Acinetobacter variabilis represents a diverse species with novel regions associated with antibiotic resistance and surface polysaccharides
Lucy L. Patterson, Eva Hatje, Mohammad Katouli, Johanna J. Kenyon, Mehrad Hamidian

TL;DR
This study explores the genetic diversity of Acinetobacter variabilis, revealing new insights into antibiotic resistance and surface polysaccharides linked to virulence.
Contribution
The paper identifies a novel transposon and structural variants in A. variabilis, highlighting its role in resistance gene dissemination.
Findings
Phylogenetic analysis showed substantial diversity in A. variabilis, including a subclade with resistance determinants.
A novel transposon, Tn6929, was identified, interrupting the comM gene and associated with resistance gene acquisition.
Integrative conjugative elements (ICEs) with varied resistance gene content were found integrated into the thyA gene.
Abstract
Acinetobacter variabilis is an opportunistic pathogen found in both clinical and environmental settings with the potential to harbour and disseminate clinically significant antibiotic resistance genes. Here, we sequenced the complete genome of NGH-QLD-N1, an A. variabilis isolate recovered from a blood sample of a patient at Nambour General Hospital located on the Sunshine Coast region of Queensland, Australia. The assembly was used as a reference to assess the diversity in available A. variabilis genome sequences and investigate the genomic context of surface polysaccharide biosynthesis genes and antibiotic resistance determinants in relation to the priority pathogen, Acinetobacter baumannii. Phylogenetic analysis revealed substantial diversity in the species, with a distinct NGH-QLD-N1 subclade containing multiple strains with important resistance determinants. We identified a novel…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
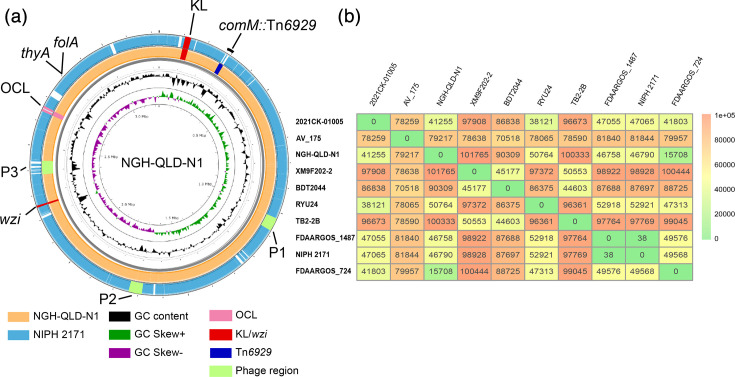 Fig. 1
Fig. 1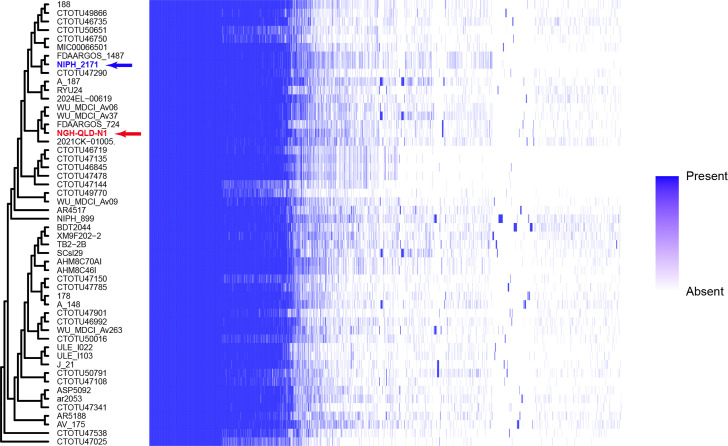 Fig. 2
Fig. 2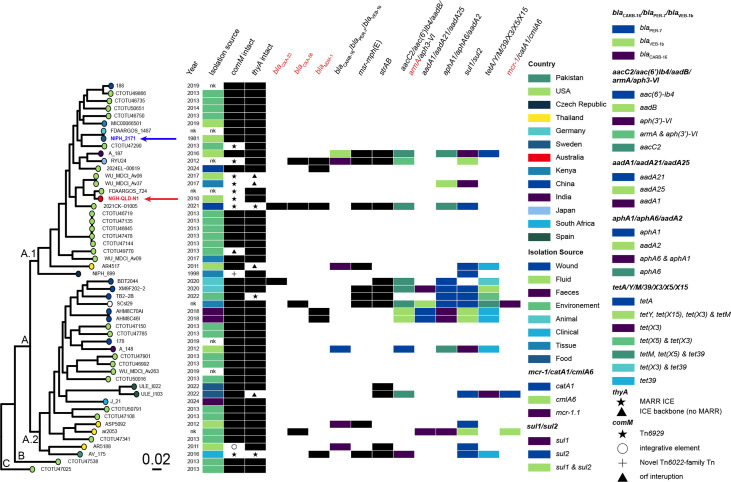 Fig. 3
Fig. 3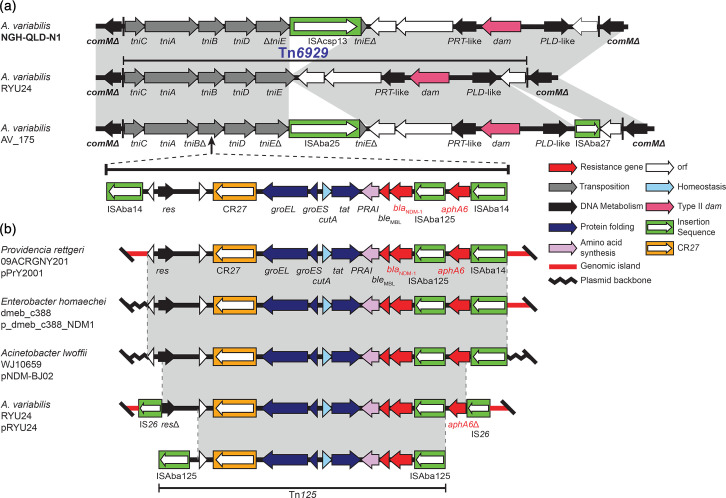 Fig. 4
Fig. 4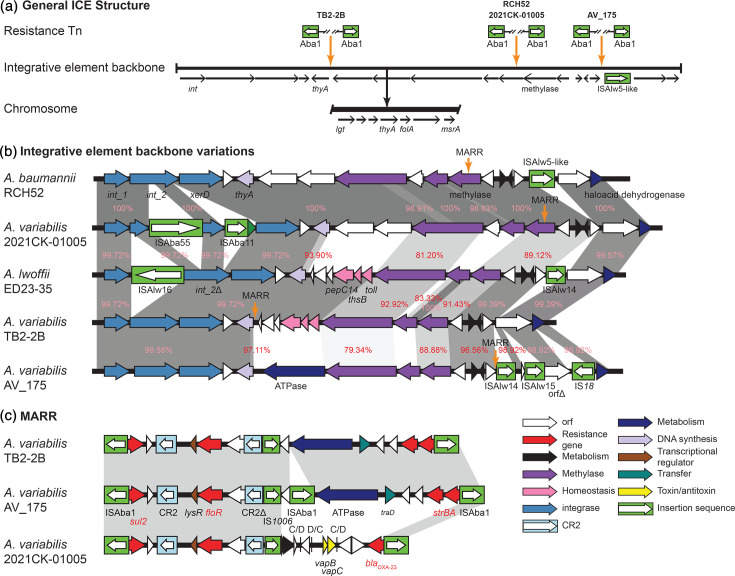 Fig. 5
Fig. 5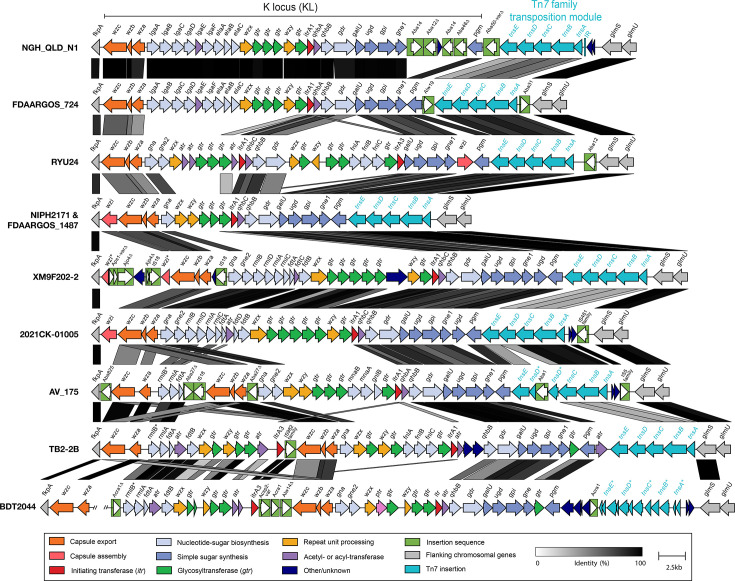 Fig. 6
Fig. 6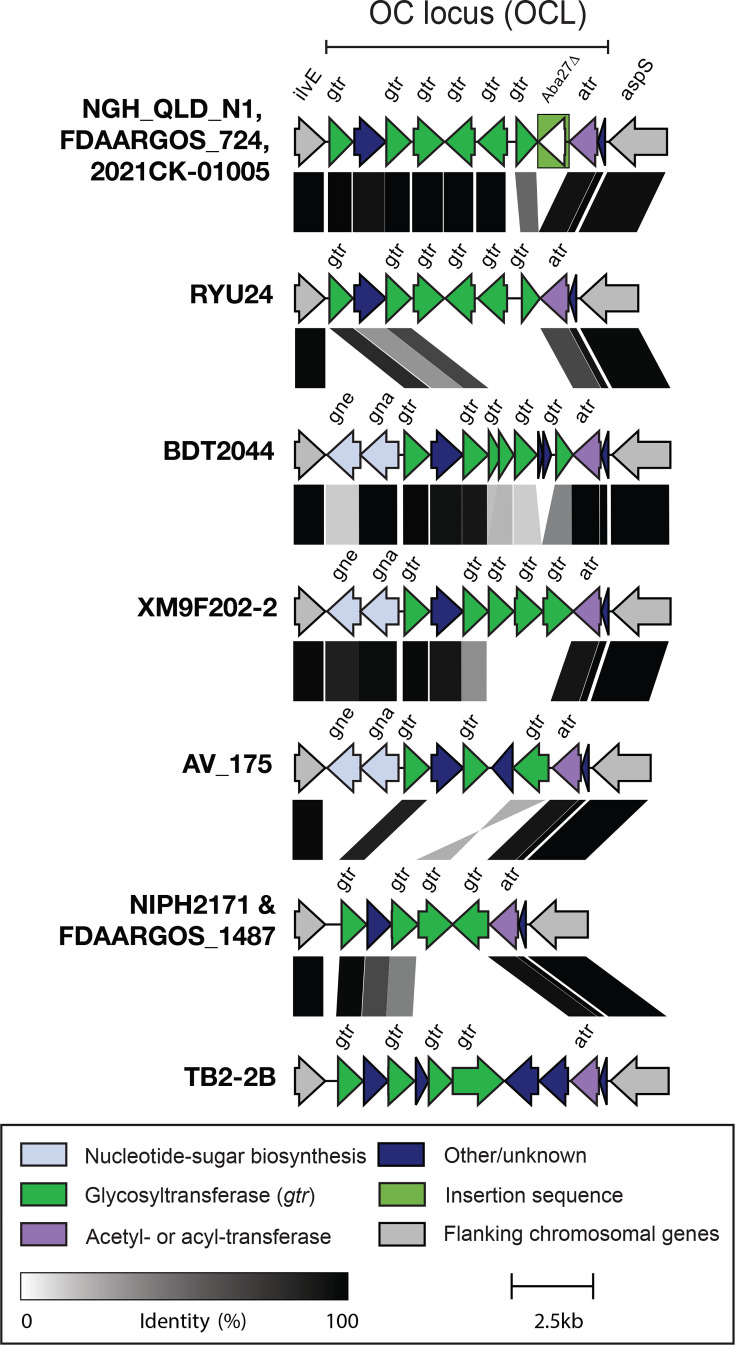 Fig. 7
Fig. 7| Strain/plasmid | Year | Country | Isolation site | Size (bp) | Resistance genes | Plasmid Rep type | Accession no. | |
|---|---|---|---|---|---|---|---|---|
|
| 1981 | Sweden | Urine | Draft | – | |||
|
|
|
|
| 3,230,105 | – | – |
| JBQTGN010000001 |
| p1NGH-QLD-N1 | 7,839 | – | R3-T18 | JBQTGN010000002 | ||||
| p2NGH-QLD-N1 | 7,851 | – | R3-T77 | JBQTGN010000003 | ||||
| p3NGH-QLD-N1 | 10,584 | – | R3-T62 | JBQTGN010000004 | ||||
| p4NGH-QLD-N1 | 12,401 | – | – | JBQTGN010000005 | ||||
| p5NGH-QLD-N1 | 37,499 | – | – | JBQTGN010000006 | ||||
| p6NGH-QLD-N1 | 210,678 | – | R3-T45 | JBQTGN010000007 | ||||
| nk† | USA | Unknown | 3,235,776 | – | – | Tn | ||
| unnamed1 | 5,829 | – | – | |||||
| unnamed2 | 214,175 | – | R3-T45 | |||||
| unnamed3 | 5,819 | – | R3-T85 | |||||
| unnamed4 | 6693 | – | R3-T83 | |||||
|
| 2021 | USA | Wound/abscess | 3,174,744 | – | Tn | ||
| unnamed1 | 185,297 | – | R3-T45 | |||||
| unnamed2 | 123,124 | R3-T28 | ||||||
