Mucin‐Inspired Filamentous Sulfated Copolymers Effectively Inhibit Human Respiratory Syncytial Virus (hRSV) Infectivity
Raju Bej, Enyu Xie, Kai Ludwig, Robert F. Schmidt, Yannic Kerkhoff, Robert Dalgliesh, Nilanjan Paul, Michael Gradzielski, Andreas Herrmann, Christian Sieben, Rainer Haag

TL;DR
Scientists created a new type of polymer inspired by mucin that can both inhibit and destroy the human respiratory syncytial virus.
Contribution
The development of virucidal mucin-inspired copolymers with both inhibitory and virucidal effects against hRSV is novel.
Findings
MIACPs exhibit strong sulfate-dependent inhibition of hRSV with low IC50 values.
MIACPs demonstrate virucidal activity confirmed through serial dilution experiments.
Filamentous MIACPs mimic mucin structures and are biocompatible.
Abstract
Virucidal compounds capable of binding to and disrupting viruses represent a promising avenue for antiviral applications. In this study, the development of high molecular weight (≈300 kDa) dendronized polyglycerol‐based mucin‐inspired amphiphilic statistical copolymers (MIACPs) is reported using the RAFT polymerization technique. These copolymers comprise ≈30% repeat units containing aliphatic C11 carbon chains with terminal carboxylate (MIACP‐1) and alkyl (MIACP‐2) functionalities, while the remaining ≈70% of the repeat units consist of dendronized polyglycerol sulfates. Structural characterization using cryo‐electron microscopy (cryo‐EM) and small‐angle neutron scattering (SANS) reveals that MIACPs form single‐chain filamentous structures, similar to natural porcine gastric mucin (PGM). These biocompatible MIACPs exhibit strong, sulfate‐dependent inhibition of human respiratory…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Scheme 1
Scheme 1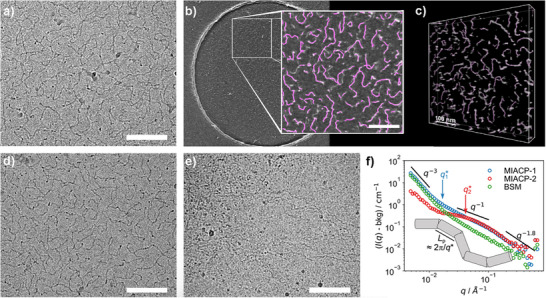 Figure 1
Figure 1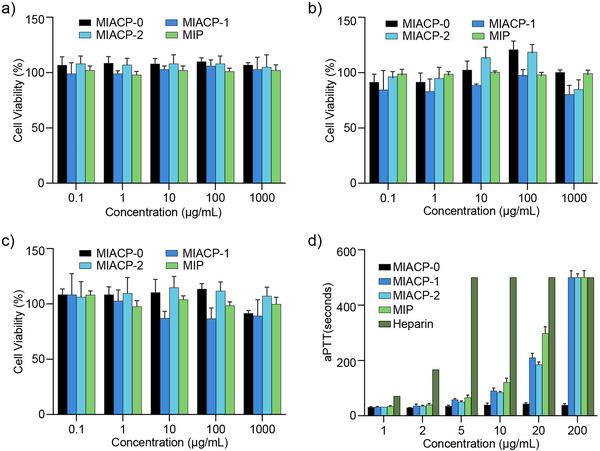 Figure 2
Figure 2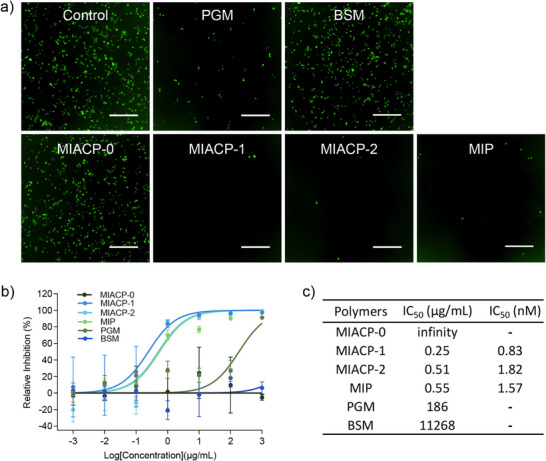 Figure 3
Figure 3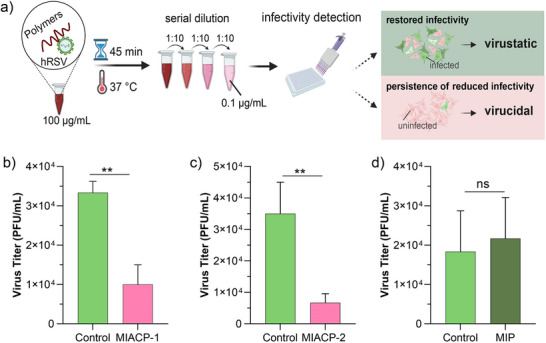 Figure 4
Figure 4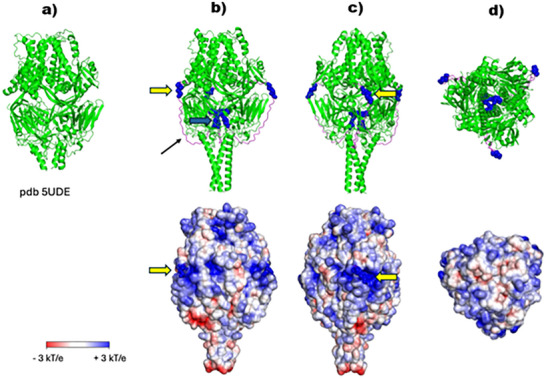 Figure 5
Figure 5| Polymers | MW | Elemental Analysis | DoF | ζ [mV] | |||
|---|---|---|---|---|---|---|---|
| Carbon (C) | Hydrogen (H) | Nitrogen (N) | Sulfur (S) | ||||
|
| 150 | 43.5 | 6.9 | 0.78 | 0.7 | 0 | −9.0 ± 1.9 |
|
| 320 | 29.2 | 3.8 | 0.59 | 13.9 | 94 | −51.2 ± 6.5 |
|
| 310 | 28.9 | 3.8 | 0.45 | 14.8 | 92 | −47.2 ± 3.4 |
|
| 350 | 29.9 | 3.5 | 0.22 | 18.6 | 94 | −56.2 ± 4.8 |
- —IIT Guwahati
