Cultivation and genomic characterization of novel methanogens from arid desert biocrust
Weitao Tian, Eva Petrová, Sanae Sakai, Julius Eyiuche Nweze, Anne Daebeler, Roey Angel

TL;DR
Scientists discovered new methane-producing microbes in dry desert soils, challenging previous ideas about where these microbes can live.
Contribution
The study reports the genomic characterization of seven novel methanogenic cultures from arid environments, revealing their genetic adaptations and ecological plasticity.
Findings
Six of the seven methanogenic cultures represent new species, but are phylogenetically close to anoxic environment organisms.
Methanobacterium spp. have fewer genes for oxygen and desiccation tolerance compared to other methanogens.
Global metagenomic analysis suggests methanogens are underdetected in dryland soils due to sequencing limitations.
Abstract
Methanogens are strictly anaerobic archaea capable of energy conservation by methane production, yet their presence in oxic and arid environments challenges existing paradigms. In this study, we enriched and genomically characterized seven methanogenic cultures from desert biocrusts, affiliated with the genera Methanobacterium, Methanosarcina, and Methanocella. Six of these new enrichment cultures represent new species. Nonetheless, phylogenomic analyses revealed close genetic relationships with organisms from anoxic environments, indicating the absence of an evolutionary distinction. Comparative genomics exposed diverse though non-unique repertories of antioxidant (e.g. catalase, superoxide dismutase and desulfoferrodoxin), and desiccation-resistance genes (including genes for maintaining osmotic pressure and repair of cell wall and membrane), with Methanobacterium spp possessing the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1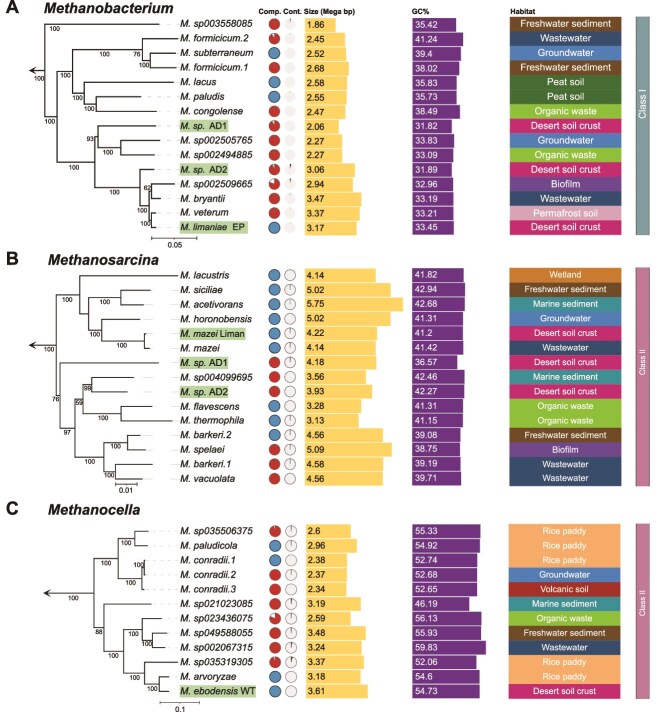 Figure 2
Figure 2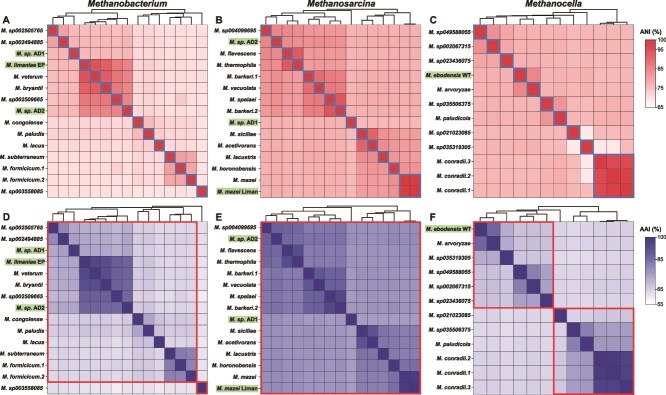 Figure 3
Figure 3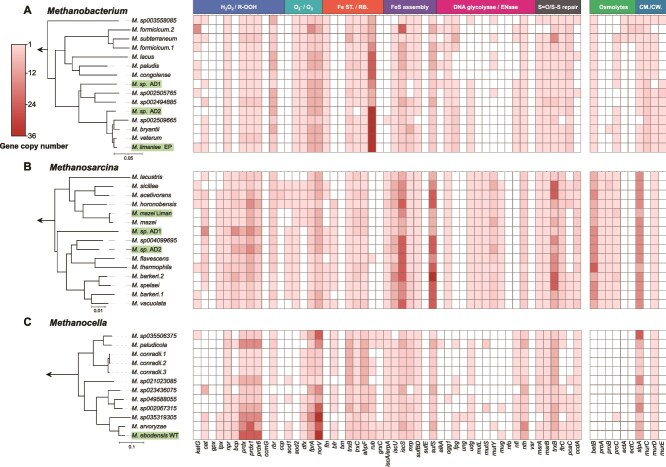 Figure 4
Figure 4- —Czech Science Foundation10.13039/501100001824
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBiocrusts and Microbial Ecology · Microbial Community Ecology and Physiology · Microbial bioremediation and biosurfactants
Introduction
Methanogens are a group of Archaea that reduce carbon dioxide (CO_2_), acetate (acetoclastic methanogenesis), or methyl compounds (methylotrophic methanogenesis) to produce methane (CH_4_) as final metabolite [1, 2]. Methane has gained increasing attention for its significant contribution to climate change, as it is a 84-fold more potent greenhouse gas than CO_2_ over a 20-year period [3]. Methanogens are the main biological source of CH_4_, and therefore play a crucial role in the global CH_4_ budget [4, 5]. They are strict anaerobes, and are commonly found in anoxic environments, such as sediments, rice paddies, and animal guts [6, 7]. Methanogenesis, which predates the appearance of O_2_ in the atmosphere [8, 9], represents a form of anaerobic respiration [1, 10]. Phylogenetic analysis based on the SSU rRNA gene and conserved ribosomal protein markers showed that methanogens are not monophyletic but comprise two distantly related groups, designated as Class I and Class II [11, 12]. Of the six recognized orders, Class I includes Methanobacteriales, Methanococcales, and Methanopyrales, and Class II, includes Methanosarcinales, Methanomicrobiales, and Methanocellales [13–15]. Additionally, recent discoveries have further expanded the known methanogen diversity, including Methanomassiliicoccales [16, 17], and novel lineages in the phyla Korarchaeota [18], Thermoproteota (Methanomethylicia) [19], and unique methanogenic lineages outside Euryarchaeota [20].
