned to a neuronal cell-based system to test key components of the proposed APOE4-HSD11B1-cortisol signaling pathway. Specifically, we investigated whether APOE4 protein modulates local cortisol activation in neuronal cells.
APOE is indeed a major apolipoprotein found on both Very Low-Density Lipoproteins (VLDL) and High-Density Lipoproteins (HDL) and contributes to lipid transport and neuronal metabolism 33. To investigate whether HSD11B1 acts as a downstream target of APOE4 in regulating cortisol activation in neuronal cells, two neuronal-like cell lines (HT-22 and SH-SY5Y) were treated with recombinant APOE3 or APOE4, together with VLDL or HDL, to increase the availability of steroid hormone precursors. 23, 34. Under these conditions, APOE4 significantly increased cortisol levels compared to APOE3 (Figure 4A), supporting the idea that APOE4 directly promotes local cortisol production in neuronal cells, potentially via upregulation of HSD11B1.
Since local cortisol activation is regulated by the balance between two enzymes, HSD11B1, which converts cortisone to cortisol, and HSD11B2, which inactivates cortisol to cortisone (Figure 4B) 35, we next examined whether APOE4 alters their expression. qRT-PCR analysis revealed that APOE4 significantly upregulated HSD11B1 mRNA levels, while having no significant effect on HSD11B2 expression in either cell line (Figure 4C and Figure S3A-S3B). These results indicate that APOE4 selectively enhances the expression of HSD11B1, thereby promoting local cortisol activation.
To further validate that APOE4 can modulate neuronal responses in the EC, we microdissected the EC and established primary neuronal cultures, which were subsequently treated with APOE4/HDL to assess HSD11B1 expression (Figure 4D and Figure S4). Consistent with our observations in HT-22 and SH-SY5Y cells, APOE4/HDL markedly increased HSD11B1 expression in EC neurons. These findings further support that APOE4 enhances HSD11B1 expression, thereby promoting local cortisol activation.
To further validate the APOE4-induced increase in cortisol mediation is mediated by HSD11B1, we treated cells with carbenoxolone (Cbxl), an HSD11B1 inhibitor predicted by SwissDock to compete with cortisone at its binding site (Figure 4E). Cbxl treatment significantly reduced APOE4-induced cortisol levels (Figure 4F; Figure S3D). Consistently, HSD11B1 knockdown produced a similar reduction in APOE4-driven cortisol elevation (Figure 4G), further supporting a requirement for HSD11B1 in this process Together, these results provide functional evidence that HSD11B1 activity is necessary for the APOE4-driven increase in cortisol. These findings identify a key molecular mechanism by which APOE4 enhances local cortisol activation in neuronal cells, offering insight into how this pathway may contribute to neurodegeneration in AD.
Transcription factor C/EBPβ mediates APOE4-driven HSD11B1 expression
To investigate how APOE4 upregulates HSD11B1 expression, we sought to identify potential transcription factors involved in its regulation. We began by analyzing the ARCHS4 RNA-seq database to identify top genes co-expressed with HSD11B136. To further refine this list, we conducted an enrichment analysis using the Enrichr database 37. This analysis identified C/EBPβ as a strong candidate transcription factor potentially regulating HSD11B1 expression (Figure 5A), supported by prior studies indicating that C/EBPα and C/EBPβ potential act as transcriptional regulators of HSD11B1 38. To further support this finding, we examined ChIP-seq datasets available through the UCSC Genome Browser and identified C/EBPβ binding sites within the HSD11B1 promoter region (Figure 5B), reinforcing its potential regulatory role in HSD11B1 expression. Collectively, we proposed a mechanistic signaling pathway in which APOE4 activates C/EBPβ, which in turn upregulates HSD11B1, leading to increased local cortisol production (Figure 5C).
To experimentally validate the proposed regulatory pathway, we performed ChIP assays in SH-SY5Y cells co-treated with HDL and recombinant APOE3 or APOE4 proteins. This approach was designed to model lipid-rich conditions relevant to neuronal environments and assess isoform-specific effects of APOE on transcription factor binding. These experiments confirmed that APOE4, but not APOE3, significantly increased C/EBPβ binding at the HSD11B1 promoter (Figure 5D). We next assessed whether C/EBPβ is required for APOE4-induced HSD11B1 expression by knocking down C/EBPβ using siRNA in the same cell system. Combined qRT-PCR, western blot, and ELISA analyses revealed that APOE4, but not APOE3, significantly increased HSD11B1, C/EBPβ, phospho-C/EBPβ (T235), and cortisol levels. Importantly, silencing C/EBPβ abrogated all of these effects (Figure 5E-G), demonstrating that C/EBPβ is essential for APOE4-mediated upregulation of HSD11B1 and subsequent cortisol activation. These findings strongly support a mechanistic pathway in which APOE4 promotes local cortisol production via activation of the C/EBPβ-HSD11B1 axis. This pathway provides a molecular explanation for our earlier observations linking APOE4 to elevated cortisol levels and EC vulnerability in AD.
