Targeting Lysosomes for Enhanced Anti-Cancer Therapeutics and Immune Response
Michal Stark, Yehuda G. Assaraf

TL;DR
This paper explores how targeting lysosomes can improve cancer treatments by enhancing immune responses and reducing tumor growth.
Contribution
The paper introduces innovative lysosome modulators and strategies to enhance cancer therapy outcomes.
Findings
Lysosomes can be targeted to reduce autophagy and improve cancer treatment efficacy.
Modulating lysosomes enhances immune response and inhibits tumor progression.
Nanoparticle technologies can specifically target tumors to minimize adverse effects.
Abstract
Cancer is a leading cause of death in Western countries. Apart from surgical resection, the primary treatment modalities chemotherapy and radiotherapy inflict serious side effects, and significantly remodel both tumor metabolism and the tumor microenvironment. This consequently compromises treatment efficacy, resulting in multiple drug resistance, immune evasion and cancer progression. Lysosomes are unique acidic intracellular organelles crucial for maintaining cellular health and homeostasis via degradation of cellular waste. Lysosomes are also required for autophagy, a stress-induced catabolic pathway that is important for cell survival. Autophagy is typically enhanced in tumor cells, as it can confer cyto-protection against the deleterious cytotoxic effects of chemotherapy, and suppress anti-cancer immune response. Owing to their acidic nature and their role in endocytosis, lysosomes…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
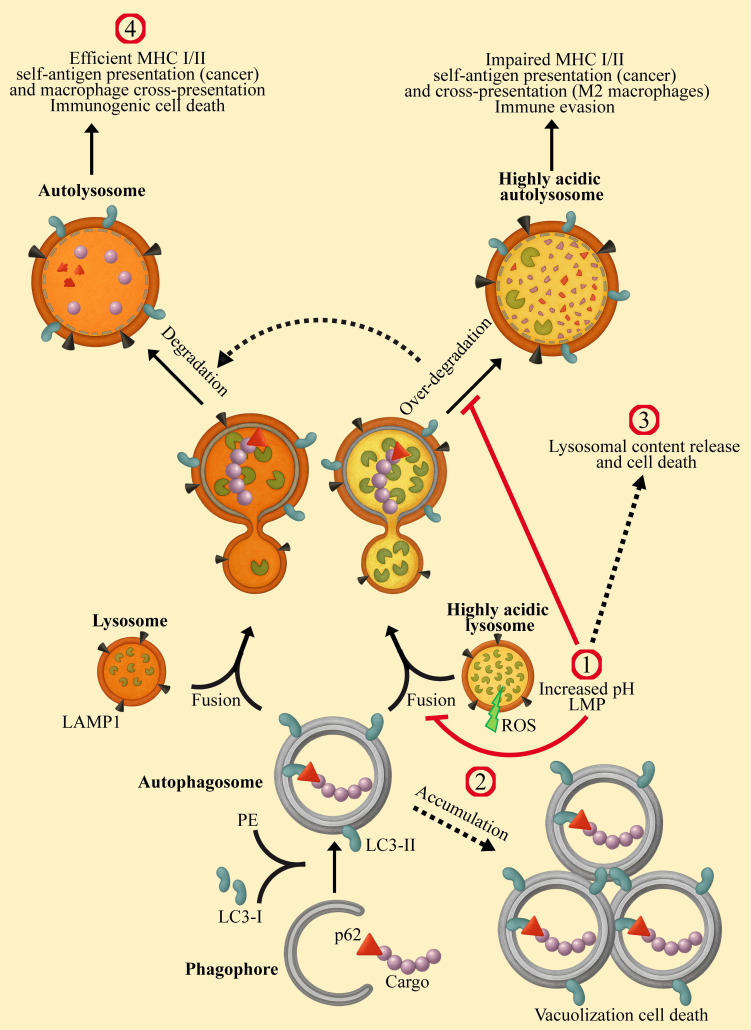 Figure 1
Figure 1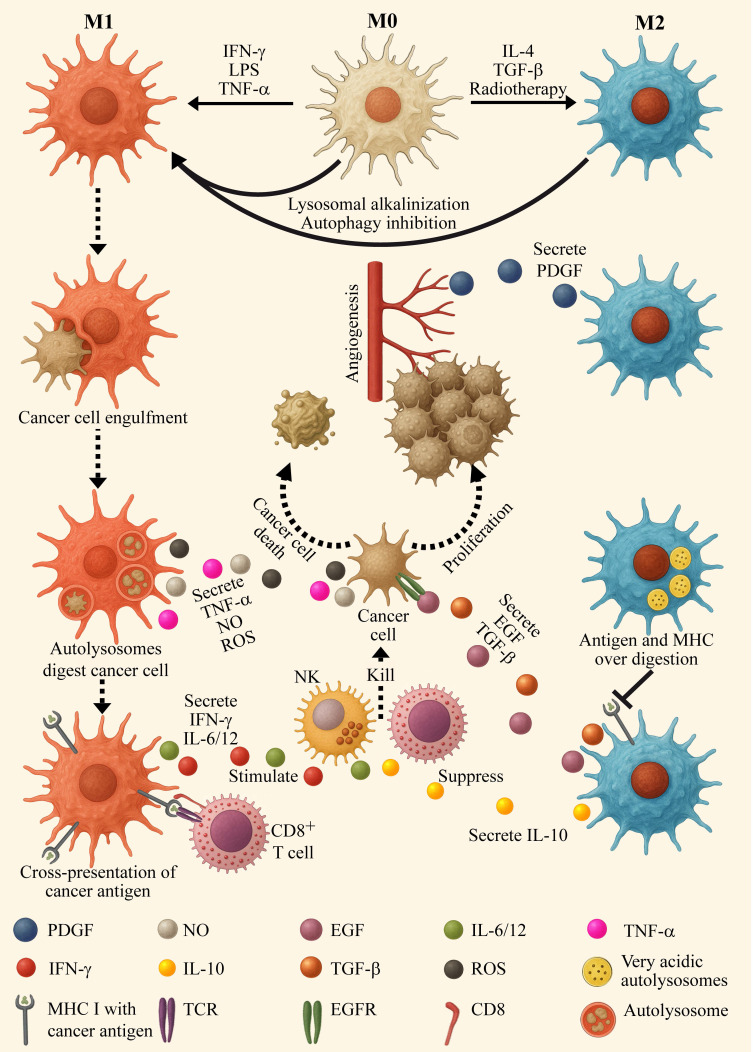 Figure 2
Figure 2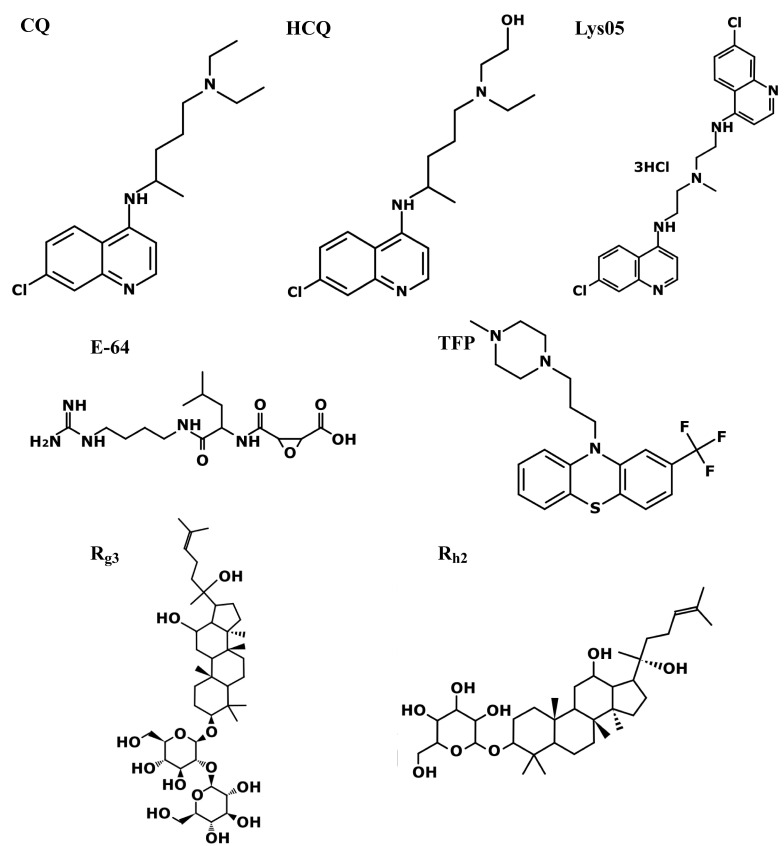 Figure 3
Figure 3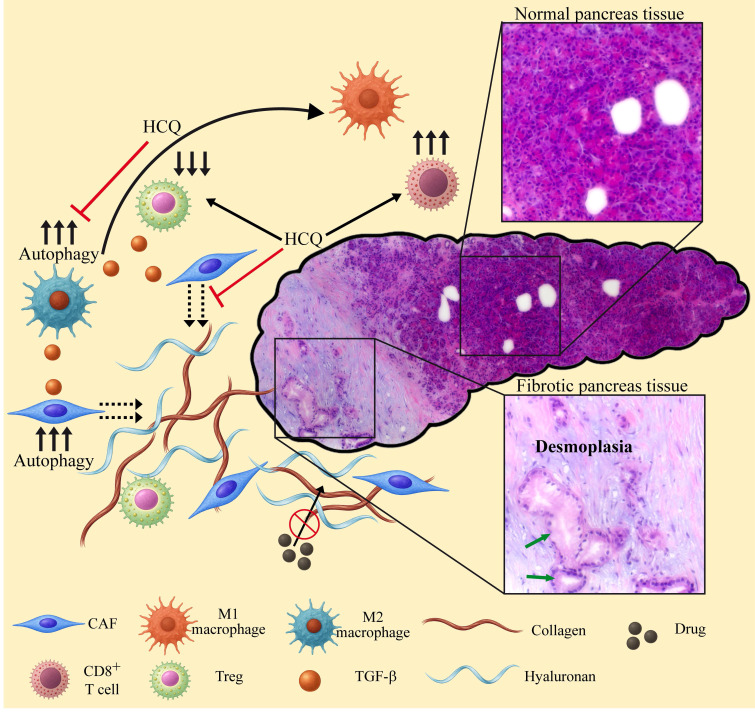 Figure 4
Figure 4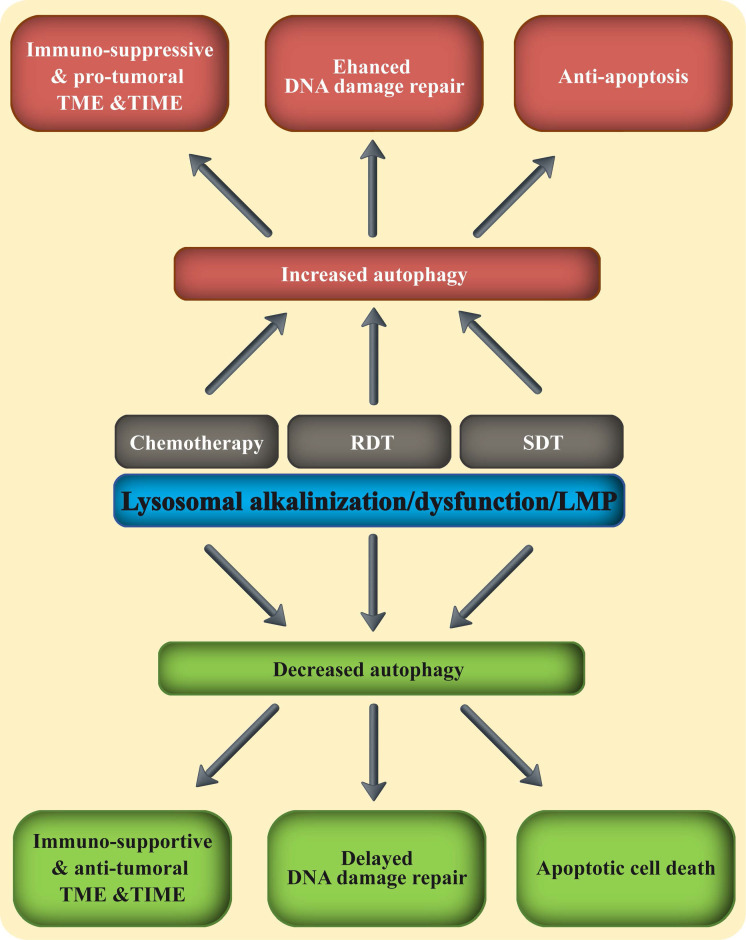 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAutophagy in Disease and Therapy · Cancer, Hypoxia, and Metabolism · Endoplasmic Reticulum Stress and Disease
Introduction
Lysosomes are eukaryotic membrane-bound cellular organelles with an acidic lumen at a pH range of 4.5-5.5 1-4, containing approximately 60 hydrolytic enzymes 5 displaying optimal activity at acidic pH 6-8. These hydrolytic enzymes, including proteases, nucleases, lipases, glycosidases, phospholipases, phosphatases, and sulfatases, are responsible for the degradation of proteins, nucleic acids, lipids, glycosides, and cellular debris, including damaged organelles, thereby maintaining cellular health and homeostasis 9-12. Apart from their major degradative role, lysosomes are central sensory hubs which respond to multiple cues to regulate metabolism, cell differentiation and division, as well as apoptosis and tumorigenesis 13-16. Furthermore, lysosomes are key components and regulators of the homeostatic autophagy process 17-19. The latter involves the sequestration of damaged organelles, misfolded proteins, intracellular pathogens and other foreign substances in double-membrane vesicles known as autophagosomes, which fuse with lysosomes for cargo degradation and recycling (Figure 1) 20-22. Autophagy