In recent years, several genera of methanogens have been detected in different environments that experience either a steady flux of oxygen, such as rice roots [15], wetlands [21], and lake water [22], or are mostly oxic, such as oxygenated sandy sediments, savannas, and even surface desert soils [23–27]. The genera Methanobacterium, Methanosarcina and Methanocella have consistently been found in above mentioned soils, including dryland areas [24, 28]. Notably, previous studies demonstrated that O_2_ tolerance in methanogens is strongly influenced by habitat, as strains form environments with fluctuating redox conditions show higher tolerance for O_2_ than strains from permanently anoxic habitats, even within the same genus [29–31]. Moreover, methanogens in aerated soils have been shown to become active and grow when oxygen levels drop, e.g. after rewetting of desiccated soil and sediment ecosystems, or when surrounding microbial community and soil texture modulating O_2_ penetration and creating micro-anoxic sites [22, 23, 28, 29, 32]. The presence and activity of methanogens in desert soils is striking, as it implies that rewetting events may create transient anoxic microenvironments facilitating methane production, highlighting an overlooked source of atmospheric methane. These upland soil and desert methanogens endure both oxygen fluctuations and the osmotic pressures of desiccation and rehydration. Accordingly, laboratory experiments have confirmed that Methanobacterium, Methanosarcina, and Methanocella can survive in oxidative environments for hours or even days [24, 28, 33, 34]. However, unlike some other strict anaerobes (e.g. Clostridium sp. and Sporomusa sp.) [35], methanogens do not form spores, and the mechanisms they use to cope with oxygen and desiccation remain largely unknown.
Several key cofactors required for methanogenesis are sensitive to the presence of oxygen (O_2_) and its derivatives, i.e. reactive oxygen species (ROS), including hydrogen peroxide (H_2_O_2_), hydroxyl radicals (-OH), and O_2_^−^ radicals [33, 36–38]. In all the methanogenic pathways, the pivotal final step of the reaction of coenzyme B with the methyl group bound to the coenzyme M (CH_3_-S-CoM) to produce CH_4_ and the heterodisulfide (CoB-S-S-CoM) is highly conserved in methanogens and the most O_2_-sensitive [39]. The [4Fe-4S]^2+^ clusters contained in key enzymes involved in the process (heterodisulfide – HDR) and the low-coordinated iron atoms bound to the substrate make this step especially susceptible to ROS damage [40, 41]. Metagenomic analyses have unveiled a range of mechanisms by which methanogens may cope with oxidative stress, including synthesing antioxidant enzymes that detoxify ROS, reducing ROS production, and repairing DNA and protein damage [42–44]. Antioxidant enzymes rely on reducing equivalents, usually supplied by small redox proteins (including rubredoxin and thioredoxin) [36, 45]. Moreover, methanogens, particularly Methanosarcina barkeri, possess functional antioxidant enzymes, including catalase and iron superoxide dismutase [46, 47], which could be upregulated under oxidative stress [48]. Both classes of methanogens encode for various antioxidant enzymes, but antioxidant-encoding genes are generally more abundant in Class II methanogens [49]. Additionally, methanogens possess repair enzymes such as MutS/MutL homologs [42], and methionine sulfoxide reductase [50], which protect against oxidative damage to nucleic acids and proteins, respectively. In response to desiccation stress, a variety of proteins in methanogens could play a protective role. Certain genera, including Methanobacterium and Methanosarcina, regulate osmotic pressure by accumulating compatible solutes, such as glycine betaine and proline, thereby maintaining cellular osmotic balance and stabilizing proteins [51, 52]. Moreover, M. barkeri has been shown to produce extracellular polymeric substances (EPS), which can help the cells retain more moisture and protect during desiccation [53]. Methanosarcina is also known to form biofilms that protect cells, with lipid synthesis upregulated to enhance membrane fluidity and aid in water retention [54]. Finally, homologues of late embryogenesis abundant (LEA) proteins, which are common in plants, have been identified in methanogens (e.g. Methanocella paludicola) [55, 56]. Such proteins may protect other proteins from being inactivated when partially dehydrated [55]. Despite these insights, most studies on methanogens in (transiently) oxic soils have focused on community-level changes in the environment, with limited molecular investigations regarding cellular mechanisms [28, 57]. Furthermore, all the studies using methanogen cultures outlined above were conducted using isolates from anoxic environments. This leaves a large gap in our understanding of the mechanisms responsible for ROS resistance in methanogens which exist and survive in oxic conditions.
In this study, we enriched and cultured new methanogens from desert biocrusts, sequenced their genomes, and performed comparative genomics with 35 genomes and metagenome assembled genomes (MAGs) from multiple closely related lineages. Our study aimed to identify the genetic traits that enable methanogens inhabiting upland soils to adapt to aerated and desiccated conditions.