Building on this mechanistic insight, we propose a model in which APOE4 promotes local cortisol activation in the EC via C/EBPβ-mediated upregulation of HSD11B1. This APOE4-specific signaling cascade leads to elevated cortisol levels, which may contribute to selective EC vulnerability and disease progression in AD.
Discussion
To address the inconsistent efficacy of HSD11B1 inhibitors observed in AD clinical trials, we performed a comprehensive, multi-level analysis integrating genetic association data, transcriptomic profiling, neuroimaging, plasma cortisol measurements, and mechanistic in vitro studies. Our findings uncover a mechanistic link between APOE4 status, HSD11B1-mediated local cortisol activation, and the selective vulnerability of the EC in AD. Specifically, we demonstrate that APOE4 enhances C/EBPβ-driven expression of HSD11B1, which catalyzes the conversion of inactive cortisone to bioactive cortisol. This APOE4 → C/EBPβ → HSD11B1 → cortisol axis may promote region-specific cortisol accumulation in the EC, thereby contributing to neurodegeneration and cognitive decline in an APOE4-dependent manner. These results provide a potential mechanistic explanation for the variability in response to HSD11B1 inhibitors and suggest that APOE genotype should be considered in the design and stratification of future AD therapeutic trials targeting cortisol dysregulation.
These findings suggest that HSD11B1 inhibition may not serve as a universally effective therapeutic strategy for AD, but rather may offer greater benefit in APOE4-positive individuals whose disease involves cortisol dysregulation in selectively vulnerable brain regions such as the EC. This genotype-dependent mechanism provides a plausible explanation for the discrepancy between the robust efficacy observed in preclinical models of HSD11B1 inhibition 14, 15 and the inconsistent outcomes reported in human clinical trials 16. Unlike preclinical models, which are typically genetically homogeneous and often lack APOE4, human populations exhibit substantial genetic variability, particularly with respect to APOE genotype, which likely influences therapeutic response. These insights highlight the importance of incorporating APOE genotype stratification into future clinical trial designs and support the development of APOE4 humanized mouse models that more accurately recapitulate human AD pathophysiology.
APOE4 has been linked to activation of stress-responsive MAPK pathways, particularly p38 and JNK, which represent canonical cellular responses to proteotoxic and lipid-associated stress 34, 39-41. Together with prior evidence that p38/JNK can phosphorylate and activate C/EBPβ, the APOE4-associated increase in C/EBPβ phosphorylation suggests a potential APOE4-p38/JNK-C/EBPβ signaling route that may contribute to HSD11B1 upregulation. In addition, APOE4-driven signaling is likely modulated by its lipidation state and the local lipid-carrier environment. In the brain, APOE predominantly exists as lipidated, HDL-like particles, and variations in lipidation degree and lipid composition can influence receptor engagement and membrane-proximal signaling 42-44. Accordingly, HDL may enhance APOE4 association with neuronal membranes and amplify downstream stress signaling, providing a potential mechanistic basis for the augmented MAPK activation and HSD11B1 induction observed under lipid-rich conditions.
Moreover, the therapeutic potential of targeting HSD11B1 or upstream regulators such as C/EBPβ should be further explored in models that more closely mimic the human disease state. In parallel, future clinical trial designs could be optimized by incorporating APOE genotype stratification. For instance, a two-arm stratified design, in which APOE4 carriers and non-carriers are separately randomized to receive HSD11B1 inhibitors or placebo, would enable direct comparison of treatment efficacy across genotypes. In addition to cognitive assessments, monitoring biomarkers such as plasma or CSF cortisol/cortisone ratios, as well as neuroimaging endpoints (e.g., EC volume measured by structural MRI or PET-based markers of neurodegeneration), would provide objective readouts of therapeutic response.