promotes either cell survival or cell death 23-27. As a stress-induced catabolic pathway, autophagy facilitates the adaptation of cells to stress conditions such as starvation, by breaking down damaged or non-essential cellular structures and macromolecules to provide essential metabolites 28,29. Mitochondrial damage triggers mitophagy (mitochondrial autophagy), by which it protects the cell against release of pro-apoptotic proteins and reactive oxygen species (ROS) generation 30. Endoplasmic reticulum (ER) stress, following accumulation of unfolded proteins can lead to ROS generation and cell death 31,32. Autophagy provides cyto-protection by degrading damaged ER, thereby attenuating ER-stress defense and apoptosis 33. Moreover, autophagy can induce anti-cancer drug resistance via several molecular mechanisms which were previously reviewed 34-36. Enhanced autophagy protects cancer cells from DNA damage 37-39, in part, by the timely degradation of proteins of the DNA damage repair (DDR) system, such as checkpoint kinase 1 (CHEK1) 40,41. This timely degradation prevents excessive retention of DDR proteins on damaged/repaired chromatin loci, allowing for their replacement by subsequent factors necessary for the next step in the DNA repair pathway. Hence, autophagy promotes enhanced DDR in cancer cells during treatment with DNA damaging modalities, such as 5-fluorouracil (5-FU) 42 and radiation therapy 43,44. Given the key role that autophagy plays in cancer development, progression and survival 35,45-48, concentrated efforts were focused on developing novel strategies for the inhibition of autophagy during cancer therapy 49-53. This development requires efficient assays for monitoring the autophagy steps (Figure 1), to identify and quantify autophagy inhibition 54,55.
Lysosomal targeting can be achieved using lysosomotropic drugs (LDs), primarily characterized by their hydrophobic weakly basic nature, which allows them to freely diffuse into cells and intracellular organelles 56-58. LDs diffuse in and out of the cellular membranous compartments, however, once they encounter the acidic lumen of the lysosome, they undergo protonation, become cationic and can no longer traverse the membrane. This results in their intercalation and accumulation within the lysosomes' membranes 59-61. This sequestration “sink” effect results in the accumulation of LDs in lysosomes at concentrations which are 1000-2500-fold higher than their extracellular drug concentrations within a few hours 62. From a mechanistic perspective, concentrated positively charged molecules at the water-interface of the membrane bilayer disrupt the electrostatic balance between the lipid headgroups, inducing repulsion between the choline groups of phospholipids and increasing the distance between neighbor lipids 59. This can lead to membrane fluidization 63-66 and lysosomal membrane permeabilization (LMP) 67. The utility of lysosomal protons (H^+^) for the protonation process and loss of protons following LMP, alkalinizes the lysosomal luminal pH 68,69. Lysosomal targeting can also be achieved by utilizing the endocytic pathway 70.