| unnamed3 | 6,631 | – | R3-T177 | |||||
| unnamed4 | 4,872 | – | R3-T26 | |||||
| unnamed5 | 4,326 | – | R3-T134 | |||||
| unnamed6 | 2,621 | – | R3-T96 | |||||
| unnamed7 | 2,366 | – | R1-T23 | |||||
|
| 2012 | Japan | Faecal swab | 3,198,423 | – | Tn | ||
| pRYU24 | 68,069 | R3-T60 | ||||||
|
| 2016 | Pakistan | Clinical | 3,252,197 | – | Tn | ||
| pAV_175-2 | 196,965 | – | R3-T90 | |||||
| pAV_175-3 | 19,844 | – | R3-T144 | |||||
| pAV_175-4 | 19,843 | R3-T10 | ||||||
| pAV_175-5 | 15,472 | – | R3-T29 | |||||
| pAV_175-6 | 4,226 | – | RP-T11 | |||||
|
| 2022 | China | Soil | 3,171,395 | – | |||
| pTB2-2B-1 | 141,842 | – | R3-T90 | |||||
| pTB2-2B-tetX | 83,648 | R3-T63 | ||||||
| pTB2-2B-3 | 18,235 | – | R3-T116 | |||||
| pTB2-2B-4 | 11,798 | – | R3-T12 | |||||
| pTB2-2B-5 | 9,336 | – | R3-T176 | |||||
| pTB2-2B-6 | 7,661 | R3-T60 | ||||||
|
| nk† | Germany | Unknown | 3,211,351 | – | – | ||
| unnamed1 | 158,619 | – | R3-T90 | |||||
| unnamed2 | 89,812 | – | R3-T80 | |||||
| unnamed3 | 13,782 | – | R3-T29 | |||||
| unnamed4 | 11,089 | – | R3-T11 | |||||
|
| 2020 | China | Chicken faeces | 3,171,535 | – | |||
| pXM9F202-2-2k | 2,301 | – | R1-T24 | |||||
| pXM9F202-2-13k | 13,130 | – | R3-T77 | |||||
| pXM9F202-2-17k | 17,946 | – | R3-T116 | |||||
| pXM9F202-2tetX-90k | 90,430 | R3-T63 | ||||||
| pXM9F202-2-186k | 186,242 | R3-T90 | ||||||
|
| 2020 | China | Pig faeces | 3,345,806 | – | |||
| pBDT2044-1 | 10,546 | – | R3-T196 | |||||
| pBDT2044-2 | 17,439 |
| – | |||||
| pBDT2044-3 | 18,754 | R3-T76 | ||||||
| pBDT2044-4 | 20.030 | – | R3-T84 | |||||
| pBDT2044-5 | 36,028 | – | – | |||||
| pBDT2044-6 | 46,646 |
| – | |||||
| pBDT2044-7 | 55,901 | R3-T20 |
| Gene | Gene present (no. genomes)* | ||||
|---|---|---|---|---|---|
| NGH_QLD_N1 | NIPH 2171 | Complete genomes ( | Draft genomes ( | ||
|
| |||||
|
| + | + | + (8) | + (38) | |
|
| − | − | + (1) | + (18) | |
|
| + | + | + (8) | + (40) | |
|
| |||||
|
| − | − | − | + (3) | |
|
| − | − | − | + (3) | |
|
| − | − | − | + (4) | |
|
| − | − | − | − | |
|
| − | − | − | − | |
|
| − | − | − | − | |
|
| − | − | − | − | |
|
| − | − | − | − | |
|
| − | − | − | + (32) | |
|
| |||||
|
| + | + | + (8) | + (38) | |
|
| + | + | + (8) | + (37) | |
|
| + | + | + (8) | + (40) | |
|
| |||||
|
| ( | − | − | − | − |
|
| |||||
|
| + | + | + (8) | + (39) | |
|
| − | − | − | − | |
|
| |||||
|
| − | + | − | − | |
|
| − | + | − | − | |
|
| − | − | − | − | |
|
| − | − | − | − | |
|
| − | − | − | − | |
| 2Fe-2S | − | − | − | − | |
|
| − | − | − | − | |
|
| − | − | − | − | |
|
| |||||
|
| + | + | + (8) | + (40) | |
|
| + | + | + (8) | + (38) | |
|
| + | + | + (8) | + (39) | |
|
| + | + | + (8) | + (40) | |
| Tn | TniC | TniA | TniB | TniD | TniE | orf4 |
|---|---|---|---|---|---|---|
| Tn | –* | – | – | – | – | – |
| Tn | 71.41% | 69.78% | 71.37% | 67.38% | 68.58% | 27.56% |
| Tn | 75.93% | 72.28% | 71.37% | 67.38% | 66.41% | 27.56% |
| Tn | 67.53% | 66.61% | 73.03% | 57.18% | 56.01% | 26.30% |
- —http://dx.doi.org/10.13039/501100000923 Australian Research Council
- —University of Technology Sydney Research Office
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntibiotic Resistance in Bacteria · Evolution and Genetic Dynamics · Genomics and Phylogenetic Studies