Materials and methods
Enrichment and cultivation, methane measurement, and growth assessment
Biocrust samples (ca. top 3 mm) were collected from two adjacent Negev Desert sites in Israel: near the ancient Nabatean city of Avdat (30°47′N, 34°46′E), and near the Liman irrigation system (30°50′N, 34°45′E). The area is an elevated, hilly region (ca. 800 m.a.s.l.) with an average annual rainfall of 80–100 mm, classified as arid [58]. The local loess soil is typically dry and exposed to air for the majority of the year [59]. Previous studies have shown the presence and activity of members of Methanosarcina and Methanocella in the biocrust, but not in the underlying soil [24].
To enrich methanogens, 1 g of biocrust was added to 5 ml of sterile deionized water in two 25 ml Balch-type tubes sealed with black butyl stoppers (VWR), crimped with an Al-ring, and flushed with a N_2_/CO_2_ (80%:20% vol/vol; see Supplementary material for more details) gas mixture. Deionized water was used as a rehydration step to reactivate microbial cells [60]. Enrichment cultures were kept in the dark at 32°C (average in situ summer temperature) in a vinyl anaerobic chamber (Coy) without shaking. Same temperature was applied subsequently throughout Methanobacterium and Methanosarcina cultivation steps. Methane production was measured weekly using a gas chromatograph (Agilent GC 6850) equipped with a ShinCarbon ST Micropacked GC column (Restek) and a Flame Ionization Detector (FID, Agilent Technologies). After one month, 1 ml H_2_ (5% final conc.) and 1 ml of deoxygenated adapted Sekiguchi medium (pH 7.0, see Supplementary material) were added [61]. After another month, CH_4_ producing enrichments were transferred (1:20) into fresh medium with N_2_/CO_2_ (80%:20%) headspace. Two enrichment lines were set up: one for Methanobacterium with an addition of 5% H_2_ in the headspace, and another for Methanosarcina with Na-acetate (0.01 g l^−1^) in the medium. Hereafter, the following antibiotics was applied alternately: kanamycin (MP Biomedicals), ampicillin, penicillin G (Merck), and vancomycin (Carl Roth) each at 50 mg ml^−1^, 1 ml per l medium for each transfer, with the final concentration at 50 μg ml^−1^. Methane production was monitored regularly. The community composition was monitored every few months via molecular methods as described below. After 10 months, 1 ml of Methanobacterium-dominated enrichment (34.4% of total community, 90.6% of total methanogens, estimated using ddPCR, see below) was transferred to 100 ml DSMZ 1523 medium [62] in 250 ml Duran bottles, capped with sterile butyl rubber stoppers, with 50% H_2_ added to the headspace. A *Methanosarcina-*dominated (91% of total community) culture was established after 16 months, for which a similar transfer was set up into adapted DSM 960 medium ([63]; see Supplementary material) with N_2_/CO_2_ (80%:20%) in the headspace.
To specifically enrich Methanocella sp., ~20 g of biocrust was pre-incubated at room temperature in a custom-made soil microcosm [28] under moist (field water-holding capacity of 9%) and anoxic (N_2_:CO_2_, 80%:20%) conditions for four weeks to activate the anaerobic microbial community. Afterwards, 5 g of pre-treated soil was mixed with 40 ml of fresh water basal medium (pH 7.0) [64] in a sterile 100 ml Duran bottle anoxically. 10% H_2_ and 5% sterile air were added to the headspace after flushing with N_2_/CO_2_ (80%:20%) and the culture was incubated anoxically at 37°C. A 5% air exposure was empirically determined during preliminary tests, in which Methanocella-containing cultures tolerated transient O_2_ levels and showed quick resumed growth. The addition of air was designed to suppress the growth of other methanogens that are assumed to be less aerotolerant than Methanocella. We followed the optimal growth temperature of M. paludicola [15] to maximize Methanocella biomass in our enrichments. Methane production was monitored as stated above on a weekly basis. A mix of penicillin and kanamycin (final concentration at 50 μg ml^−1^ for each) was applied to all media to suppress bacterial growth. More details of media and enrichment lines are given in the Supplementary material 1. Images of the enriched methanogenic strains were obtained using a scanning electron microscope (Supplementary material 2).
DNA extraction, sequencing, and genome assembly
Soil biocrust DNA was extracted with the FastDNA™ SPIN KIT for SOIL (MP Biomedicals), and culture DNA was extracted using a phenol-chloroform extraction method [65]. The proportion of methanogens to all prokaryotic community members in the cultures were determined through droplet digital PCR (ddPCR) on a Bio-Rad QX200 Droplet Digital PCR System using the following assays: universal prokaryotes (general 16S rRNA gene primers), universal methanogens metahnogens (mcrA primers), and specific methanogen genera (genus-specific 16S rRNA primers). Details of the primers used and the ddPCR assays are given in Table S1. The genus-specific ddPCR values were corrected to methanogen cell numbers by dividing by the number of 16S rRNA gene copies in the genomes of each target genus: 2 copies in Methanobacterium [66], 3 copies in Methanosarcina [67], and 2 copies in Methanocella [15].