- —China Scholarship Council10.13039/501100004543
- —Helmholtz‐Gemeinschaft10.13039/501100001656
- —Deutsche Forschungsgemeinschaft10.13039/501100001659
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRespiratory viral infections research · Antimicrobial agents and applications · Dendrimers and Hyperbranched Polymers
Introduction
1
Human respiratory syncytial virus (hRSV) is a leading cause of acute lower respiratory tract infections in infants, as well as in elderly individuals and high‐risk adults with chronic lung condition or a compromised immune system.^[^ 1, 2, 3 ^]^ Despite its clinical significance,^[^ 4 ^]^ there remains a lack of effective vaccines^[^ 5 ^]^ or widely available antiviral treatments for hRSV, highlighting the urgent need for new treatment options. As a result, the development of broad‐spectrum, potent, and biocompatible antiviral inhibitors remains a critical research priority.
Preventing hRSV infection at its early stages represents an alternative strategy to inhibit viral progression. hRSV initiates infection by interacting with cell surface heparan sulfate proteoglycans (HSPGs) or other sulfated proteoglycans.^[^ 6, 7 ^]^ Additionally, several membrane proteins have been implicated in facilitating hRSV entry into host cells.^[^ 8 ^]^ On the viral side, the envelope glycoprotein (G protein) and fusion protein (F protein) mediate attachment to the target cell membrane and promote membrane fusion.^[^ 9, 10 ^]^ At the molecular level, the negatively charged sulfate or carboxyl groups of HSPGs or heparin bind to clusters of positively charged basic amino acids within the linear heparin‐binding domain (HBD) of hRSV glycoproteins.^[^ 11 ^]^ This HSPG‐HBD interaction is a common and critical feature in hRSV‐mediated infection, making it an attractive target for the development of negatively charged antiviral agents.
In this context, heparan sulfate‐mimetic polymers and peptides have emerged as promising inhibitors of hRSV due to their ability to bind viral glycoproteins.^[^ 12 ^]^ Inspired by these findings, our group explored heparin‐mimetic linear and hyperbranched polyglycerol sulfates, which showed strong potential as antiviral agents owing to their biocompatibility.^[^ 13 ^]^ These results further motivated us to design functional polyglycerol sulfates with varied molecular weights, architectures, and structural compositions, aiming to broaden the scope of polysulfate‐based inhibitors with enhanced anti‐hRSV activity.