In this review we focus on strategies, from the last decade, for lysosomal targeting, leading to the induction of lysosomal dysfunction, LMP, lysosomal rupture and lysosomal cell death (LCD), with emphasis on autophagy inhibition, and immunogenic cell death (ICD). We discuss lysosomal targeting with single agents and in combination with other drugs (i.e., chemotherapeutics) or bona fide therapy modalities (i.e., radiotherapy and sonodynamic therapy) for the eradication of cancer cells and tumors. While targeting lysosomes with photosensitizers for efficient photodynamic therapy (PDT) is a rapidly growing research field, it has been widely reviewed in recent years 71-78 and hence will not be discussed herein.
Targeting lysosomes for anti-tumor immune response
Macrophage polarization
Macrophages are the predominant immune cell population present in cancer tissues 79,80. While macrophages have tumor-cell killing capacities, most experimental and clinical reports describe macrophages as protumor cells attenuating antitumor immune responses 81-84. Following macrophage polarization, they assume two main phenotypes designated M1 (pro-inflammatory) and M2 (anti-inflammatory) depicted in Figure 2 81-85. M1 macrophages are stimulated by interferon gamma (IFN-γ) and toll like receptor (TLR) ligands, including lipopolysaccharide (LPS) and lipoteichoic acid (LTA). Following stimulation, they express high levels of pro-inflammatory cytokines, including IL-1, IL-6, IL-12, IL-23, tumor necrosis factor α (TNFα) and IFN-γ, as well as inducible nitric oxide synthase (iNOS) 86. While M1 macrophages eliminate malignant cells, they also promote the antitumor cytotoxic activity of other leukocytes. Their enhanced tumor antigen presenting ability activates cytotoxic T-lymphocytes (CTLs, killer T cells, CD8^+^ T cells), and the cytokines they secrete stimulate and boost the function of natural killer (NK) cells.
In contrast, protumoral M2 macrophages act as immune suppressors 87. They have an impaired tumor antigen presentation ability, and secrete anti-inflammatory suppressor factors such as IL-10, hence negating any antitumor immune response. Furthermore, M2 macrophages release tumor promoting growth factors, such as platelet-derived growth factor (PDGF), transforming growth factor β (TGF-β) and vascular endothelial growth factor (VEGF), thereby encouraging tumor cell proliferation, angiogenesis and metastasis 81-85. Collectively, high prevalence of M2 tumor associated macrophages (TAMs) predicts dismal prognosis in various cancers, including lung 88, breast 89, pancreatic 90, and prostate cancer 91. It is well established that one of the most paramount characteristics that distinguishes between M1 and M2 macrophages is their lysosomal status. Lysosomes of M2 macrophages are more acidic than those of their M1 counterparts (pH ~4.5 vs ~5.3, respectively) 92-94. This affects the degradation process of biomolecules within the lysosomal lumen and the phagocytotic cascade, since many lysosomal proteases possess acidic pH optima below 4.5 6. M2 lysosomes display elevated hydrolase activity 95,96, leading to enhanced degradation of proteins (as well as lipids and nucleic acids), resulting in excessive antigen processing and diminished peptide presentation by MHC 97,98. This enhanced lysosomal activity also promotes an elevated autophagic flux in M2 macrophages 99,100.
In this respect, autophagy promotes lysosomal degradation of MHC I, thereby decreasing cancer-related antigen presentation, and facilitating immune evasion 101,102. Alkalinization of lysosomal pH with chloroquine (CQ) 101 or bafilomycin A1 (BafA1) 102 attenuated both the lysosomal enzymatic activity and the autophagic flux, resulting in higher surface presentation of MHC-I and an improved immune response against pancreatic cancer. This was also demonstrated by inhibiting the activity of the lysosomal protease cathepsin B (CTSB) 103.
Strategies are being developed to convert M2 to M1 macrophages, many of which utilize compounds that attenuate the lysosomal acidity and enzymatic activity. Naphplatin, a conjugation product of cisplatin to the core of the topoisomerase II inhibitor amonafide 104, was shown to localize in lysosomes of macrophages and increase their luminal pH 94. In turn, lysosomal alkalinization promoted the release of Ca^+2^ via the lysosomal cation channel mucolipin (Mcoln1), resulting in activation of the MAPK p38 signaling pathway. This led to the transformation of bone marrow derived macrophages (BMDMs) and M2-BMDMs to the pro-inflammatory M1-phenotype 94. In mice transplanted with murine colorectal cancer (CRC) CT-26 cells, treatment with cisplatin monotherapy induced an immune-suppressive response by increasing the percentage of tumoral M2 macrophages to 40% (p < 0.01). In contrast, naphplatin decreased the tumoral M2 macrophage population below 5%, while increasing the M1 population to ~80% (p < 0.01 and p < 0.001, respectively). This change was accompanied by a ~50% decrease in the levels of regulatory T cells (Tregs) in the tumor, and myeloid-derived suppressor cells (MDSCs) in the blood, further alleviating immune suppression. Naphplatin treatment boosted TAMs to inhibit CRC growth (final tumor weight was 10% of control) and pulmonary metastasis in CT-26 cells bearing mice, resulting in an 82.7% increase in survival time. To assess the contribution of macrophages to the anti-tumor effect of naphplatin, mice were also pretreated with the macrophage depleting agent clodronate 105, which abolishes mitochondrial ATP generation via inhibition of mitochondrial ADP/ATP translocase leading to apoptotic cell death. Pretreatment with clodronate diminished the anti-tumor effect of naphplatin, resulting in only 35% reduction in tumor weight. These findings establish the importance of lysosomal alkalinization to the immune response 94.
The same mechanism of increased lysosomal pH, consequent Ca^+2^ release and activation of MAPK p38 was demonstrated following treatment of mouse BMDMs and human macrophages with CQ 92. M2-TAMs were converted to the M1 phenotype, resulting in tumor growth inhibition and prolonged survival of B16 melanoma-bearing mice. Furthermore, following CQ treatment, mouse models of lung metastasis displayed a decreased number of tumor nodules in the lungs, and H22 hepatocarcinoma malignant ascites mouse models displayed reduced volume of ascites as well as reduced number of tumor cells 92.
pH-gated nanoparticles (PGNs) that self-assemble from amphiphilic copolymers were designed to accumulate within- and distinguish between lysosomes of M2-like BMDMs and other cell types, based on subtle lysosomal pH deviations 93. When conjugated to the TLR7/8 agonist imidazoquinoline (IQ), the now termed pH-gated nanoadjuvant (PGN_4.9_) selectively increased lysosomal pH in M2 macrophages, decreased cathepsin activity and converted these cells to the M1-phenotype. This in turn increased antigen presentation and activation of CTLs, resulting in tumor regression in a mouse 4T1 breast cancer model and further circumvented the formation of lung metastasis 93.
Yue Chen et al., took a different approach by harnessing the increased expression/activity of CTSB in tumor cells 106-109 and M2 macrophages 95,110 to ignite a newly designed lysosomal “nanorocket” 111. The latter, termed UIOQM-IQ, consists of an ultrasmall iron oxide (UIO) nanoparticle (NP) conjugated to a CTSB-cleavable peptide, an aggregation-induced emission fluorophore QMTPA, and surface IQ. Following IQ-dependent endocytosis and internalization into lysosomes, CTSB cleaves the peptide within UIOQM, thus releasing QMTPA and UIO NPs with exposed -NH_2_ and -SH termini which drive cross-linking and aggregation of both QMTPA and UIO NPs. Both aggregates allow tumor visualization, with QMTPA activating a fluorescent “on” switch, and UIO inducing a distinct MRI contrast shift, enabling deep-tissue imaging. Most importantly however, the bulky UIO aggregates within lysosomes lead to elevated osmotic pressure, and consequent LMP 111. LMP can lead to lysosomal alkalinization and dysfunction, or to the release of lysosomal content into the cytosol, leading to LCD 112,113. The former will result in autophagy inhibition and macrophage polarization, and the latter should release cancer specific antigens and elicit an immune response. In fact, LMP is regarded as a mechanism driving ICD which triggers an intact antigen-specific adaptive immune response 114,115. Murine mammary carcinoma 4T1 cells express higher levels of CTSB than normal 3T3 murine fibroblast cells 111,116, hence UIOQM-IQ elicited a specific cytotoxic effect on 4T1 but not on 3T3 cells (surviving fractions ~20% and > 90%, respectively) 111. In vivo evaluation of UIOQM-IQ was conducted using 4T1 cells bearing mice. The lysosomal “nanorocket” promoted a robust anti-tumor immune response, represented by a remarkable increase in M1:M2 macrophage ratio from ~2 to > 30 (p < 0.0001), > 4-fold increase in secretion of TNFα and IFN-γ as well as IL-6 and IL-12. Moreover, an increase in the percentage of mature dendritic cells in tumor-draining lymph nodes (TDLN), and an increase in infiltrating CTLs was also noted, along with a decrease in the levels of Tregs. The combination of the anti-tumor immune response and the cytotoxic effect of UIOQM-IQ resulted in 5-fold smaller tumors and longer survival time (~35 days vs. > 60 days for control and UIOQM-IQ treated mice, respectively). Notably, UIOQM-IQ reduced lung metastases by ~8-fold 111.