When the cell numbers of Methanobacterium, Methanosarcina, and Methanocella reached of 1.9 × 10^6^, 2.1 × 10^6^, and 1.3 × 10^6^ per ml, respectively as determined by ddPCR, DNA was extracted from the enrichment cultures using the Quick DNA HMW Magbead Kit (Zymo Research) according to the manufacturer’s instruction after an overnight treatment of harvested cells in DNA/RNA shield solution (Zymo Research) at −20°C. Total DNA was sent to Novogene Co., Ltd. (Munich, Germany) for library preparation and HiFi long-read sequencing on a PacBio Revio platform. The FASTQ sequences were extracted from the BAM file format using the “bam2fastq” tool from the PacBio BAM toolkit [68]. The extracted reads were then de novo assembled with the “—meta” command of the metaFlye algorithm (v2.9.3; [69]). The assembly outputs were visualized with the Bandage software (v0.8.1; [70]), and the contigs were exported in FASTA format. Circular genomes underwent further quality evaluation using QUAST (v5.2.0; [71]) to assess the assembly metrics, BUSCO (v5.7.0; [72]) to evaluate genome completeness based on conserved single copy orthologs, and CheckM (v1.2.2; [73]) to assess the level of contamination and completeness. Method details for DNA extraction and sequencing on an Illumina platform of an additional set of four enrichment cultures are given in Supplementary material 3.
Phylogenomics and comparative genomics
PacBio- and Illumina-derived genomes were classified using GTDB-Tk (v1.4) with the archaeal reference database [74, 75]. To provide context, 35 reference genomes and MAGs (completeness >80, contamination <5) from methanogens of the same respective families (i.e. Methanobacteriaceae, Methanosarcinaceae, and Methanocellaceae) were obtained from Genome Taxonomy Database. For each class, a set of 122 archaeal single-copy marker genes [75] was separately aligned, resulting in three alignment files [76–78] and trimmed using trimAl (v1.5; removed gaps >50%) [79]. Afterwards, three maximum-likelihood trees were generated with IQ-TREE (v2.1.1) applying the WAG evolutionary model and with 1000 bootstrap iterations [80]. One outgroup genome was included per tree (M. mazei for Methanobacterium and Methanocella, and M. veterum for Methanosarcina). The phylogenetic trees were visualized and annotated using iTOL [81]. Subsequently, the open reading frames (ORFs) of all genomes and MAGs were predicted using Prodigal [82]. A custom set of 60 HMM marker profiles (oxic and desiccation stress genes) was searched using “hmmsearch” [83] with an e-value cutoff of 1.0 × 10^−10^. The antioxidant gene set was selected according to Lyu & Lu [42] and Johnson & Hug [84]. These included genes involved in combating oxidative stress (peroxides removal, free radical removal, iron storage / redox buffer, Fe-S cluster assembly, DNA glycolyase / endonuclease, and S=O / S-S recovery), and desiccation related damage (osmotic pressure regulation and membrane and cell wall repair) in methanogens. A complete list of all 61 marker proteins is given in Table S2. Methods for ordination analysis and a global search in publicly available metagenomes are given in Supplementary material 4.
To allow species delineation, genome-wide Average Nucleotide Identity (ANI) and Average Amino Acid Identity (AAI) values were calculated between the seven newly enriched strains and selected genomes of closely related methanogens for Methanobacterium, Methanosarcina, and Methanocella, according to [85, 86] respectively. ANI and AAI heatmap matrices were visualized with ChiPlot [87].
Pangenome construction and annotation
To further compare the genomic composition of methanogens obtained in this study with closely related ones, we conducted a pangenome analysis using Anvi’o (v8; [88]). Unique and shared genes were identified as described in Eren, 2025 ([89]; detailed in Supplementary material 5). Through the pangenome analysis we identified distinct genes in each of the newly sequenced genomes (hereafter referred to as unique genes). These gene lists were manually explored for the presence of genes related to stress tolerance. Selected genes were quantified across all genomes presented in this study and gene neighbourhoods of these stress-associated genes were visualized with the “Gene Graphic” online tool [90]. For further methods regarding protein analyses, please see Supplementary material 6.
Results and discussion
Cultivation and methanogenic activity
The occurrence of active methanogens in environments experiencing oxygen or periodic desiccation have been known for years [23, 24, 28, 34], but the genetic basis has not been systematically studied. In part, this is due to the lack of isolates and their genomes from such environments in public repositories. Here, we partially filled this gap by culturing and genomically characterising desert biocrust methanogens affiliated with genera systematically found in upland soils, namely with Methanobacterium, Methanosarcina, and Methanocella.
It is believed that cultures of methanogens require strictly anoxic conditions. These classical headspace conditions, i.e. under H_2_/N_2_/CO_2_, or N_2_/CO_2_, excluding O_2_, were applied in this study to successfully enrich Methanobacterium and Methanosarcina. However, members of the genus Methanocella are notoriously difficult to culture owing to their slow growth and reduced competitiveness with other H_2_-utilizing methanogens [15, 91]. Although O_2_ tolerance can vary depending on habitat characteristics, all Methanocella strains isolated to date were from rice paddy soils [15, 92, 93], where periotic expose to O_2_ through root-mediated transportation. Based on this fact which suggests that members of Methanocella genus may exhibit relatively higher O_2_ tolerance than many other methanogens [24], we regularly spiked the headspace of the Methanocella enrichment cultures with 5% lab air in order to favour them over the faster-growing Methanosarcina and Methanobacterium species.
The abundances of Methanobacterium spp, Methanosarcina spp, and Methanocella spp in the original soil crust samples were 7.0 × 10^4^, 6.5 × 10^2^, and 1.3 × 10^2^ cells per gram dry weight, representing 2.4 × 10^−4^%, 2.6 × 10^−6^%, and 5.2 × 10^−7^% of the total prokaryotic communities, calculated and normalized from ddPCR results. Although abundances are extremely low, similar findings have been reported by Hall et al., who showed that aerotolerant methanogens present at low copy numbers in coastal sediments can nevertheless sustain ecologically significant methane emissions [23]. It suggests that rare methanogens, even when close to the detection limit, may still support critical ecosystem processes under favourable conditions [24, 32].