Mucin, a high molecular weight glycoprotein, is a key component of the protective mucus layer.^[^ 14 ^]^ Inspired by its structure and function, various mucin‐inspired biomaterials have been developed, demonstrating excellent inhibitory activity against a range of pathogens.^[^ 15, 16, 17, 18, 19 ^]^ This effectiveness is attributed to their unique characteristics, including high molecular weight, filamentous structure, and strong electronegative charge. Recently, our group developed a series of mucin‐inspired dendronized polyglycerol sulfates with varying molecular weights and found them to be potent inhibitors of sulfate‐binding viruses such as HSV‐1, SARS‐CoV‐2, and their variants.^[^ 20, 21, 22 ^]^ In those studies, polymers with longer filamentous structures and higher overall electronegative charge emerged as the most effective inhibitors. Their antiviral activity was comparable to that of antibodies,^[^ 23 ^]^ making them among the most promising synthetic inhibitors reported to date. These findings motivated us to investigate functionalized, high molecular weight, filamentous polymeric architectures for the inhibition of hRSV.
Another important consideration is that virus binding mediated by electrostatic interactions may be a reversible process, potentially allowing virus particles to detach from the inhibitors and proceed to infect host cells‐thereby limiting their therapeutic efficacy. This limitation can be addressed by employing inhibitors with virucidal properties that irreversibly deactivate viruses.^[^ 24 ^]^ This insight further motivated us to enhance the previously developed mucin‐inspired platform by introducing a defined percentage of virucidal functionality, forming the basis for the design of new copolymers. In selecting the virus‐disrupting moieties, alkyl chains with aliphatic C11 carbon lengths have been shown to irreversibly disrupt viral membranes through hydrophobic interactions,^[^ 25, 26, 27, 28 ^]^ which guided our development of amphiphilic copolymers (Scheme 1a).
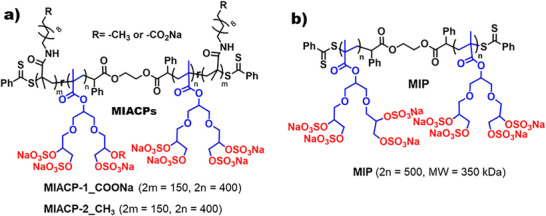
Chemical structure of the investigated mucin‐inspired a) amphiphilic copolymers (MIACPs) and b) polymer (MIP). MIACPs contain ≈30% repeat units of virucidal functionality: aliphatic C11 carbons chain with terminal carboxylate functional groups (MIACP‐1), alkyl functional group (MIACP‐2). All cases remaining ≈70% repeat units’ functionality from dendronized polyglycerol sulfates. MIP contains only dendronized polyglycerol sulfates as repeat units without any aliphatic functionality.
Amphiphilic polymers often tend to self‐assemble into various aggregated structures.^[^ 29, 30, 31, 32, 33 ^]^ However, in our research, we aimed to retain a filamentous morphology instead of promoting other ways of aggregation, in order to leverage the structural advantages of mucin‐like filaments.^[^ 22 ^]^ Previous studies have reported that other types of aggregation can be minimized by carefully tuning the hydrophilic‐hydrophobic balance of amphiphilic polymers. Notably, systems containing up to ≈30% hydrophobic units have been shown to be very positive in that aspect.^[^ 34, 35, 36, 37 ^]^ Guided by this, we designed amphiphilic copolymers composed of ≈70% dendronized sulfated groups and ≈30% C11 hydrophobic units (Scheme 1a). The sulfated groups are intended to inhibit viral binding via electrostatic interactions with positively charged regions on viral surface proteins, while the hydrophobic C11 domains are expected to provide virucidal activity by disrupting the viral membrane.
In this work, we present the synthesis and comprehensive characterization of mucin‐inspired amphiphilic copolymers (MIACPs) incorporating C11 aliphatic chains, along with their homopolymer counterpart (MIP) of similar molecular weight (Scheme 1b). We investigate their morphology in aqueous solution using cryo‐electron tomography (Cryo‐ET) and small‐angle neutron scattering (SANS), assess their biocompatibility, and evaluate their antiviral and virucidal efficacy against hRSV.