Data Summary
The Acinetobacter variabilis NGH-QLD-N1 genome sequenced in this study is deposited in GenBank and available under the BioProject PRJNA1304695. All 64 publicly available A. variabilis genomes used in this study were downloaded from GenBank with their accession numbers listed in Table S1.
Introduction
The global spread of multidrug-resistant (MDR) bacterial pathogens poses a significant threat to public health and has complicated the treatment and management of infectious diseases across healthcare settings [12]. Among these, members of the genus Acinetobacter, particularly Acinetobacter baumannii, have been extensively studied due to their high levels of antibiotic resistance and global dissemination [35]. As of July 2025, the Acinetobacter genus includes 119 species (https://lpsn.dsmz.de/genus/acinetobacter). While some have found their way into hospitals, causing serious healthcare-associated infections, the majority of species are commonly found in natural and non-clinical environments (e.g. soil, water, food production and domestic animals) [6]. Despite emerging reports of their role in human infections and their potential to carry clinically significant antibiotic resistance genes (ARGs) and virulence genes, non-baumannii species within the Acinetobacter genus remain relatively understudied.
Non-baumannii species have been suggested to serve as reservoirs for ARGs, increasing diversity and spread, particularly through mobile genetic elements. Recently, we showed that plasmids play an important role in the dissemination of ARGs across the Acinetobacter genus [78]. These plasmids have been implicated in the spread of carbapenemases, aminoglycoside-modifying enzymes and other significant resistance determinants. Characterizing the structure, content and phylogenetic context of plasmids within less-studied Acinetobacter species is therefore important to understand their contribution to the potential for interspecies ARG acquisition and spread.
In 2015, Acinetobacter variabilis sp. nov. (formerly known as genospecies 15 sensu Tjernberg and Ursing) was officially named as a novel species isolated from a variety of human clinical specimens and animals [9]. Since the identification of this species, few studies have reported A. variabilis strains recovered from a variety of clinical, environmental and animal samples in different countries in Asia, Europe and the USA [1013] with no reports from Australia. The earliest reported isolate is the type strain, * A. variabilis* NIPH 2171, which had been recovered from the urine of a patient in Malmö, Sweden, in 1980–1981 (GenBank accession number APRS01000000) [14]. However, despite early isolation and numerous reports [1013], analysis of genomic data for * A. variabilis* remains limited, with only 64 whole-genome sequences, and none from the Oceania region, currently available in NCBI databases (as of July 2025). There have also been a limited number of studies reporting the genetic context of antibiotic resistance determinants [101213], or that have investigated the broader diversity in virulence genes across the species, including for the capsular polysaccharide (CPS) and lipooligosaccharide (LOS).