E64-DNA, a DNA nanodevice composed of the small-molecule cysteine protease inhibitor, E-64 (Figure 3) 117, conjugated to a 38-base pair DNA duplex, was developed to selectively enter TAMs via endocytosis and accumulate within their lysosomes. Once there, E64-DNA inhibited lysosomal-specific cysteine proteases which are elevated in M2-TAMs, thereby increasing the cells' ability for antigen cross- presentation and effective CTL activation 95. When E64-DNA was combined with the widely used alkylating chemotherapeutic cyclophosphamide 118, sustained tumor regression was achieved in a triple negative breast cancer (TNBC) mouse model 95.
Panax ginseng ginsenosides
In an effort to potentiate anti-tumor immunity with minimal chemotherapy-inflicted adverse effects, as well as to improve the quality of life of cancer patients, strategies are being developed for the combined treatment of herbal agents along with chemotherapy 119-122. One of the most commonly used roots that has been subjected to extensive anti-cancer research is ginseng, primarily its pharmacologically active constituents ginsenosides 123-127.
Ginsenosides are triterpene saponins (Figure 3), which include major ginsenosides and their secondary metabolic derivatives 128, of which R_g3_ 129,130 and its deglycosylated derivative R_h2_ 131,132, exhibit the most beneficial biological activities in various human pathologies. Regarding the present review, various ginsenosides were shown to increase lysosomal pH 42,133, induce LMP 133,134, inhibit autophagy flux 42,133,135-138, and enhance immunity 126,139-141. These resulted in sensitization to chemotherapy/immunotherapy 42,135,137-139,142,143, and most importantly enhanced in vivo anti-tumor activity 136,139,141,143,144.
It should be emphasized that although ginsenosides affect lysosomal function, they are not LDs according to their physicochemical properties, endowing them with both low water solubility and poor membrane permeability 145. Ginsenosides R_o_ 146,147 and R_h2_ were shown to increase cytosolic ROS levels in cancer cells, either via the estrogen receptor 2 (ESR2)-neutrophil cytosolic factor 1 (NCF1)-ROS axis 42, or mitochondrial ROS production 133. Extralysosomal ROS can damage the lysosomal membrane, and induce LMP with consequent lysosomal alkalinization, as was demonstrated upon mitochondrial ROS generation 148,149. This was also the case with R_o_ and R_h2_ 42,133,134. As abovementioned, lysosomal alkalinization reduces the activity of resident hydrolases; consistently, treatment with R_o_ reduced the activity of CTSB and CTSD 42. Lysosomal alkalinization can also inhibit autophagosome-lysosome fusion 150. Both decreased lysosomal enzymatic activity and autophagosome-lysosome fusion result in a reduction in the autophagic flux, as was observed following R_o_ or R_h2_ treatment 42,133. The ability of ginsenoside R_h2_ to inhibit autophagy was utilized to reverse the phenotype of RAW264.7 derived M2 macrophages to the M1 subtype in vitro 140. While an R_h2_ liposome (R_h2_-lipo), where R_h2_ functioned both as a cholesterol substitute membrane stabilizer, and a chemotherapy adjuvant, successfully polarized macrophages to the M1 phenotype in vivo 141. The increase in M1 macrophages in an orthotopic breast cancer 4T1 tumor bearing mouse model injected with R_h2_-lipo, was accompanied by residual levels of IL-10 and consequently high levels of activated CTLs and a dramatic decline in Tregs. The resulting potentiation of the immune response led to a 40% decrease in tumor volume, an impact which was enhanced to 80% via the co-encapsulation of R_h2_ with paclitaxel (PTX) 141. Utilizing R_h2_ as a component of the liposome membrane allowed tumor targeting via the glucose transporter 1 (GLUT1) which recognizes ginsenosides as substrates 151, and is overexpressed in tumors 152.
Ginsenoside R_g3_, another late-stage autophagy inhibitor 137,138, was also incorporated as a cholesterol substitute in the membrane of liposomes (R_g3_-LPs) 139. In this study, the affinity of ginsenoside-liposomes for GLUT1 was exploited to both penetrate the blood-brain-barrier (BBB), and deliver PTX for the targeted eradication of C6 glioma brain tumors in both mice and rat models. R_g3_-LPs, and to a greater extent R_g3_-PTX-LPs, polarized M2 macrophages to the M1 phenotype, both in vitro and in vivo, along with an 80% decline in the levels of TGF-β. The immune activation in the tumor microenvironment (TME) drastically reduced the presence of MDSCs and Tregs, while increased the abundance of CTLs from 5% to 30-40%. Collectively, these favorable activities dramatically increased the survival of C6 glioma bearing mice from 21 days to 32 and 54 days for R_g3_-LPs and R_g3_-PTX-LPs, respectively (p < 0.01). In the rat model, the survival was even longer, with the R_g3_-PTX-LPs group exceeding the experiment time of 60 days 139.
A systematic review analyzed the impact of combining first-line chemotherapy with the ginsenoside R_g3_ in the treatment of advanced non-small cell lung cancer (NSCLC) 153. Analysis of 2,200 NSCLC patients from China revealed excellent results highlighting the beneficial effects of R_g3_ for cancer patients. When compared to the control group receiving first-line chemotherapy alone, the R_g3_ supplemented patients exhibited higher response rates (p < 0.00001), higher Karnofsky performance status index (p < 0.00001), higher one- and two-year survival rates (p = 0.01, p = 0.006), higher rate of weight improvement (p = 0.02), reduced VEGF levels (p = 0.02), less gastrointestinal adverse effects (p = 0.02) as well as lower rates of myelosuppression (p < 0.00001) 153. An additional review analyzed the benefits of ginsenosides R_h2_, R_g3_ and compound K 154,155 as adjuvant therapy in 1,448 hepatocellular carcinoma patients, demonstrating similar remarkable results 156. It should be conveyed, that since ginsenosides display pleiotropic activities 129-132, the beneficial effects they elicit as adjuvants cannot be solely attributed to their lysosomotropic effects.
Radiotherapy
Radiation therapy aka radiotherapy (RDT) is an established hallmark of cancer treatment, along with chemotherapy, immunotherapy, hormone therapy, and surgery 157-159. RDT employs high-energy ionizing radiation in order to elicit two major antitumor activities 160,161. The first is the obvious eradication of the irradiated target cells by inducing DNA damage, mitotic catastrophe and apoptosis 162. The second, is referred to as in situ tumor vaccination 163,164 which stimulates a systemic immune-mediated antitumor response. Following RDT-induced cell death, the local release of tumor-derived antigens promotes their cross-presentation by various antigen presenting cells (APCs), which in turn instigates an immediate and prolonged immune response via the activation of NK cells, CTLs and B-cells 165-168. This “vaccination” can elicit an abscopal effect, where irradiation of a small tumor area induces a systemic antitumor immune response throughout the body, resulting in regression of tumors in remote, untreated parts of the body 169-171. Inasmuch as this sounds promising, RDT is actually effective in only a fraction of cancer cases, while in others it has the exact opposite effect. In this respect, various studies have shown that following RDT, a burst in immune-suppressive stimuli occurs 162, leading amongst others, to an elevation in pro-tumoral M2 macrophages 162,172,173. Moreover, using the lung colonization model of transplanted murine 4T1 breast cancer cells, RDT was shown to enhance lung metastasis in mice 173,174, and the pro-metastatic impact required the presence of macrophages 174. Although studies have shown that lysosomes and autophagy are main contributors to this RDT-resistance 159,175,176, only a few studies have attempted to target lysosomes to overcome this radioresistance.