A key question arises from this model: if HSD11B1 inhibitors are predicted to be most effective in APOE4 carriers, why do they also show strong efficacy in preclinical models lacking human APOE4? One explanation is that endogenous murine Apoe shares greater structural and functional similarity with human APOE4 than with APOE3. Evolutionarily, APOE4 is the ancestral allele, and murine APOE retains arginine residues, pro-inflammatory properties, and glucocorticoid response profiles that resemble APOE4 45, 46. These features may render mice more responsive to HSD11B1 inhibition, thereby partially explaining the discrepancy between preclinical and clinical results. However, most AD mouse models rely on overexpressed amyloid or tau pathology, which may override subtle genotype-specific effects, and drug efficacy could reflect alternative or compensatory pathways.
Several limitations should be acknowledged. Our genetic analyses are population-based and may be influenced by confounding factors such as sex, age, or comorbidities. In addition, while cell culture experiments provide causal evidence, they do not capture the full complexity of in vivo brain environments, including neuron-glia interactions. Future studies using brain region-specific HSD11B1 knockouts and APOE-genotype-specific mouse models will be important to validate this pathway in vivo. More specifically, APOE4-humanized mouse models in which HSD11B1 is either knocked out or overexpressed could provide direct evidence for the causal role of this pathway in disease progression. Such models would allow rigorous testing of whether APOE4-driven upregulation of HSD11B1 is sufficient to accelerate EC vulnerability and tau pathology in vivo.
Conclusions
This study uncovers a novel mechanistic link between the APOE4 genotype, cortisol metabolism, and the pathogenesis of AD. We show that HSD11B1-mediated glucocorticoid dysregulation contributes to entorhinal cortex vulnerability, offering a potential explanation for the persistent translational gap between preclinical findings and clinical outcomes in AD therapeutics. The identification of an APOE4-specific pathway highlights cortisol metabolism as a promising target and points toward genotype-stratified therapeutic strategies that could advance precision medicine in AD while informing broader approaches to aging and neurodegeneration.
Supplementary Material
Supplementary figures and table.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chen Y Jin H Chen J Li J Gaman MA Zou Z The multifaceted roles of apolipoprotein E 4 in Alzheimer's disease pathology and potential therapeutic strategies Cell Death Discov 2025113124062871610.1038/s 41420-025-02600-y PMC 12238274 · doi ↗ · pubmed ↗
- 2Belaidi AA Bush AI Ayton S Apolipoprotein E in Alzheimer's disease: molecular insights and therapeutic opportunities Mol Neurodegener 202520474027532710.1186/s 13024-025-00843-y PMC 12023563 · doi ↗ · pubmed ↗
- 3Peskind ER Wilkinson CW Petrie EC Schellenberg GD Raskind MA Increased CSF cortisol in AD is a function of APOE genotype Neurology 200156109481132018510.1212/wnl.56.8.1094 · doi ↗ · pubmed ↗
- 4Dammann C Stapelfeld C Maser E Expression and activity of the cortisol-activating enzyme 11beta-hydroxysteroid dehydrogenase type 1 is tissue and species-specific Chem Biol Interact 201930357613079690510.1016/j.cbi.2019.02.018 · doi ↗ · pubmed ↗
- 5Di Vincenzo M Pellegrino P Schiappa G Campanati A Del Vescovo V Piccirillo S Role of 11beta-Hydroxysteroid Dehydrogenase and Mineralocorticoid Receptor on Alzheimer's Disease Onset: A Systematic Review Int J Mol Sci 20252610.3390/ijms 26031357 PMC 1181839939941125 · doi ↗ · pubmed ↗
- 6Lesuis SL Weggen S Baches S Lucassen PJ Krugers HJ Targeting glucocorticoid receptors prevents the effects of early life stress on amyloid pathology and cognitive performance in APP/PS 1 mice Transl Psychiatry 20188532949136810.1038/s 41398-018-0101-2PMC 5830444 · doi ↗ · pubmed ↗
- 7Burke MR Sotiropoulos I Waites CL The multiple roles of chronic stress and glucocorticoids in Alzheimer's disease pathogenesis Trends Neurosci 202447933483930762910.1016/j.tins.2024.08.015PMC 11563862 · doi ↗ · pubmed ↗
- 8Hernandez-Frausto M Vivar C Entorhinal cortex-hippocampal circuit connectivity in health and disease Front Hum Neurosci 20241814487913937219210.3389/fnhum.2024.1448791 PMC 11449717 · doi ↗ · pubmed ↗