A. variabilis isolate NGH-QLD-N1 was recovered in 2010 from a blood sample of a patient at the Nambour General Hospital (NGH) on the Sunshine Coast region of Queensland, Australia. Here, we sequenced the complete genome of NGH-QLD-N1 using short- and long-read sequencing data and confirmed the isolate as a member of the A. variabilis species. We examined its genomic features and phylogenetic relationship with other A. variabilis genomes and analysed the genomic context of ARGs and loci for major virulence determinants, including the CPS and LOS. Our findings show that transposons related to the Tn6022 family, which are frequently found in A. baumannii, integrative conjugative elements (ICEs) and plasmids found in A. variabilis may serve as a reservoir and play a significant role in driving antimicrobial resistance spread across the genus.
Methods
Bacterial isolate and antibiotic resistance profile
NGH-QLD-N1 was recovered in a blood sample of a patient at NGH, Queensland, Australia, in 2010. The antibiotic resistance profile of NGH-QLD-N1 was determined against 23 antibiotics (shown in Table S1, available in the online Supplementary Material) as we described previously [15] using the standard Calibrated Dichotomous Sensitivity disc diffusion method [16].
Whole-genome sequencing and assembly
Whole-cell DNA was isolated from a single colony of A. variabilis NGH-QLD-N1 cultured in Luria–Bertani medium at 37 °C overnight using the PureLink^™^ Genomic DNA mini kit (Thermo Fisher Scientific). Concentration was measured using the Invitrogen Qubit system and purity by spectrophotometry (NanoDrop, Thermo Fisher Scientific). The whole-genome sequence was obtained via hybrid sequencing: Illumina MiSeq at the Australian Genome Research Facility, Australia, and Oxford Nanopore Technology at Plasmidsaurus, USA. Raw read quality was assessed with FastQC [17], and adapter trimming was performed using Btrim v0.2.0 [18]. De novo assembly of the complete genome was completed using Autocycler v0.1.2 (https://github.com/rrwick/Autocycler/tree/v0.2.0), followed by polishing with the Illumina data using Polypolish (https://github.com/rrwick/polypolish). The complete genome assembly consisted of a 3,230,105 bp chromosome and 6 plasmids ranging in size from 7.8 kb to over 210 kb. The final assembly was submitted to NCBI and is available under BioProject PRJNA224116 and BioSample SAMN50566246, and accession number JBQTGN000000000.
Collation and annotation of available A. variabilis genome sequences
As of July 2025, a total of 64 genome assemblies were available in NCBI for the A. variabilis taxon (ID: 70346). Genome quality, including completeness and contamination metrics, was evaluated using CheckM v1.2.3 [19] for all genomes analysed, including NGH-QLD-N1 sequenced in this study and those retrieved from NCBI. This led to 13 genomes being removed from further analyses due to a CheckM completeness score of zero. Hence, a total of 52 genomes, including A. variabilis NGH-QLD-N1, were analysed in this study (accession numbers listed in Table S1). Whole-chromosome sequence alignments were constructed using Proksee [20] or pyGenomeViz (https://github.com/moshi4/pyGenomeViz) using the blast workflow with default parameters.
ARGs were detected using the AMRFinderPlus v.4.0 program [21]. Plasmid replicon types (rep/Rep) were identified using the Acinetobacter plasmid typing scheme [22]. Insertion sequences were identified using ISFinder [23] and Standalone blast v2.16.0 [24]. Prophage elements were detected using PHASTER (PHAge Search Tool Enhanced Sequence Translation; https://phaster.ca/). Virulence genes, including CPS (KL) and LOS outer core (OCL) biosynthesis loci, were manually identified by searching for homologues of A. baumannii proteins using BLASTp and were annotated consistent with the nomenclature system described for A. baumannii [25]. Protein-coding sequences were functionally annotated via UniProt [26] and InterPro [27]. Average nucleotide identities (ANIs) of the complete genomes were calculated using FastANI v1.34 (available at https://github.com/ParBLiSS/FastANI). Genomic regions of interest were visualized using SnapGene^®^ v6.0.5 or Clinker v0.0.28 (available at https://github.com/gamcil/clinker) and then annotated in Illustrator^®^ v26.2.1.
Phylogenetic analysis and pangenome reconstruction
Maximum-likelihood phylogenetic (MLP) trees were constructed (using the NIPH 2171 genome as a reference) by aligning genome sequences using Panaroo [28] and analysed with IQ-TREE v2.3.6. The high-quality core-genome SNPs were identified and exported for further refinement. The final MLP tree was inferred from the alignment of substitution mutations rooted using an A. baumannii outgroup (strain ATCC 17978; GenBank no. CP012004). The final MLP tree was visualized using FigTree v1.4.4. Pangenome analysis was performed using Panaroo [28] in ‘strict mode’ with the core defined as genes present in 100% of genomes, the soft core as >95% and the shell genome as 15–95%. Additionally, antimicrobial resistance genes were plotted against the recombination-free tree using the plotTree v1.0 (https://github.com/katholt/plotTree) and ggplot2 [29] v3.5.1 packages in R.
Results and discussion
Resistance profile, genomic features of NGH-QLD-N1 and A. variabilis genomes
A high-quality hybrid genome assembly (Illumina and Oxford Nanopore) was obtained for NGH-QLD-N1, an isolate recovered from a blood infection at NGH in Queensland, Australia. The complete genome sequence was found to be a total of 3,517,317 bp with an average G+C content of 42.19 %, which included a 3,230,105 bp chromosome (Fig. 1a; NCBI accession number CM126357.1) and 6 plasmids ranging in size from 7.8 kb to over 210 kb (Table 1). NGH-QLD-N1 lacked the intrinsic ampC and oxaAb genes, which are characteristic for A. baumannii, suggesting that it is non-baumannii. To determine the species, we compared the DNA sequences of several chromosomal genes, including the recA and the 16S rRNA genes, with those of other known Acinetobacter species and found 100% identity with A. variabilis genomes. To confirm the species, phylogenetic analysis was performed with reference genomes of all Acinetobacter species (over 90 species), showing NGH-QLD-N1 clustering with the representative A. variabilis genome (Fig. S1). This was further validated by ANI analysis, which showed 95.23% identity across the genome.
(a) Acinetobacter variabilis NGH-QLD-N1 genome, showing BLAST comparison to A. variabilis NIPH 2171 and the location of the KL/wzi and OCL loci, as well as Tn6929 and phage regions. (b) Average nucleotide identity of nine complete and one near complete A. variabilis genomes. The number of single nucleotide differences are shown and also indicated by the coloured scale.