A very recent study that targeted lysosomes, followed the example of successfully enhancing immunotherapy by alkalinizing the lysosomes of macrophages, and implemented this strategy following RDT treatment 173. Using the mouse 4T1 orthotopic breast cancer model, Bei Li et al., demonstrated that an immune-suppressive TME was established following RDT. This included a high prevalence of M2 macrophages and poor tumor infiltration of CTLs 173. Post-RDT macrophages were stimulated with cytidine monophosphate guanosine oligodeoxynucleotide (CpG), a TLR 9 agonist, resulting in the rewiring of their central carbon metabolism. This promoted these macrophages to engulf and eradicate tumor cells for antigen cross-presentation 177. However, since M2 macrophages have extremely acidic lysosomes with enhanced hydrolytic activity, as mentioned above, the antigens were over-processed, abolishing antigen-presentation by MHC I. Thus, the authors administered a MgAl-based hydrotalcite (bLDH) alkaline nanoadjuvant to the peritumoral area post-RDT. Hydrotalcite is an antacid currently used to neutralize stomach acidity 178,179. While the combination of CpG and bLDH was sufficient to increase surface localization of antigen presenting MHC I in macrophages, the effect was even stronger post RDT. A previous study with hydrotalcite-embedded magnetite NPs, showed the accumulation of these NPs in lysosomes 180. Consistently, bLDH induced lysosomal alkalinization in BMDMs. The enhanced cross-presentation following co-administration of CpG and bLDH resulted in priming of antigen-specific CTLs and tumor infiltrating NKs leading to the consequent suppression of the primary tumor and lung metastasis in the mouse 4T1 breast tumor model 173.
The brain tumor glioblastoma multiforme (GBM) is a highly aggressive and fulminant malignancy which displays immune-evasion 181, chemoresistance and radioresistance 182,183. In the latter respect, to surmount this RDT resistance, Xin Zhang et al., utilized trifluoperazine (TFP, Figure 3) 184, an antipsychotic phenothiazine from the 1950s 185. TFP has been shown to inhibit proliferation, migration, and invasion of GBM cells, however it failed to extend the survival time of orthotopic U87MG xenograft bearing mice 186. In contrast, when combined with RDT, TFP significantly increased the survival of orthotopic xenograft GBM mouse models with P3 cells (median survival 46.0 vs 29.7 days for combined treatment vs. radiation alone, p < 0.01) 184. TFP is a highly hydrophobic weak base compound 187, like the majority of anti-psychotic drugs, and thus highly accumulates in lysosomes 59,188. Indeed, Xin Zhang et al., demonstrated lysosomal alkalinization following treatment with TFP, along with a decrease in the activity of lysosomal cathepsin proteases, and a consequent decrease in the autophagic flux 184. In a follow up paper, this group further revealed that TFP induced lysosomal swelling and LMP, resulting in reduction of the autophagic flux 189. However, the radio-sensitization achieved by TFP was attributed to impaired homologous recombination during radiation-induced DDR, with no mention of abrogating RDT-induced immune-suppressive responses 184.
Qi Xu et al., developed a core-shell copper selenide-coated gold (Au@Cu_2-x_Se) NPs which were shown to minimally affect lysosomal pH, and to prominently block the autophagic flux 190. This resulted in radio-sensitization and tumor eradication, leading to prolonged survival of orthotopic mouse xenograft GBM model harboring human U-87MG cells (median survival 42 vs 29 days, for combined Au@CS + X-Ray treatment vs. X-Ray alone) 190. These NPs required focused ultrasound (US) to better traverse the BBB and reach the intracranial tumor site.
Chloroquine
The anti-malarial drug CQ (Figure 3) has been widely studied in the treatment of various pathological disorders 191 including cancer 192,193. Its lysosomotropic properties have been exploited for lysosome alkalinization (as discussed in the immunotherapy chapter) and autophagy inhibition 194. These properties have also been exploited to sensitize tumors to RDT, primarily GBM.
Glioma initiating cells (GICs) 195 are radio-chemo-resistant stem-like cells responsible for relapse following treatment of GBM with RDT 196,197. Early studies demonstrated that enhanced autophagy promotes differentiation of GICs 198, decreases their tumorigenicity 199 and restores their radio-sensitivity 200. In contrast, in recent years, autophagy inhibitors were shown to increase sensitivity of GICs to both RDT 201 and chemotherapy 202. Chenguang Li et al., settled the dispute by showing that inhibition of autophagy at different stages of the process has distinct effects 203. Blocking the autophagic flux at the end of the process, i.e., autophagosome-lysosome fusion and/or content degradation, leads to the accumulation of degradative vacuoles, and resensitizes GBM cells to anti-cancer treatments.
Autophagy inhibition by CQ at the stage of autophagosome-lysosome fusion 194 potentiated the radio-sensitivity of GICs isolated from the human glioma cell line U87 201. The combination of CQ and X-ray markedly decreased the clonogenic surviving fraction of GICs by ~10-fold compared to X-ray alone, and increased apoptosis by a factor of > 2-fold. Finally, the combination exhibited a synergistic activity on GICs generated tumor spheres, decreasing both their numbers and diameters 201.
The potential of CQ in restoring radio-sensitivity to GBM has been tested in several clinical trials over the past 20 years with encouraging outcomes (Table S1) 204-209. When presenting increased progression free survival (PFS) and median survival time after surgery for CQ-treated patients, it emerged that these clinical trials could have been the initiators of routine CQ administration in GBM treatment. However, the patient numbers in all these trials were too small to attain statistical significance and draw definitive conclusions. For example, the Sotelo group published the results of a prospective controlled randomized trial, where nine patients receiving an additional daily dose of 150-mg CQ to the radiochemotherapy, were compared to nine control patients. The mean survival time was 31±5 and 10.6±2 months, respectively,* p*<0.0002 204. Later, the same group published a post-surgery median survival time of 24 months for CQ-treated patients (n=15) and 11 months for control patients (n=15), with double the number of survivors in the CQ-treated patients at the end of observation (p = 0.139) 205. Furthermore, a study on the treatment of recurrent GBM (rGBM), retrospectively compared 33 patients in a control group receiving only adjuvant-radiochemotherapy (aRCT) to those receiving aRCT+ bevacizumab (BEV, n = 5) or the triple combination: aRCT+BEV+CQ (n = 4). Median post recurrence survival times were 9.63, 12.97 and 23.92 months, respectively, p = 0.022 209.
Some beneficial effects were clinically attributed to the addition of CQ in combination with standard chemotherapy, in case of advanced or metastatic anthracycline-refractory breast cancer 210 and metastatic or unresectable pancreatic cancer 211 (Table S1). The maximal tolerable CQ dose in clinical trials was found to be 200-250 mg/day 208,209,212. Higher doses of CQ elicited multiple adverse effects, including irreversible blurred vision and vomiting 208. Although CQ is an excellent lysosomal alkalinizing agent, its tolerable dose might not be sufficient to allow for effective autophagy inhibition required for chemo/radio/immuno-sensitization. One plausible modality to circumvent these adverse effects of high dose CQ is its encapsulation and tumor targeting 213.
CQ encapsulation
Temozolomide (TMZ), a DNA alkylating and methylating agent, is the first-line chemotherapeutic drug in the treatment of GBM and anaplastic astrocytoma 214,215. However, like most cytotoxic drugs, TMZ inflicts adverse effects with up to 20% of glioma patients suffering from thrombocytopenia 216. The combination of TMZ and CQ has been shown to bear a synergistic effect in eradicating GBM cells 217,218, primarily via lysosomal dysfunction and autophagy modulation 218. Furthermore, this combination displayed promising results in clinical trials 205,208. Hence, the combined encapsulation of these drugs in targeted NPs has the potential to exert synergistic anti-tumor effects without harming healthy tissues.
Mesoporous silica NPs (MSNs) have been extensively used experimentally for drug delivery in vivo 219. The incorporation of polydopamine (PDA) into MSNs allows to modify the surface of the NPs and attach specific ligands for cancer targeting 220. PDA also adds a pH-responsive element to the NPs as it undergoes degradation under acidic conditions, enhancing drug release in lysosomes or in the acidic TME 221,222. The arginyl-glycyl-aspartic acid (RGD) tripeptide 223,224 is an established ligand of α_v_β_3_ integrin used for in vivo tumor mapping and targeting 225-227. The surface integrin α_v_β_3_ is crucial for tumor angiogenesis and is highly expressed predominantly in new blood vessels 228,229, as well as various tumors 230-234 including GBM 230,235. TMZ and CQ were co-loaded into MSNs coated with PDA decorated with RGD, designated TMZ/CQ@MSN-RGD 236. In vitro growth inhibition assays, using the α_v_β_3_ integrin expressing human GBM cell line U87 237, showed a 4-fold lower IC_50_ value for TMZ/CQ@MSN-RGD compared to free TMZ (24.5 vs. 104.3 µg/ml, respectively), while TMZ/CQ@MSN-RGD had little effect (< 15%) on the viability of rat cortical neuronal cells, even at a high concentration of 1 mg/ml. These results suggest the specificity of the NPs for GBM cells. By accumulating in lysosomes (following endocytosis), inhibiting autophagy, and enhancing apoptosis, TMZ/CQ@MSN-RGD inhibited tumor growth in U87 cells xenograft bearing mice, twice as much as free TMZ, with no apparent toxicity to healthy organs 236.