After eight, six, and two transfers of the Methanobacterium, Methanosarcina, and Methanocella cultures (i.e. on days 586, 503, and 129 of cultivation in each lines), respectively, a strong dominance of each of the target methanogens was achieved. The enrichment levels of the target methanogen genus, expressed as a percentage of total methanogens, were 99.98% for Methanobacterium, 99.96% for Methanosarcina, and 98.05% for Methanocella, and of the entire prokaryotic communities were: 22.4% for Methanobacterium, 91% for Methanosarcina, and 16.3% for Methanocella. Methane production rates during enrichment cultivation were (1.13 ± 1.10) × 10^−3^, (7.37 ± 7.75) × 10^−3^, and (1.05 ± 1.19) × 10^−3^ nmol d^−1^ cell^−1^ for Methanobacterium, Methanosarcina, and Methanocella enrichment cultures, respectively. The methane production observed in these biocrust enrichments were within the lower range of reported laboratory pure-culture rates when normalized per cell [15, 94, 95], but community and matrix effects (e.g. soil macromolecules fermentation) in enrichment cultures can alter per-cell activity. Therefore, direct comparisons with pure cultures are difficult to make at this point.
The microscopic images obtained from scanning electron microscopy (SEM) analysis confirmed the typical cell (single-cell forms for Methanosarcina) morphologies and sizes for each methanogenic genus (Fig. 1).
Scanning electron microscopy (SEM) images of the three newly enriched methanogens from desert biocrusts. Methanobacterium sp. (A) with a long rod shape, Methanosarcina sp. (B) with aggregated, coccoid cell clusters, and likely Methanocella sp. (C) with a shorter rod shape. The white line scale bars denote 1 μm.
Although the presence and even activity of methanogens in upland soils in general, and desert biocrust in particular, has been confirmed in the past [24, 28], the detection of Class I Methanobacterium spp is particularly striking. This is because Class I are thought to possess a smaller repertoire of antioxidant enzymes [42], and more [4Fe-4S] clusters [44, 96], making them more vulnerable to transient oxidative conditions than Class II methanogens, such as Methanosarcina and Methanocella [42, 44]. Additionally, unlike Class II, Class I methanogens do not possess cytochromes, which allow for energy conservation via membrane-bound electron transport chains and fuel protective mechanisms linked to increased oxygen resistance [96]. The successful enrichment and cultivation of methanogens including a cytochrome-lacking Methanobacterium from an environment which is mostly oxic points to the possibility of the existence of yet-undescribed oxygen survival and detoxification strategies in these organisms. These new enrichment cultures provide a valuable addition to the known methanogen cultures, which have all been obtained from anoxic habitats. They highlight a wider-than-expected ecological plasticity of methanogens and present a unique tool to further investigate potential oxygen tolerance and detoxification mechanisms.
Genome characteristics and phylogeny
Using de novo assembly, we obtained seven distinct high-quality methanogen genomes from our enrichment cultures, four of which were circular: three classified as Methanobacterium spp. and three as Methanosarcina spp. (each two MAGs from the Illumina NovaSeq sequencing and one closed genome from PacBio sequencing), as well as one closed genome classified as belonging to a Methanocella sp. (closed genomes are shown in Fig. S1). Quality assessment using QUAST showed genome completeness of non-circular MAGs were 92.8, 93.6 (Methanobacterium spp.), and 99.7, 99.8% (Methanosarcina spp.), with minimal contamination levels evaluated by CheckM at 0, 4 (Methanobacterium spp.), and 0.98, 0 (Methanosarcina spp.), respectively. Likewise, minimal contamination levels of closed genomes were determined with 0.8, 0.65, and 0% for the Methanobacterium sp., Methanosarcina sp., and Methanocella sp., respectively.
Phylogenomic analysis confirmed the affiliation of the organisms in our enrichment cultures with the Methanobacteriales, Methanosarcinales, and Methanocellales orders (Fig. 2, Table S3). Unexpectedly, the new methanogenic enriched cultures from a desert environment did not fall into separate clusters, but showed to be closely related to several known taxa from generally anoxic habitats such as wastewater, groundwater, permafrost, sub-sea sediments, or rice paddies. Moreover, we did not detect any phylogenomic clustering correlating with the environmental conditions in which the organisms live. The average genome sizes and G + C contents across the examined Methanobacterium, Methanosarcina, and Methanocella genera were 2.62 Mb and 35.9%, 4.39 Mb and 40.9%, 2.39 Mb and 50.4%, respectively. The variations in genome size and GC content among these methanogens may reflect their distinct metabolic strategies and environmental niches. Commonly, a lower GC content, such as found for the Methanobacterium spp., is associated with adaptation to anaerobic conditions [97, 98], which aligns with the classical view on Class I methanogens.
Phylogenomics and basic genome-assembly properties of the newly enriched methanogens and known representatives. Genome-based, maximum-likelihood trees for the genera Methanobacterium (A), Methanosarcina (B), and Methanocella (C) are shown with the newly enriched methanogens of this study highlighted in green. Numbers on branches indicate bootstrap support value (%) on 1000 replicates. The scale bars denote 0.05, 0.01, and 0.1 estimated substitutions per amino acid, respectively. The arrow at the base of each tree denotes rooting based on an outgroup (see Materials and methods for specifications). Comp., completeness (in red; blue solid circles represent circular genomes); Cont., contamination.