The general features of NGH-QLD-N1 were compared with eight other complete A. variabilis genomes identified in the NCBI database and one near-complete draft genome (contigs n=23) for the A. variabilis type strain NIPH 2171 (Table 1). Like NGH-QLD-N1, the average chromosome size of these genomes ranged from 3.1 Mb to just over 3.3 Mb, and their G+C content ranged from 42.04 to 42.38%, indicating that the average G+C content for A. variabilis is just over 42%. In comparison to A. baumannii, the average chromosome size for A. variabilis is significantly smaller (typically 4 Mb in A. baumannii), while the G+C content is higher (e.g. 39.01% for A. baumannii ATCC 17978 and 39.17% for A. baumannii ATCC 19606; determined here). Pairwise genome comparisons among the 10 available complete genomes (Fig. 1b) revealed between 38 and 101,765 single-nucleotide differences (SNDs), corresponding to roughly 99.99% down to ~97.1% genome-wide nucleotide identity across a ~3.5 Mb genome. While based on a limited dataset, this range of divergence is comparable to levels of within-species genomic heterogeneity reported for other Acinetobacter species, including A. baumannii [3031], and is consistent with the genus-level pattern of an open pangenome and high genomic plasticity [32]. The observed SND range is indicative of genomic heterogeneity among the currently sampled members of this species. However, as additional genomes become available, the extent and structure of this diversity can be further evaluated and refined.
The NGH-QLD-N1 chromosome was therefore aligned with the earliest recovered isolate, NIPH 2171 (Fig. 1a), and several regions of difference were identified. One of these was found to be a prophage region (P3 in Fig. 1a), whereas others indicated differences at comM, a common location disrupted by antibiotic resistance islands in A. baumannii [3433], and a locus predicting synthesis of a surface polysaccharide (see below). NGH-QLD-N1 was also found to include several IS, including six copies of ISAcsp13, four copies of ISAba25 and many novel IS, including one distantly related to ISAba59 (IS5 family) with 87% DNA identity and a second novel IS distantly related to ISAlw33 (IS30 family), also with 87% DNA identity. The latter two IS (related to ISAba59 and ISAlw33) were also present in NIPH 2171.
To further identify whether the regions of difference were associated with the locations of resistance determinants, the NGH-QLD-N1 genome was searched for ARGs. However, none were identified. The isolate was therefore tested against a panel of 23 antibiotics and found to be susceptible to all antibiotics tested except for trimethoprim (Table S1). Further analysis of the genome revealed that NGH-QLD-N1 carried a single chromosomal resistance gene (locus tag ACTNFC_13365) encoding a FolA homologue (97.65% aa identity to DfrA40) that would account for resistance to trimethoprim [34]. This gene was also identified in the NIPH 2171 chromosome (Fig. 1a) and the other complete genomes, suggesting that the species may be intrinsically resistant to trimethoprim.
A. variabilis species has a diverse pangenome
A total of 64 genome assemblies, including NIPH2171 and the 8 complete genomes, were identified in NCBI under the A. variabilis taxon (ID: 70346). Completeness and contamination of all 64 genomes were assessed using CheckM, and 13 draft assemblies did not pass our quality metrics and were therefore removed from the dataset. The final pool of 52 genomes (including NGH-QLD-N1) represented diverse isolates recovered from a range of clinical and non-clinical samples (i.e. soil, water, food and animals) from several countries across Asia, Europe and the USA. However, none from NCBI were found to originate from South America or Australia. Hence, NGH-QLD-N1 represents the first A. variabilis genome to be reported from the Oceania region. Pangenome analysis of the A. variabilis set identified a total of 8,410 unique genes, with a relatively small core genome of just 15.4% (1,294 genes across 52 genomes) (Fig. 2). The accessory genome was extensive, comprising 905 soft core genes, 1,329 shell genes and 4,882 cloud genes (Fig. 2). Our dataset included a diverse collection of A. variabilis genomes, likely representing multiple sequence types, comparable to previous studies of diverse A. baumannii genomes, which reported core genomes of ~13% [3536]. The genomic diversity identified across the A. variabilis population supports the original phenotypic heterogeneity observations, which inspired the species name ‘variabilis’, reflecting its variable nature [9].
Phylogenetic tree and the pangenome heatmap of 52 Acinetobacter variabilis genome sequences. The pangenome contains 8, 410 unique genes with gene presence/absence indicated by the blue scale shown.
Phylogenetic analysis of A. variabilis genomes
To assess the population structure of the species, an MLP tree was constructed using NGH-QLD-N1 and the 51 publicly available A. variabilis genomes. The phylogeny (Fig. 3) revealed three major clades, named A, B and C, with clade A comprising the majority of isolates in two subclades (A.1 and A.2). However, clades B and C each contain a single genome and were regarded as provisional lineages as their distinct phylogenetic placement may reflect under-sampled population structure rather than well-defined clades. Both NGH-QLD-N1 and the earliest isolate, NIPH 2171, were found in A.1 (highlighted in Fig. 3), which is hereafter referred to as the NGH-QLD-N1 subclade. The eight other complete genomes were found across multiple clades in the phylogeny. Genomes in clade A originated from diverse geographical regions, including the USA, Kenya, South Africa, Pakistan and several European and Asian countries, with all but two (NIPH 2171 from 1981 and NIPH 899 from 1998) recovered after 2010 (Fig. 3).
Maximum likelihood phylogenetic tree of Acinetobacter variabilis (n=52) genome sequences. Next to each A. variabilis strain the year, country and source of isolation are shown. Presence of antimicrobial resistance genes are also indicated, by the colour scheme shown.
Each genome was assessed for known ARGs, and this information was overlaid on the phylogeny. While ARGs were not detected in NGH-QLD-N1 and several other isolates predominately within the NGH-QLD-N1 subclade, 21 genomes from diverse geographical regions contained at least 1 resistance gene. Except for a few related genomes (e.g. see Fig. 3), these genomes generally belonged to unrelated phylogenetic sub-lineages and were distributed across the phylogenetic tree. However, despite the relatively low abundance of resistance genes in A. variabilis genomes in general, several clinically significant carbapenemase genes, including blaOXA-23, blaOXA-58 and blaNDM, the 16S rRNA methyltransferase gene armA, conferring high-level resistance to all aminoglycoside antibiotics, and the *mcr-*1 colistin resistance genes were detected. Several strains from diverse geographical regions, including China, Thailand, Pakistan and the US, also contained tetA(A), tetY, tetM, tet39, tetX3, tetX5 and tetX15 tetracycline resistance genes (Fig. 3). Overall, with the exception of five strains from China that were grouped together (XM9F202−2, BDT2044, TB2−2B, AHM8C46I and AHM8C70AI; Fig. 3), the remaining genomes with multiple resistance genes were distributed across the phylogenetic tree without a distinct geographical or temporal pattern.
Within the NGH-QLD-N1 subclade, two closely related strains, RYU24 (Japan) and 2021CK−01005 (USA), along with two more distantly related genomes, AV_175 (Pakistan) and A_187 (China), contained multiple resistance genes, including those for carbapenems, aminoglycosides, sulphonamides and tetracyclines. Notably, RYU24 carried two carbapenemase genes, blaOXA-58 and blaNDM, while 2021CK−01005 carried three, with the addition of blaOXA-23. Eight genomes outside the NGH-QLD-N1 subclade also exhibited a similar profile, containing several clinically important resistance genes (Fig. 3).