A cyclic RGD peptide was also used to coat lipid NPs (LNPs) co-loaded with CQ and the anti-malarial drug dihydroartemisinin (DHA) 238. Apart from integrin α_v_β_3_ being a target, RGD is also an established ligand of integrin α_v_β_6_ 224,239,240, which is highly overexpressed in CRC, where it enhances tumor aggressiveness 241-244. DHA is considered a sensitizing agent shown to increase ROS levels in cancer cells 245-247, however, its pharmacologic effect as a single agent is usually abrogated by the activation of protective autophagy 248-250. Hence, it was determined that DHA should be combined with other cytotoxic agents 251, preferably an autophagy inhibitor 252,253. This was the rational for the design of LNPs co-loaded with CQ and DHA and decorated with RGD (RLNP/DC) for the treatment of CRC 238. Even when loaded with a relatively low CQ concentration of 7.5 µM, RLNP/DC exhibited superior potency in inhibiting colony formation, increasing ROS production, inhibiting autophagy, and increasing apoptosis in CRC HCT116 cells compared to free CQ+DHA. The results obtained from in vitro invasion and migration assays suggested an anti-metastatic potential for RLNP/DC. Consistently in vivo, murine HCT116 cell models of CRC and metastasis, presented with ~3-fold less CRC tumors, which were ~3-fold smaller upon treatment with RLNP/DC, compared to free CQ+DHA. RLNP/DC-treated mice also developed half the number of liver metastases than those treated with free drugs, and exhibited a 25% increase in survival rates, with all mice being alive at the end of the 60 days experiment. RLNP/DC-treated mice also maintained the highest body weight throughout the experiment 238.
Encapsulation of hydroxychloroquine
The devastating pancreatic ductal adenocarcinoma (PDAC) 254,255 has a unique TME characterized by hyperactivated stromal fibroblasts, effective immunosuppression, and an elevated dense extracellular matrix (ECM) deposition known as desmoplasia 256 (Figure 4). This physical barrier, promoted by high levels of autophagy 257-259, limits the delivery and efficacy of chemotherapy 260 and immunotherapy 261, while autophagy supports immune evasion 101,102. Drug encapsulation has been tested to help penetrate this dense desmoplastic ECM barrier and target stromal cancer associated fibroblasts (CAFs) and tumor cells 262,263. PDAC cells overexpress integrins α_v_β_6_ 264 and α_v_β_3_ 234,265 and thus are good targets for RGD-decorated NPs. The CQ derivative hydroxychloroquine (HCQ, Figure 3) has similar lysosomotropic properties and anti-cancer modes of action to CQ by alkalinizing lysosomes and inhibiting autophagy 193,266. HCQ has been shown to be ~40% less toxic in animals 267 and have less adverse effects in humans 268, and has been clinically explored in the treatment of PDAC (Table S1) 269-274. For example, pre-operative treatment of PDAC patients with gemcitabine and HCQ markedly increased the overall survival in patients who had a > 51% increase in the autophagy marker LC3-II in circulating peripheral blood mononuclear cells (34.8 vs. 10.8 months, p < 0.05) 275. Moreover, HCQ was shown to possess antifibrotic activity, by reducing collagen levels and inhibiting ECM synthesis in 4T1 mouse tumor models 258. These findings led to the design of TR-PTX/HCQ-Lip, liposome-based NPs decorated with a multifunctional tandem peptide TH-RGD (TR), loaded with a combination of HCQ and PTX 276. TR consists of a targeting cyclic RGD tripeptide and a pH-responsive cell-penetrating peptide (CPP) 277. CPP should become protonated under the acidic pH of the TME, thus converting its charge from negative to positive and facilitating its membrane penetration by electrostatic forces, particularly between the positive charge of the CPP and the negatively charged polar head groups of membrane phospholipids 278,279. This enhances the RGD-based specificity of the liposomes to tumors. The ability of TR-PTX/HCQ-Lip to penetrate the dense fibrotic stroma and target the PDAC tumor was verified using a murine BxPC-3/NIH 3T3 heterogenous pancreatic tumor model 276. The heterogenous tumor model consisting of both CAFs and tumor cells is utilized to mimic the complex PDAC architecture and desmoplastic components 280. In comparison with free HCQ and non-targeted HCQ-containing NPs (PEG-HCQ-Lip, PEG-PTX/HCQ-Lip), TR-PTX/HCQ-Lip completely disrupted lysosomal accumulation of Lysotracker, and robustly inhibited autophagy in BxPC-3 and NIH 3T3 cells 276. Autophagy inhibition and reduction in ECM deposition were also verified in vivo in harvested tumors. Moreover, TR-PTX/HCQ-Lip was superior in inhibiting migration and invasion of BxPC-3 cells. In an orthotopic BxPC-3 tumor bearing mouse model, administration of free PTX+HCQ had little effect on tumor growth, eliciting a reduction of ~15% in tumor growth. In contrast, TR-PTX/HCQ-Lip exerted a dramatic growth inhibitory effect, resulting in an ~85% reduced tumor weight (p < 0.001). Consistent results were obtained with the heterogenous tumor mouse model, suggesting good response by CAFs. Moreover, TR-PTX/HCQ-Lip completely eliminated any surface liver metastases, compared to 50-90 metastatic nodules on livers of mice treated with free PTX+HCQ (p < 0.001) 276. In accord with the loss of body weight as a hallmark of PDAC 281, all orthotopic BxPC-3 tumor bearing mice exhibited weight loss with disease progression, while those treated with TR-PTX/HCQ-Lip largely retained their original weight. Importantly, the encapsulation of PTX and HCQ prevented hepatic toxicity induced by the free drugs 276. Collectively, these findings reveal a good response to HCQ as a drug adjuvant, and the advantages of drug encapsulation and tumor targeting.
Taking advantage of autophagy a step further, Yang Wang et al., decided to not only prevent the beneficial impacts of autophagy in tumors, but to also use autophagy as a tumor killing approach. By both enhancing the first step and inhibiting the last stage of autophagy, the researchers induced autophagic catastrophic vacuolization and death of both tumor cells in vitro and mouse tumor models in vivo 282. This was achieved by utilizing a TAT-Beclin 1 peptide (T-B) along with HCQ-loaded liposomes (HCQ-Lip). The T-B peptide consists of the transduction domain of the CPP TAT protein, linked to the HIV-1 Nef-binding domain of Beclin 1 required to initiate autophagy 283. Indeed, inducing the generation of autophagosomes by T-B and preventing their fusion with lysosomes via HCQ-Lip, led to the overwhelming synergistic accumulation of autophagosomes (6-7-fold over free HCQ or T-B) in four different tumor cell lines. This led to 95% apoptosis/necrosis of cancer cells 282. Treatment of 4T1 xenograft-bearing BALB/C mice with an intra-tumoral injection of T-B and an intravenous injection of HCQ-Lip resulted in ~90% smaller tumors than in the control group (p < 0.001), which were 3.3-fold smaller than those of mice treated with each component alone (p < 0.001). Examination of the resected tumors revealed vast autophagic vacuolization and necrotic areas at the center of the tumors 282. Since HCQ is less toxic than CQ, all currently ongoing clinical trials, utilizing autophagy inhibitors to improve the outcome of cancer therapy, include HCQ (Table 1).
Lys05
A CQ derivative that has gained much interest is Lys05 (Figure 3), a bisaminoquinoline dimeric form of CQ which is 10-fold more efficacious as an autophagy inhibitor than HCQ in human GBM LN229 cells 284. At high doses, Lys05 is such a potent autophagy inhibitor, that it elicited in mice an intestinal phenotype resembling genetic defects in the autophagy gene ATG16L1 284. Unlike HCQ, Lys05 was shown to have single-agent antitumor activity without untoward toxicity in mice bearing HT-29 CRC xenografts at low doses (i.e., 10 mg/kg and 40 mg/kg), hence achieving the goal of preventing adverse effects 284. The mode of action of Lys05 was demonstrated using the GBM U251 and LN229 cell lines 44. Following its accumulation in lysosomes, Lys05 induced LMP, resulting in lysosomal alkalinization and content release, resulting in mitochondrial depolarization and tumor cell death. While Lys05-induced lysosomal dysfunction did not prevent the fusion of lysosomes with autophagosomes, the degradation within autolysosomes was impaired, hence inhibiting the autophagy flux 44. In accord with the immune-suppressive effects of irradiation detailed above, irradiation of U251 and LN229 cells resulted in increased CTSB activity. In this respect, Lys05 was shown to robustly enhance the cytotoxic effect of irradiation in vitro via LMP and elevated irradiation-induced DNA damage 44.