Species delineation and distribution in global oxic drylands
Genome-wide average nucleotide identity (gANI) and average AAI based analyses were performed on the same Methanobacterium, Methanosarcina, and Methanocella datasets to classify the newly enriched desert methanogens on the species and genus levels and higher taxonomic ranks (Fig. 3). Based on the gANI analysis and a species level threshold of 96.5% [99] the three newly enriched Methanobacterium organisms represent separate species from each other and from M. veterum and M. bryantii (Fig. 3, Table S4) as their closest cultured relatives. After meeting the minimal standards proposed by Prakash et al. [100], for the species from which we obtained a closed genome, we therefore tentatively propose the name “Candidatus Methanobacterium limaniea EP”. We refrain from proposing new species names for the other two enrichment cultures, since they were only transiently present in the enrichment culture and moreover, we did not obtain circular genomes from them. Henceforth, we refer to these organisms as Methanobacterium sp. AD1 and AD2. Likewise, two of the newly enriched Methanosarcina organisms are separate species from each other and from Methanosarcina flavescens and M. barkeri 2, as their closest cultured relatives. We refer to these organisms as Methanosarcina sp. AD1 and AD2 from hereafter. The third Methanosarcina organism was not a separate species from M. mazei and we therefore give the strain name “Liman” to this organism. Finally, the newly enriched Methanocella organism was shown to represent a new species, distinct from its closest cultured relative Methanocella aevoryzae. We therefore tentatively propose [100] the name “Candidatus Methanocella ebodensis WT”. In summary, we have enriched and identified six novel methanogen species through genomic analysis of enriched organisms. The difficulty in cultivating and isolating methanogens, especially those with specialized metabolic requirements, has long been a challenge in microbial ecology. The identification of new species from three genera inhabiting oxic desert biocrusts in this study highlights the underappreciated diversity of methanogens in oxic habitats.
Genome-wide average nucleotide identity (gANI) and average amino acid identity (AAI) heatmaps for Methanobacterium (A, D), Methanosarcina (B, E), and for Methanocella (C, F). Names of genomes from this study are highlighted in green. Genomes within the same species in ANI heatmaps are framed in blue boxes, and genomes belonging to the same genera in AAI heatmaps are framed in red boxes.
To assess the distribution of methanogens in arid oxic soils worldwide, we surveyed 24 metagenomic datasets collected from the NCBI Bioproject database generated from samples from Europe, the Americas, Asia, Africa, Oceania, and Antarctica. Methanogens were detected in only five of the 24 projects (44 datasets, Table S5 and S6). Among the detected methanogens, members of the class Methanosarcinia were most frequently observed. In addition, members of other methanogen classes including Methanomicrobia, Methanococci, Methanobacteria, and Methanopyri were all present in more than one dataset. However, the relative abundances were consistently low, representing max. 0.28% of the total microbial communities. Considering the discrepancy of apparent methanogen absence as assessed by metagenomic sequencing with the proven presence of methanogens via targeted approaches in this and previous work (e.g. for Negev desert biocrust [28]), we assume that the low detection rate of the global search reflects insufficient sequencing depth rather than their absence from these systems. Metagenomic surveys can underdetect low-abundance taxa due to limited sequencing depth, assembly and binning biases, however, low-abundance organisms may play disproportionate ecological roles [101, 102]. Therefore, targeted enrichment and deeper sequencing can help reveal their presence and potential function.
Genomic potential for antioxidant and desiccation-resistance mechanisms in the newly enriched methanogens
To identify genes associated with oxidative stress resistance and desiccation tolerance in the newly enriched methanogens, we conducted a targeted comparative genomic analysis across the genomes of our new enrichment cultures and those of their closely related relatives. There was a clear distinction in the encoded diversity of oxygen and desiccation stress genes between the three investigated species, indicating their different potential stress response strategies (Fig. 4, Fig. S2, Table S7). Notably, Methanosarcina genomes exhibited higher completeness in Fe-S cluster assembly and DNA glycosylase / endonuclease genes (Fig. 4B), which may confer an adaptive advantage to maintain genomic integrity even under harmful conditions that cause DNA lesions. Interestingly, the antioxidant gene set in Methanocella species was not the most complete among the three genera (Fig. 4C). This finding was unexpected, considering the generally attributed highest relative tolerance to oxygen of Methanocella and their ecological niche as rice-root methanogens [92, 103].
Distribution of distinctive genes related to antioxidant and desiccation stress traits in methanogens of the Methanobacterium (A), Methanosarcina (B), and Methanocella (C) genera. Genomes used in the analysis and phylogenomic trees on the left correspond to those in Fig. 2. A homology-based search for functional genes was performed by using HMMER’s hmmsearch and manual examination. Solid and open squares indicate the presence and absence of the genes, respectively. The presence of multiple copies is indicated by colour intensity. Gene names are given at the bottom (see Table S2 for full names), and the corresponding functions are annotated at the top. Fe ST., iron storage; RB., redox buffer; CM./CW., cell membrane/cell wall.
Surprisingly, the enriched methanogens from this study did not show a higher diversity, higher gene copy numbers, or specific patterns compared to their sister taxa within the genera. However, “Ca. Methanocella ebodensis WT” showed a modest increase in average copy number of antioxidant genes, ranking highest among the Methanocella genomes in this study. It aligns with our cultivation experiment, in which Methanocella demonstrated comparatively greater tolerance to O_2_ exposure than members of the Methanobacterium. The overall trends on the genus level remained apparent for our newly sequenced genomes with the three Methanosarcina genomes containing the highest diversity and copy numbers and the Methanobacterium genome containing the least (Fig. 4, Table S7). Previous studies showed that O_2_ tolerance can vary among strains belonging to the same genus depending on environmental exposure [29, 30]. Our findings likely reflect class and lineage level tendencies in antioxidant and desiccation-related gene repertoires. However, realized O_2_ tolerance is determined by gene regulatory differences, expression under stress, microhabitat protection, and community interactions, not only gene presence or absence. Future transcriptomic experiments and microcosm studies are required to resolve these layers.