A. variabilis genomes were further assessed for common virulence determinants in A. baumannii [37], and the results are summarized in Table 2. Most isolates encoded the Blc and SmpA outer membrane proteins, though OmpA homologues were found in less than half the genome pool. Very few isolates carried genes predicting pilin or siderophore biogenesis. However, almost all isolates encoded proteins associated with CPS biosynthesis and assembly [25], as well as a PglL homologue required for O-glycosylation of proteins with CPS glycan units [38]. A homologue of the WaaL ligase required for the attachment of an O-antigen polysaccharide to form LPS could not be detected, suggesting that, like A. baumannii, A. variabilis does not produce LPS with O-antigen but rather LOS consisting of the lipid A-core oligosaccharide components only [2539]. Consistent with this hypothesis, the majority of genomes encoded a homologue of Wzi required for mediating CPS assembly on the cell surface [40]. As for A. baumannii, the gene coding for this protein is located far from the CPS biosynthesis K locus (Fig. 1a) and, in * A. variabilis* genomes, is found between a lysine--tRNA ligase and tryptophan--tRNA ligase gene. An additional copy of wzi was also identified in some genomes (see below).
As resistance and virulence genes are often found on plasmids or in genomic islands that can be highly fragmented in bacterial genomes, analysis of their genetic context was completed using the NGH-QLD-N1, NIPH 2171 and eight other complete genomes (Table 1).
Plasmids
Similar to NGH-QLD-N1, most of the complete genomes contained at least four plasmids (Table 1). Interestingly, several plasmids were found to lack the R1, R3 or RP plasmid Rep types, which is also common in A. baumannii [7822]. However, variants of the R3-type (Rep_3) plasmids were among the most widespread. Among these R3-type Rep variants, R3-T80, T83, T85, T90, T116, T134, T173, T176 and T196 have not been detected in A. baumannii, while others, such as R3-T144 and T177, have been reported only in A. variabilis [7]. In contrast, variants such as R3-T11, T12, T20, T23, T29, T45, T60, T76 and T84 were found in multiple A. variabilis complete genomes (Table 1) [7]. In addition, we found several plasmids lacking R1, R2 or RP rep/Rep, but belonging to plasmid families we recently defined (e.g. the pA207-3 type and its variants) [7]. Among all complete plasmids, only pBDT2044-7 was predicted to be potentially conjugative, as it appeared to encode a complete set of conjugative transfer functions belonging to the MPF^T^ [41] system (locus IDs MOV98_17755–MOV98_17800 in GenBank accession CP094253).
Plasmids identified in 2021CK-01005, RYU24 and BDT2044 genomes carry several important ARGs, including the carbapenem resistance genes blaOXA-58, blaOXA-23 and blaNDM, as well as armA and the amikacin resistance gene, aphA6 (Table 1). For plasmid pBDT2044-6, the blaOXA-23 carbapenem resistance gene was located in the Tn2008 transposon, which is one of the common transposons that disseminate this gene in Acinetobacter [34]. For the MDR isolate RYU24, the genetic context of blaNDM, blaOXA-420 and armA genes in the 68, 069 bp plasmid, pRYU24, has been described previously [13], and each was reported to be surrounded by complete or remnant IS elements. Here, we identified that the blaNDM gene was in a variant of Tn125 (a transposon known to mediate the spread of blaNDM) with an IS91 interrupting the groL gene, and we detected a remnant of aphA6 interrupted by an IS26 (immediately upstream). The archetypal form of Tn125 carrying blaNDM and bleMBL was instead identified in the unnamed2 plasmid from 2021CK-01005. However, in this plasmid, aphA6 was located immediately downstream of Tn125 and was bounded by ISAba125 elements. The blaOXA-58 gene was located ~2–3 kb upstream of Tn125. Overall, in most complete A. variabilis genomes, plasmids appear to play an important role in carrying ARGs, as most ARGs were plasmid-borne rather than chromosomal.
Variants of Tn6929, a novel transposon related to Tn6022-family transposons, carry the blaNDM carbapenem resistance gene
In A. baumannii strains belonging to major global clones, the chromosomal comM gene is often disrupted by large antibiotic resistance islands (AbaR-type in ST1, AbGRI-type in ST2 and AbaR4 in various other STs), which comprise transposon backbones (e.g. Tn6019 and Tn6022, known as class III transposons) and a complex resistance region [33]. Analysis of the comM gene in NGH-QLD-N1 revealed disruption by a 17,051 bp element (bases 279,811–296,861 in GenBank accession no. JBQTGN010000001) showing characteristics like Tn6019 and Tn6022, and other transposons belonging to this family [33]. This region (Fig. 4a) was found to include genes that encode a set of transposition functions (TniCABDE) related to those found in Tn6019 and Tn6022. It was also found to be flanked by 26 bp inverted repeats (IRl: TGTCATATACTATAATAAAAGCTAGT and IRr: ACTAGCTTTTATTTTTGTAAATGACA), generating 5 bp (GCCAC) target site duplications (TSDs), and inserts precisely at the same location in the comM gene where variants of Tn6019 and Tn6022 and other large transposons belonging to this family (i.e. AbaR-type and AbGRI islands) are found. The tni module is more closely related to those encoded by Tn6022 [42], Tn6021 [33] and Tn6173 [33], with 56–75% aa identities (Table 3), than to those of Tn6019 [33] with no significant matches. However, the tni module shares other characteristics (i.e. IRs and TSDs) with all other known class III transposons.
Genetic structure of a novel transposon in A. variabilis. (a) Variants of the novel Tn6929 in three Acinetobacter variabilis genomes, including the resistance region containing the blaNDM carbapenemase gene and the aphA6 amikacin resistance gene in AV_175. (b) Comparison of the genetic structure of the AV_175 resistance region identified in closely related genera. Arrows indicate orientation and genes are coloured by their function. Open boxes coloured green show insertion sequences with their transposase gene indicated inside the boxes. Colour scheme is shown.
In NGH-QLD-N1, the backbone of this Tn is interrupted by an ISAcp13. However, an intact version without this interruption was identified in the complete genome of isolate RYU24 (bases 265,584–280,057; GenBank accession no. AP024524). Hence, this intact version was named Tn6929. The sequence was found to encode a putative methylase (annotated as Type II dam gene in Fig. 4) that belongs to the IPR000241 protein family (Table 1). The comM gene was found intact in NIPH2171, BDT2044, XM9F202-2, FDAARGOS_1487 and TB2-2B. However, additional variants of Tn6929 were identified in three of the other complete genomes (Table 1). For example, FDAARGOS_724 included a version interrupted by ISAlw34, ISAba12 and ISAba22, and 2021CK-01005 contained another version interrupted by ISAba125 and ISAba16.