Overcoming resistance to tyrosine kinase inhibitors
Tyrosine kinase inhibitors (TKIs) have truly revolutionized the treatment of human malignancies 285-287. However, the efficacy of cancer treatment with TKIs has been hampered by the frequent emergence of multiple mechanisms of TKI resistance 288-290. In this respect, various TKIs are hydrophobic weak bases which highly accumulate in lysosomes, thereby being sequestered away from their kinase target 291,292. In fact, many TKIs were shown to enhance protective autophagy 293-299 which constitutes a major resistance mechanism 293,298.
Clear cell ovarian carcinoma (CCOC) is a subtype of ovarian cancer characterized by intrinsic chemoresistance, including to established TKIs 300,301. Sunitinib, a small-molecule multi-targeted TKI 302,303, initially elicited a good response in two CCOC patients 304, but had little effect in a clinical trial 305. A suggested mechanism of sunitinib resistance was its accumulation and sequestration in lysosomes 306-308, a mechanism that could be exploited for LCD by PDT 309,310. At low concentrations, sunitinib was shown to impair autophagy, however at clinically relevant cytotoxic drug levels, sunitinib increased the autophagy flux 294,295. Thus, several studies have shown the benefit of combining sunitinib treatment with an autophagy inhibitor 311-314, as was the case with CCOC 315. The combination of sunitinib and Lys05 exerted a synergistic growth inhibitory effect on three CCOC cell lines compared to each drug alone; an effect that was recapitulated by combining sunitinib with autophagy protein 5 (ATG5) siRNA. The effect of the combined treatment was further explored in vivo in heterotopic murine models bearing the human CCOC cells TOV21G and OVTOKO. Mice receiving this combination treatment exhibited a substantial reduction of 45% (p < 0.01) and 54% (p < 0.0001) in tumor growth compared with mice treated with monotherapy of sunitinib or Lys05, respectively 315.
Chronic myeloid leukemia (CML) is successfully treated with TKIs including imatinib, nilotinib, dasatinib, bosutinib and the newer TKI asciminib 316,317. However, leukemic stem cells (LSCs) are insensitive to TKIs and persist as a minimal residual disease (MRD) source, resulting in relapse 318-320. In this respect, inhibition of autophagy has been shown to sensitize CML cells to TKIs 321,322. Using a CML patient-derived xenograft model, Pablo Baquero et al., showed that hematopoietic LSCs exhibit an increased autophagic flux compared to non-leukemic cells 323. Remarkably, treatment of these leukemic mice with Lys05 resulted in autophagy inhibition, while HCQ had no inhibitory effect. The latter results were recapitulated in stem cell-enriched (CD34^+^) cells isolated from CML patients. Lys05 induced autophagy inhibition, which reduced LSCs quiescence and promoted myeloid cell expansion and maturation in the CML mouse model 323. Lastly, the combination of Lys05 and nilotinib, a second generation TKI 324,325 that was shown to induce autophagy 296,297, resulted in a significant additive therapeutic effect by reducing the fraction of human CD45^+^ cells in the bone marrow of these CML mouse model (p = 0.05), while HCQ had no additive pharmacological effect 323. CD45 is a pan-leukocyte marker expressed on nearly all hematopoietic cells, including hematopoietic stem cells 326.
The most common type of leukemia, chronic lymphocytic leukemia (CLL), accounts for ~33% of newly diagnosed leukemias in the US 327,328. Survival of CLL cells relies on B-cell receptor (BCR) signaling 329,330, which is conveyed through various kinases, including the pivotal Bruton's tyrosine kinase (BTK) 331,332. While the TKI ibrutinib, an irreversible inhibitor of BTK 333,334 which induces autophagy 335,336, is considered to have revolutionized CLL treatment, patients still present acquired drug resistance and low complete remission rates 337,338. Various studies have demonstrated the hypersensitivity of CLL cells to LDs in comparison to healthy B-cells 339-341. Hence, the combination of ibrutinib with lysosome-sensitizing agents has been explored 342,343. This included the repurposing of widely used cationic amphiphilic antihistamines (CAAs) which have recently been recognized as LDs 60,63,69,343-347. These CAAs, including for example desloratadine, clemastine, and ebastine, bear a specific chemical structure containing hydrophobic rings and a hydrophilic amine group. This structure allows them on the one hand to traverse cell membranes and on the other hand undergo accumulation in acidic lysosomes upon amine group protonation.
Sonodynamic therapy
RDT using X ray has an advantage of deep tissue penetration, enabling tumor targeting throughout the body 348. However, irradiation has inevitable side effects including secondary tumors induced by this mutagenic treatment 349, radiation-induced vasculopathy 350, cardiovascular disorders 351, and serious fatigue 352, all of which limit the biomedical application of RDT. PDT, which combines light energy with a wavelength compatible photosensitizer, is considered a well-established method for cancer treatment, including via lysosomal damage 71-76. However, PDT has many limitations and disadvantages 353,354; primarily, near-infrared-based PDT laser has poor tissue penetration (~1-5 mm) 355,356, requires photosensitizers at specific wavelengths and induces serious photosensitivity of healthy tissues like the skin 357.
In comparison to RDT and PDT, US-based sonodynamic therapy (SDT) has high tissue penetration capacity (> 10 cm) 358 and displays negligible side effects 359. SDT is useful for drug delivery 360,361, including for the temporary opening of the BBB 362 to facilitate delivery of drugs for the treatment of various CNS malignancies 363 including GBM 364 and brain metastases 365. SDT is also used with various sonosensitizers to generate cytotoxic ROS for cancer therapeutics 359,365,366, including immunotherapy 367-371, eliciting the desirable abscopal effect 366. However, as observed with other anti-cancer treatment modalities, SDT can induce autophagy which mitigates the anti-cancer therapeutic effects 372-375. Thus, combining SDT with lysosome-targeted autophagy inhibitors restores tumor sensitivity to treatment 376-379, and induces LMP-driven ICD, stimulating an adaptive immune response 114.
Achieving both inhibition of autophagy and LMP using SDT was demonstrated by Yong Liu et al. 379. This group implemented an innovative NP strategy by using a piezoelectric material, which can convert mechanical pressure into electrical energy 380,381, to generate Ba_0.85_Ca_0.15_Zr_0.1_Ti_0.9_O_3_ (BCZT) NPs 379. Following endocytosis, these BCZT NPs localized within lysosomes of murine B16 melanoma cells, where they had only minor deleterious effects on lysosomal function. However, during a 6 min application of 1.5 W/cm^2^ US, the US-mediated mechanical forces 382 caused the charge centers within the BCZT NPs to shift, resulting in a dipole moment 383 and consequent formation of (e-) electrons on their surface 379. These electrons interacted with the abundant protons (H^+^) in the lysosomes to produce hydrogen gas (H_2_), thereby drastically alkalinizing the lysosomes. This resulted in a 75% reduction in lysosomal acid phosphatase activity and robust LMP, leading to autophagy inhibition and rapid cell death. The combination of BCZT NPs + US was also tested in vivo on B16 tumor-bearing mice, where these NPs consistently accumulated in the tumor's lysosomes. The combined therapy with BCZT NPs + US displayed an efficacious 86.1% tumor suppression rate (p < 0.0001), while the BCZT NPs alone elicited a non-significant 10% reduction in tumor volume. This allows for US-directed targeted therapy, even if the NPs themselves are not tumor specific 379.