The absence of distinctive genomic signatures uniquely associated with our desert strains suggests that these methanogens persistence under oxic and desiccating conditions may rely on mechanisms beyond gene content alone. One possibility is that the fine-structured matrix of biocrusts generates transient anoxic micro-niches, enabling methanogens to remain metabolically active during brief windows of O_2_ depletion [104]. Another explanation is regulatory plasticity, where antioxidant and repair pathways are rapidly upregulated in response to oxidative stress, even without an expanded gene repertoire. Additionally, physicochemical protection, such as encapsulation within soil aggregates, extracellular polymeric substances, or metabolic coupling with oxygen-scavenging community members, may help buffer methanogens from oxygen exposure [105–107]. Strains from habitats experiencing periodic oxygenation, even if phylogenetically close to strictly anaerobic strains, can have elevated tolerance for O_2_ [29, 30]. Such hypotheses requires transcriptomic and proteomic analysis, however, obtaining sufficient biomass for Methanocella sp. in particular remains extremely challenging. Future work combining controlled oxidative stress transcriptomic, targeted proteomics, and in situ microscale O_2_ profiling will be essential for resolving how methanogens tolerate and recover from oxygen and desiccation stress in desert.
Additionally, we specifically investigated the presence of LEA protein homologs (late embryogenesis abundant proteins) that were previously reported in M. paludicola genome and annotated as such by Campos et al. [55]. In plants, LEA proteins protect against dehydration stress by stabilizing proteins and membranes [108, 109], and have been discussed to provide similar functions in methanogens. However, our search using multiple archaeal LEA protein sequences from UniProt found zero hits across all Methanocella genomes including M. paludicola. A BLASTx search on the NCBI server showed that these M. paludicola LEA proteins annotated by Campos et al. have a high sequence similarity with KGG domain-containing proteins (Pfam domain PF10685) and general stress proteins rather than with LEA proteins (Table S8A). However, we did not find stress-induced KGG domain in those identified as “KGG domain-containing protein” based on domain analysis. To further examine this discrepancy, we compiled 19 LEA protein sequences from plants, bacteria, archaea, the five Methanocella putative LEA proteins, et al. bacterial KGG domain-containing proteins (uncharacterized protein YciG and Putative virulence-regulating protein), and an outgroup protein (hypersensitive response domain-containing protein), and constructed a neighbor-joining phylogenetic tree (Fig. S3A). In this tree, the five putative LEA proteins from Methanocella formed in a single separate cluster, sister to a plant LEA protein cluster. Sequence alignment likewise showed low similarity between these five proteins and canonical LEA proteins, as well as between them and KGG domain-containing proteins (Fig. S3B). Additionally, all five putative LEA proteins are highly hydrophilic and intrinsically disordered, which aligned with features of plant LEA proteins but not with archaeal LEAs. Finally, AlphaFold2 predicted the five M. paludicola proteins form extended, unstructured polypeptides with intermittent coiled α-helical segments, a feature of plant LEA proteins under stress-induced conformational changes [110, 111]. However, unlike archaeal LEA proteins exhibiting stable tertiary folds, the M. paludicola putative proteins lack any defined or persistent three-dimensional structures (Fig. S3C). Despite the physicochemical similarity with plant LEA proteins, these results raise doubts that Methanocella sp. truly harbour LEA proteins. We therefore caution against annotation of these sequences as LEA proteins or KGG domain-containing proteins without functional validation.
Pangenomic analysis and potentially non-functional genes
To further explore genomic differences between the newly enriched desert methanogens and their close relatives, we conducted a pangenome analysis. The analysis identified both core genes shared across all genomes and unique genes specific to single genomes. All seven genomes of our newly cultivated methanogens contained unique genes, with the genome of “Ca. Methanocella ebodensis WT” containing the largest set (Fig. S4, Table S9). Strikingly, many (16%) of these unique genes in all our genomes were associated with membrane and cell wall biogenesis. In comparison, such genes accounted for 15% in Methanobacterium, 7% in Methanosarcina, and 10% in Methanocella among published genomes included in this study. The relative enrichment of such genes in our desert-derived strains may indicate lineage-specific strategies for maintaining cell integrity in the multi-factor extreme environment.
Additionally, several functionally intriguing genes were found, including those annotated as encoding subunits of glycosyltransferase (rfaB, wcaA, and wcaE) and the nitrous oxide reductase subunit D (nosD). Glycosyltransferases are typically involved in exopolysaccharide biosynthesis and cell envelope modification, processes that may support stress resistance in bacteria [112, 113], but are poorly characterized in methanogens. The, nosD genes encodes for an accessory protein associated with the Nos operon for N_2_O reduction in denitrifiers [114], yet methanogens are not known to perform denitrification. To assess the distribution of these four genes, we quantified their copy numbers across all selected genomes in this study based on the anvi’o annotation (Fig. S5). The glycosyltransferase genes were widespread without genus-specific patterns. In contrast, nosD was enriched in Methanosarcina genomes only, implying a genus-specific functional adaptation possibly related to protein maturation. However, the putative nosD genes were not enriched in the genomes of the new desert methanogens, pointing towards a more general role.
To further understand the potential function of the gene annotated as nosD in our newly enriched methanogens, we performed a BLAST search against the NCBI nr protein database and out of the seven genes annotated as NosD, only three gave hits designated as NosD, albeit unverified (Table S8B). A Neighbor-joining phylogenetic tree built with bona fide NosD sequences showed that our putative NosD protein sequences formed a distinct cluster, separate from the other known ones (Fig. S6A). Additionally, synteny analysis of the putative nosD genes showed that other nos operon genes were absent (Fig. S7), suggesting that these genes may have alternative, yet unknown functions. Domain analysis showed that in addition to a distant periplasmic copper-biding (NosD-like) β-helix domain, these putative NosD proteins included pectin lyase folds, carbohydrate-binding / sugar-hydrolysis domains, and multiple parallel β-helix domains. Although the putative NosD proteins share similar domains with canonical ones, they were not classified within the representative “nitrous oxide reductase family maturation protein” by InterPro/PFAM databases (Fig. S6B). While the precise functions remain to be determined, these putative NosD proteins showed potential roles in cell-envelope modification, extracellular polymer processing, or general stress response, rather than denitrification [115–117].