Notably, A. variabilis AV_175 (GenBank no. CP078028) includes a much larger version of Tn6929, containing a 14,275 bp resistance region that carries the blaNDM carbapenemase gene and the aphA6 amikacin resistance gene (Fig. 4a). This region is bounded by two copies of ISAba14 and flanked by 3 bp TSDs characteristic of ISAba14 insertion. While the internal segment of the blaNDM-aphA6 region was identical to Tn125, it was different to the forms found in pRYU24 and the 2021CK-01005 unnamed2 plasmid (described above). Hence, it appears that the 14 kb ISAba14-bounded resistance region was acquired from another variant of this region, also associated with ISAba14. Supporting this hypothesis, we identified several closely related regions in distantly related genera (Fig. 4b), including the pPrY2001 plasmid (Providencia rettgeri strain 09ACRGNY201; GenBank no. KF295828), the p_dmeb_c388_NDM1 plasmid (Enterobacter hormaechei strain dmeb_c388; GenBank no. CP095673) and the pNDM-BJ02 plasmid (Acinetobacter lwoffii strain WJ10659; GenBank no. JQ060896). These findings highlight the potential for ISAba14-associated resistance regions to disseminate across diverse bacterial hosts, posing a risk for interspecies transfer of the most clinically significant carbapenem resistance gene, blaNDM. It also demonstrates the potential for Tn6929 to spread to other Acinetobacter species, including A. baumannii, given the proven ability of class III transposons (i.e. Tn6022 and Tn6019) [433] to capture and mobilize resistance genes within globally disseminated, high-risk clones.
Amongst the draft A. variabilis genomes, 37 included an intact comM gene (Fig. 3), and 8 contained a variant of Tn6929. Variants of Tn6929 were identified in distinct branches of the phylogenetic tree, indicating that these acquisitions occurred independently on multiple occasions. A single genome, NIPH 899, included a novel cryptic transposon (29,543 bp Tn, located at bases 262,001–291,543 in GenBank no. APPE01000087) belonging to the Tn6019–Tn6022 family. This region encodes several proteins involved in various metabolic pathways and many hypothetical proteins. The comM gene was also interrupted by a novel ICE in AR5188 (GenBank no. JAZHCM000000000) and CTOTU47290 (GenBank no. DAISMA000000000). These findings highlight the role of transposons related to Tn6019, Tn6022 and Tn6929 to spread a wide range of genetic material and the comM gene as a hotspot to capture these elements in different Acinetobacter species, including A. baumannii and A. variabilis.
ICEs as potential hotspots for ARG acquisition in A. variabilis
We previously characterized a novel ICE in an A. baumannii strain (RCH52, recovered in Australia) [43], which contained a 129 kb ISAba1-bounded multiply antibiotic resistance region (MARR), integrated into the chromosomal thyA gene (Fig. 5a). This ISAba1-bounded MARR comprised two clusters of resistance genes separated by a large segment of the type 1 IncC plasmid backbone [43]. Our earlier work showed that several resistance genes originating from IncC plasmids had been brought together through an IS26-mediated deletion of the original plasmid [43]. This composite unit was then incorporated into an ISAba1-bounded segment containing blaOXA-23 [43]. Here, we identified several related ICE variants in the chromosomal thyA gene in the three complete A. variabilis genomes, TB2-2B, AV-175 and 2021CK-01005, with different ISAba1-bounded regions that lacked the IncC plasmid segment. The thyA gene was found uninterrupted in NGH-QLD-N1, NIPH 2171 and the five other complete genomes.
Integrative conjugative element (ICE) in Acinetobacter variabilis. (a) General ICE structures, (b) backbone variations in A. variabilis and other Acinetobacter spp. and (c) variants of ISAba1-bounded multiply antibiotic resistance regions (MARR) in A. variabilis. Gene clusters are drawn to scale and genes coloured by their predicted functional groups; arrows show the gene orientation. Open boxes coloured green show insertion sequences with their transposase gene indicated inside the boxes.
The elements identified in TB2-2B, AV-175 and 2021CK-01005 included several differences in the ICE backbones (i.e. additional/missing segments and regions with different identities; Fig. 5b), indicating that these ICE elements belong to a diverse family. The backbone in the TB2-2B genome was also found to be similar to one identified in the genome of A. lwoffii strain ED23-35 (Fig. 5b), indicating that they are also widespread across the Acinetobacter genus. All three variations identified in A. variabilis also included different variants of an ISAba1-bounded MARR integrated at different positions in the ICE backbone (Fig. 5c) compared to the MARR previously described for RCH52 [43]. However, the variant found in A. variabilis 2021CK-01005 also contained a blaOXA-23 carbapenem resistance gene. Genomes containing ICE variants were found in distinct phylogenetic branches and originated from diverse geographical regions, suggesting that these variants were acquired independently.
The thyA gene was further assessed in all draft A. variabilis genomes and was found interrupted by an ICE backbone in a further five genomes, ULE_I103 (GenBank no. JBIMAH000000000), WU_MDCI_Av06 (GenBank no. JAHPPI000000000), WU_MDCI_Av37 (GenBank no. JAHPQG000000000), AR4517 (GenBank no. JAZHCO000000000) and CTOTU47290 (GenBank no. DAISMA000000000). However, these included a cryptic version of the ICE, as a MARR could not be identified in any of them (not shown in [Fig. 5](#F5 F5) as structures span multiple contigs). The variations observed in the ICE elements further indicate that they belong to a diverse family that can capture and disseminate important resistance genes (i.e. blaOXA-23) in A. variabilis, and potentially other Acinetobacter species.
Locus for CPS biosynthesis
The CPS biosynthesis K locus has been studied extensively in A. baumannii [254445] and is located between conserved fkpA and lldP genes on the chromosome. A region including CPS-associated genes was similarly located immediately upstream of the fkpA gene in the chromosome of NGH-QLD-N1 (Fig. 6). The organization of this region is largely analogous to that of * A. baumannii*, with a module of genes (wza/wzb/wzc) for CPS export at one end of the locus, and a module of genes (galU–pgm) for simple sugar synthesis on the other side. The central region included genes predicting synthesis of 8-epilegionaminic acid (lgaABCDEF/elaABC) [46] and bacillosamine (qhbA/qhbB/gdr) sugar precursors [25], as well as wzx and wzy genes, indicating that CPS biosynthesis in A. variabilis follows the Wzy-dependent pathway as in A. baumannii [25]. The galU–pgm module is known to include insertions between gpi and pgm in A. baumannii [25], and this region in NGH-QLD-N1 was found to include IS remnants that flank a gene coding for a hypothetical protein related to type IV pilus proteins.
Capsular polysaccharide biosynthesis loci identified in A. variabilis complete genomes. Loci are drawn to scale using Clinker, and grey shading indicated sequence identity with scale shown below. Arrows show gene orientation, and genes are coloured by their predicted functional groups with the key shown below. Genes are annotated consistent with the nomenclature scheme for A. baumannii. A * suffix indicates gene interruption. The large sequence insertion in BDT2044 was removed and the position of the break is indicated.