An additional piezo-sonosensitizer was designed by Xianbo Wu et al., who synthesized a novel O_2_ self-sufficient Ir-C_3_N_5_ nanocomplex, composed of a nitrogen-rich carbon nitride (C_3_N_5_) nanosheet and 30% iridium(III) 384. Ir-C_3_N_5_ exhibited a strong dipole moment and consequent high piezoelectric catalytic performance under US. The surface electrons reacted with O_2_ to generate singlet oxygen (^1^O_2_), and the intermediate ·O_2_^-^ reacted with additional electrons to form H_2_O_2_, followed by H_2_O_2_ decomposition to generate ·OH, thus producing high ROS levels. Moreover, as a self-sufficient O_2_ producer, Ir-C_3_N_5_ can be used under hypoxic conditions, which exist in both solid and hematological malignancies 385. When incubated with human A-375 melanoma cells, Ir-C_3_N_5_ was shown to accumulate in lysosomes, though no explanation was given to this specific organelle targeting 384. However, since the accumulation in lysosomes was not time dependent, this probably did not occur via endocytosis, and the asymmetric structure of the C_3_N_5_ component with positive and negative charge centers, probably conferred lysosomotropic characteristics. Upon US activation (0.5W, 1 MHz, 3 min), lysosomal Ir-C_3_N_5_ generated high levels of ROS leading to LMP. The latter induced robust autophagy inhibition and cell death, ~70% apoptosis and necrosis for Ir-C_3_N_5_ + US vs ~10% for Ir-C_3_N_5_ and only ~3% in the control group. To verify that Ir-C_3_N_5_ + US induced ICD, the authors conducted an in vitro transwell macrophage polarization assay. As controls for M1 and M2 polarization, mouse J774A.1 macrophages were incubated with the canonical stimuli LPS and IL-4 for M1 and M2, respectively. Apoptotic A-375 cells following treatment with Ir-C_3_N_5_ + US, stimulated M1 polarization as strong as LPS, including the secretion of the pro-inflammatory cytokines TNFα and IL-6. Using subcutaneous murine B16-F10 tumor models, Ir-C_3_N_5_ + US was shown to stimulate CTLs infiltration into the tumor and lymph nodes. This stimulation resulted in the complete inhibition of both the primary tumor and distant tumor growth as well as eliminated lung metastases, suggesting an efficient abscopal effect. The combined treatment with Ir-C_3_N_5_ + US increased the survival rate of the mice beyond the scope of the experiment (> 50 days) 384.
Future perspectives
The current review highlights the burning necessity of simultaneously targeting tumor cells as well as the TME and tumor immune microenvironment (TIME) 386. Targeting only tumor cells often results in chemoresistance, as the TME and TIME actively promote tumor cell survival, growth, invasion, immune evasion and metastasis 387-391. Lysosomal modulating agents that impair autophagy simultaneously target the tumor and its TME and TIME, while minimizing adverse effects. Targeting lysosomes of TAMs and stroma cells can convert an immunosuppressive microenvironment into an immune-supportive one, enhancing the efficacy of immunotherapies. Several clinical trials using CQ/HCQ in combination therapy, have shown great promise (Table S1) 53 and warrant further dedicated studies. This is particularly relevant in high mortality cancer types with no efficacious therapy like PDAC. As abovementioned, PDAC is characterized by a highly dense desmoplasia. The latter is present in other types of tumors, e.g., cervical cancer 392, breast cancer 393, lung cancer 394, squamous cell carcinoma 395 and small intestine neuroendocrine tumors 396, leading to chemoresistance and dismal prognosis 392,395,397-399. Thus, it is paramount to overcome this desmoplastic barrier for efficient cancer eradication. In this regard, recent studies reveal that desmoplasia is promoted by autophagy 257-259. Hence, we find autophagy inhibition via lysosomal targeting a promising therapeutic strategy. Figure 4 illustrates the role of autophagy in shaping the TME and TIME of PDAC, and the reversal effect of autophagy inhibition by lysosome targeting agents. The anti-fibrotic activity of HCQ inhibits ECM synthesis by reducing the secretion of collagen and hyaluronan by stromal fibroblasts 258,276. The polarization of macrophages to the M1 phenotype reduces the levels of TGF-β which promotes autophagy and ECM stiffness 400. Reduction in desmoplasia along with immune stimulation restores chemotherapeutic drug accessibility to the tumor and chemosensitivity, as well as an enhanced anti-tumor immune response. It should be noted that HCQ is used in this demonstration since it was shown to reduce desmoplasia in murine models 258,276. However, since an HCQ dose of 1,200 mg/day is required to induce inhibition of autophagy in cancer patients 275,401, a dose that can elicit grade 3-4 adverse effects 401-403, other more potent lysosome disrupting agents should be tested, or HCQ should be encapsulated. A potential candidate as an HCQ substitute is the FDA approved TKI nintedanib 404. From a mechanistic perspective, nintedanib blocks multiple tyrosine kinase receptors including VEGFR, PDGFR, and FGFR, which are paramount in the signaling pathways which culminate in pathological lung fibrosis. Nintedanib accumulates in the membrane of lysosomes and disrupts their integrity and function 59,405,406, leading to autophagy inhibition and autophagic cell death 405,407. Consistently, nintedanib has displayed potent antifibrotic activities that reprogram the TME by reducing ECM secretion by CAFs as well as enhancing immunity and reducing TGF-β levels 408-410. While these abilities are exploited for the treatment of lung fibrosis 411,412, nintedanib is still primarily referred to as a TKI and not as a bona fide lysosomotropic agent. Based on the established anti-fibrotic activity of nintedanib, clinical trials are warranted that will explore its plausible antitumor activity as a desmoplasia inhibitor, as monotherapy or in combination with other chemotherapeutics, in desmoplastic cancers like PDAC. In this respect, a clinical trial has been already conducted in PDAC (NCT02902484).
In a recent review, Stephanie Nagy et al., retrospectively analyzed the impact of CAAs on the efficiency of immune checkpoint inhibitors (ICIs)-based immunotherapy in cancer patients 413. The six studies included in this analysis, encompassing 4,171 patients with different types of malignancies, showed great potential. Collectively, patients who received a combination of CAAs and ICIs displayed a significant improvement in overall survival rates and longer progression-free survival rates compared to patients who did not receive antihistamines 413. Considering the minimal adverse effects of second generation CAAs 414, and their ability to induce lysosomal alkalinization and LMP 69,343,345,347, further clinical evaluation should be performed. The same can be conveyed about ginsenosides which exhibit multiple beneficial properties, including health improvement, anti-tumor activity and enhanced immunity. Thus, drug repurposing to generate efficacious combination therapies targeting the tumor as well as the stroma should be in the epicenter of future innovative therapeutic drug development efforts (Figure 5).
Supplementary Material
Supplementary table.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ohkuma S Poole B Fluorescence probe measurement of the intralysosomal p H in living cells and the perturbation of p H by various agents Proc Natl Acad Sci U S A 19787573327312852410.1073/pnas.75.7.3327 PMC 392768 · doi ↗ · pubmed ↗
- 2Johnson DE Ostrowski P JaumouilléV Grinstein S The position of lysosomes within the cell determines their luminal p HJ Cell Biol 20162126677922697584910.1083/jcb.201507112 PMC 4792074 · doi ↗ · pubmed ↗
- 3Webb BA Aloisio FM Charafeddine RA Cook J Wittmann T Barber D Lp HLARE: A new biosensor reveals decreased lysosome p H in cancer cells Mol Biol Cell 2021322131423323783810.1091/mbc.E 20-06-0383 PMC 8120692 · doi ↗ · pubmed ↗
- 4Hu Y Wang X Lu K Cheang C Liu Y Zhu Y Aggregation-induced emission of DNA fluorescence as a novel pan-marker of cell death, senescence and sepsis in vitro and in vivo Theranostics 20261621063814135679910.7150/thno.122009 PMC 12675146 · doi ↗ · pubmed ↗
- 5Schröder BA Wrocklage C Hasilik A Saftig P The proteome of lysosomes Proteomics 201010224053762095775710.1002/pmic.201000196 · doi ↗ · pubmed ↗
- 6Ratto E Chowdhury SR Siefert NS Schneider M Wittmann M Helm D Direct control of lysosomal catabolic activity by m TORC 1 through regulation of V-AT Pase assembly Nat Commun 202213148483597792810.1038/s 41467-022-32515-6PMC 9385660 · doi ↗ · pubmed ↗
- 7Yoon MC Hook VO'Donoghue AJ Cathepsin B Dipeptidyl Carboxypeptidase and Endopeptidase Activities Demonstrated across a Broad p H Range Biochemistry 202261171904143598150910.1021/acs.biochem.2c 00358 PMC 9454093 · doi ↗ · pubmed ↗
- 8Abe A Shayman JA Purification and characterization of 1-O-acylceramide synthase, a novel phospholipase A 2 with transacylase activity J Biol Chem 199827314846774952596010.1074/jbc.273.14.8467 · doi ↗ · pubmed ↗