In summary, although certain genes with intriguing annotations such as nosD and glycosyltransferases were found among the unique genes, their unclear functional roles, lack of operon context, and phylogenetic divergence suggest that they may represent non-functional remnants or genes with alternative, yet-to-be-elucidated roles in methanogens.
Conclusion
The occurrence of methanogens of the Methanbacterium, Methanosarcina, and Methanocella genera in the investigated biocrusts defied basic assumptions about their physiology and left open questions regarding the restricted occurrence of only these genera and their genomic machinery to persist under the stressful conditions of these habitats. Our characterisation of newly-enriched methanogens showed that while they are equipped, to some extent, to handle ROS and desiccation, their genomes fail to reveal any unique mechanisms that would set them apart from their relatives living in anoxic and wet environments. Moreover, we found that Methanocella, which is regarded as the most oxygen tolerant methanogen, is in fact depleted in many known antioxidant genes compared to its sister genera. Concomitantly, we found that Methanobacterium, which was predicted to be particularly oxygen sensitive, is not only prevalent in oxic soils but also well-equipped to handle oxygen and desiccation stressors. Given the lack of strong genomic signals in the newly enriched methanogens from desert biocrust, it is possible that differences in gene expression contribute to their apparent stress tolerance. Therefore, in addition to ongoing cultivation and genomic sequencing attempts, further work on gene expression under physiologically-relevant stress conditions is required to elucidate the survival mechanisms of methanogens living in oxic and dry environments.
Taxonomic consideration of “Candidatus Methanobacterium limanae” sp. nov
Li.ma’nae. N.L. gen. n. limanae, belonging to the Liman, from Greek λιμήν through Turkish and Russian, originally meaning a bay or a port, but in this context refers to a constructed runoff catchment, from which the strain has been isolated.
Phylogenetically affiliated with the genus Methanobacterium, phylum Methanobacteriota. The genome consists of one circular chromosome of 3 168 381 bp. The DNA G + C content is 33.45 mol%. Strain “Ca. Methanobacterium limanense EP” was cultivated from biocrust of the Negev desert, Israel. Soil hydrogenotrophic methanogen with a rod-shaped morphology. Cells are slender and elongated, typically measuring ~0.5 μm in diameter and 4–5 μm in length. The strain was routinely cultured with 80% H_2_ at 32°C in DSM17711 medium.
Taxonomic consideration of “Candidatus Methanocella ebodensis” sp. nov
E.bo.den’sis. N.L. fem. adj. ebodensis, named after Eboda, a significant ancient Nabataean city situated along the “Incense Route,” close to the location where the strain was isolated. The Nabataeans were renowned for their sophisticated water collection techniques, which enabled them to thrive in the desert environment.
Phylogenetically affiliated with the genus Methanocella, phylum Halobacteriota. The genome consists of one circular chromosome of 3 614 771 bp. The DNA G + C content is 54.73 mol%. Strain “Ca. Methanocella ebodensis WT” was cultivated from biocrust of the Negev desert, Israel. Soil hydrogenotrophic methanogen with a rod-shaped morphology. Cells typically measure ~0.8 μm in diameter and 7–8 μm in length. The strain was routinely cultured with 10% H_2_ at 37°C in fresh water basal medium and subjected to 5% air regularly.
Supplementary Material
Supplementary_material_revised_ycag013
Supp_table_revised_ycag013
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Blaut M . Metabolism of methanogens. Antonie Van Leeuwenhoek 1994;66:187–208. 10.1007/BF 008716397747931 · doi ↗ · pubmed ↗
- 2Garcia PS, Gribaldo S, Borrel G. Diversity and evolution of methane-related pathways in archaea. Ann Rev Microbiol 2022;76:727–55. 10.1146/annurev-micro-041020-02493535759872 · doi ↗ · pubmed ↗
- 3Masson-Delmotte V, Zhai P, Pirani AA. et al. (eds.). IPCC. Climate change 2021: The physical science basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge, United Kingdom and New York, NY, USA: Cambridge University Press, 2021.
- 4Kirschke S, Bousquet P, Ciais P. et al. Three decades of global methane sources and sinks. Nat Geosci 2013;6:813–23. 10.1038/ngeo 1955 · doi ↗
- 5Reeburgh WS . Oceanic methane biogeochemistry. Chem Rev 2007;107:486–513. 10.1021/cr 050362 v 17261072 · doi ↗ · pubmed ↗
- 6Boone DR, Whitman WB, Rouvière P. Diversity and taxonomy of methanogens. In: Ferry J.G. (ed.), Methanogenesis: Ecology, Physiology, Biochemistry & Genetics. Boston, MA: Springer US, 1993, 35–80.
- 7Garcia JL, Patel BKC, Ollivier B. Taxonomic, phylogenetic, and ecological diversity of methanogenic archaea. Anaerobe 2000;6:205–26. 10.1006/anae.2000.034516887666 · doi ↗ · pubmed ↗
- 8Battistuzzi FU, Feijao A, Hedges SB. A genomic timescale of prokaryote evolution: insights into the origin of methanogenesis, phototrophy, and the colonization of land. BMC Evol Biol 2004;4:44. 10.1186/1471-2148-4-4415535883 PMC 533871 · doi ↗ · pubmed ↗