For NIPH 2171 and the other complete genomes, a different combination of genes was found at this location. While most carried a unique configuration, some shared modules of genes for the synthesis of complex sugars, which have also been reported in * A. baumannii* previously, e.g. gna/gne2 for N-acetyl-d-galactosaminuronic acid, fnlABC for N-acetyl-l-fucosamine, rmlBDAC for l-rhamnose and qhbA or qhbC/qhbB/gdr for bacillosamine variants [44]. Six of the loci included additional genes between fkpA and wzc (Fig. 6). Three of these carried a wzi gene at this location, though this was interrupted by several IS and IS remnants in XM9F202-2. The three other loci included additional CPS biosynthesis genes in this region, including a second copy of wza and wzc export genes. However, in the BDT2044 chromosome (NCBI accession number CP094246.1), a large insertion (~250.5 kb) interrupting the locus was identified at this location (Fig. S2). This insertion included two predicted prophage regions and several segments homologous to known Acinetobacter plasmids encoding R3-T90 [7]. A large island within this region that includes 13 variant copies of tet(X3) conferring tigecycline resistance had been described for BDT2044 previously, and these elements were shown to be potentially mobilizable via the finding of circular intermediates [10].
Unlike A. baumannii, lldP was not found at the distal end of the locus in any of the A. variabilis genomes studied. Instead, several variations of a region, including five genes, were identified downstream between pgm and glmS/glmU. These five genes encode proteins distantly related to TnsABCDE of Tn7, with 39%, 40%, 49%, 38% and 29% amino acid identity, respectively. We also found a 28 bp sequence (TTCTTGTTCGTAATCTACTACTAGTCAC) located 426 bp upstream of the 5′ end of the tnsA gene in NGH-QLD-N1 that was absent in other variants. However, we could not detect the other end of the transposon in NGH-QLD-N1 (or any other genome), indicating that it is a remnant of a novel Tn7-family transposon. The other end may have been deleted due to an IS-mediated adjacent deletion, given the presence of several IS elements downstream of the tnsE gene. In several variants, tnsA was located immediately adjacent to the 3′ end of the glmS gene, the target site for Tn7 and related transposons [47]. We also identified this Tn7-family transposition module in several draft genomes (including SCsl29, WU_MDCI_Av37, WU_MDCI_Av06, A_187, AR4517, WU_MDCI_Av09, ASP5092 and Ar2053), indicating an early entry into A. variabilis.
Locus for biosynthesis of the LOS outer core
In species such as A. baumannii that produce LOS, rather than an LPS inclusive of an O-antigen moiety, genes for the outer core (OC) component of the core oligosaccharide of the LOS have been shown to vary substantially [254849]. In NGH-QLD-N1, NIPH 2171 and the eight complete genomes, a locus including genes predicted to be responsible for OC synthesis was identified between ilvE and aspS genes. As this is the same location for the OC locus in A. baumannii [254849], this region was also designated as the OC locus in A. variabilis. Seven different OC loci were found amongst the ten genomes (Fig. 7), and in general, each was predominately composed of glycosyltransferase genes. NGH-QLD-N1 had the same genetic arrangement as FDAARGOS_724 and 2021CK-01005, whereas NIPH2171 had a different locus type that was also shared with FDAARGOS_1487. The five other genomes carried different locus variations, and all arrangements were different to those previously reported in A. baumannii [49].
Outer core oligosaccharide biosynthesis loci identified in A. variabilis complete genomes. Loci are drawn to scale using Clinker. Grey shading shows sequence identity and scale is below. Genes are coloured by their predicted functional groups with the key below and are annotated consistent with the nomenclature scheme for A. baumannii.
Conclusions
This study presents the complete genome sequence of the earliest clinical Australian A. variabilis strain, recovered in 2010, and examines its phylogenetic relationship and other genetic features compared with all publicly available A. variabilis genomes. We identified major genomic regions, including the CPS and OC biosynthesis loci. In addition, despite harbouring only one resistance determinant, we characterized Tn6929, a class III transposon related to the Tn6022 family, and its variants in the chromosomal comM gene of NGH-QLD-N1 and several A. variabilis genomes*.* One of these carried blaNDM and aphA6 within an ISAba14-bounded variant region of Tn125, while other variants of this transposon were identified on A. variabilis plasmids suggesting mobilization of this region and acquisition from diverse bacterial hosts. Multiple ICE variants were also identified in the chromosomal thyA gene, differing in backbone structure and resistance gene content from those previously described for * A. baumannii*. These findings highlight Tn6929 and ICEs as important genomic elements for the capture of resistance genes in * A. variabilis*, particularly in the chromosomal thyA and comM genes, with potential to spread to other species within the Acinetobacter genus. This study serves as a foundational genomic analysis of A. variabilis for future studies of this species.
Supplementary material
10.1099/mgen.0.001643Uncited Supplementary Material 1.
10.1099/mgen.0.001643Uncited Table S1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Murray CJL Ikuta KS Sharara F Swetschinski L Robles Aguilar G et al Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis The Lancet 202239962965510.1016/S 0140-6736(21)02724-0PMC 884163735065702 · doi ↗ · pubmed ↗
- 2Sartorius B The burden of antimicrobial resistance in the americas in 2019: a cross-country systematic analysis Lancet Reg Health Am 2023251005613772759410.1016/j.lana.2023.100561 PMC 10505822 · doi ↗ · pubmed ↗
- 3Hamidian M Nigro SJ Emergence, molecular mechanisms and global spread of carbapenem-resistant Acinetobacter baumannii Microb Genom 2019510.1099/mgen.0.000306 PMC 686186531599224 · doi ↗ · pubmed ↗
- 4Cain AK Hamidian M Portrait of a killer: Uncovering resistance mechanisms and global spread of Acinetobacter baumannii P Lo S Pathog 202319 e 101152010.1371/journal.ppat.101152037561719 PMC 10414682 · doi ↗ · pubmed ↗
- 5Zarrilli R Pournaras S Giannouli M Tsakris A Global evolution of multidrug-resistant Acinetobacter baumannii clonal lineages Int J Antimicrob Agents 201341111910.1016/j.ijantimicag.2012.09.00823127486 · doi ↗ · pubmed ↗
- 6Nemec A Krizova L Maixnerova M van der Reijden TJK Deschaght P et al Genotypic and phenotypic characterization of the Acinetobacter calcoaceticus-Acinetobacter baumannii complex with the proposal of Acinetobacter pittii sp. nov. (formerly Acinetobacter genomic species 3) and Acinetobacter nosocomialis sp. nov. (formerly Acinetobacter genomic species 13TU)Res Microbiol 201116239340410.1016/j.resmic.2011.02.00621320596 · doi ↗ · pubmed ↗
- 7Tobin LA Lam MMC Hamidian M Pan-genus analysis and typing of antimicrobial resistance plasmids in Acinetobacter NPJ Antimicrob Resist 202536510.1038/s 44259-025-00133-z 40696136 PMC 12284255 · doi ↗ · pubmed ↗
- 8Lam MMC Hamidian M Examining the role of Acinetobacter baumannii plasmid types in disseminating antimicrobial resistance NPJ Antimicrob Resist 20242110.1038/s 44259-023-00019-y 39843567 PMC 11702686 · doi ↗ · pubmed ↗