Results and Discussion
2
Synthesis and Characterization of Mucin‐Inspired Amphiphilic Copolymers (MIACPs)
2.1
The chemical structures of the investigated polymers are depicted in Scheme 1. We report three different dendronized polysulfates with similar molecular weights (Table 1). The first group consists of copolymers (MIACPs) comprising two components: ≈70% repeat units of sulfated dendronized oligoglycerol methacrylate (OGMA), and ≈30% repeat units containing an aliphatic C11 carbon chain terminated with either a carboxylate group (MIACP‐1) or a purely aliphatic group (MIACP‐2). The second is a homopolymer (MIP), which lacks any hydrophobic functionality and serves as a control to assess the impact of virucidal groups in MIACPs. Due to their amphiphilic nature, MIACPs carry a risk of particle aggregation. To minimize this, hydrophilic and hydrophobic units were statistically distributed within the copolymer chains.^[^ 38 ^]^
For synthesis, we first optimized the polymerization conditions to prepare a relatively high‐molecular‐weight precursor copolymer, P1 (pOGMA‐co‐pNHSMA), on a gram scale (Scheme S1, Supporting Information). Synthesis and characterization of P1 are provided in the Supporting Information. N‐Hydroxysuccinimide methacrylate (NHSMA) units were selected as pre‐functional groups, which can later be replaced by amine functionalities to install virucidal moieties.^[^ 39 ^]^ The composition of copolymer P1 was determined by ^1^H NMR analysis (Figure S1, Supporting Information), which confirmed that ≈30% of the repeat units were NHSMA. Considering ≈80% conversion of individual monomers, the molecular weight of P1 was estimated to be 183 kDa, closely matching the size exclusion chromatography (SEC) measurement (M W = 128 kDa) (Figure S2, Supporting Information), indicating successful controlled radical copolymerization. The NHS units in P1 were then substituted with either commercially available 11‐amino undecanoic acid or 11‐amino undecane via standard nucleophilic substitution, yielding P2a and P2b, respectively. The disappearance of the NHS peak at δ = 2.88 ppm and the appearance of additional peaks at δ = 1.35–0.85 ppm region (corresponding to ‐CH_2_ protons from the aliphatic chain) for P2a and P2b in the ^1^H NMR spectrum confirmed successful substitution (Figures S3 and S4, Supporting Information). The integration of aliphatic protons relative to glycerol units further supported ≈30% incorporation of hydrophobic C11 units.
The acetonide groups of P2a were deprotected to yield P3a which was then sulfated to produce MIACP‐1 (Scheme S1, Supporting Information). The disappearance of the δ = 1.4–1.35 ppm proton peak in the ^1^H NMR spectrum of MIACP‐0 (Figure S5, Supporting Information) confirmed deprotection. Additionally, the shift in the methylene proton peak adjacent to the sulfate groups in the ^1^H NMR spectrum of MIACP‐1 (Figure S7, Supporting Information), compared to P3a, and confirmed the successful introduction of electronegative sulfate groups.
Elemental analysis of MIACP‐1 (Table 1) showed >90% sulfation of the hydroxyl groups in P3a. The molecular weight of MIACP‐1, estimated from ^1^H NMR, was ≈320 kDa, matches with the molecular weight (M W = 290 kDa) obtained from size exclusion chromatography (SEC) (Figure S8, Supporting Information) and within the molecular weight range for the large, glycosylated regions of mucin polymers.^[^ 40 ^]^
MIACP‐2 was synthesized using similar reaction conditions as described for MIACP‐1. For that, the acetonide groups of P2b were deprotected to yield P3b (MIACP‐0) which was then sulfated to produce MIACP‐2 (Scheme S1, Supporting Information). The characterization data are shown in Table 1 and in the Supporting Information (Figures S6, S9, and S10, Supporting Information). The investigated homopolymer, MIP, was synthesized using a previously reported protocol from the group, with slight modifications (Scheme S2, Supporting Information). Characterization data are provided in Table 1 and in the Supporting Information (Figures S11–S15, Supporting Information).
Morphology Investigation: Cryo‐EM and SANS Analysis
2.2
The overall electronegative charge of sulfated MIACPs and MIP was confirmed by their negative zeta potential values, attributed to the highly sulfated functionalities (Table 1). Cryo‐EM was measured to investigate the morphology of MIACPs in aqueous solution. Both MIACP‐1 and MIACP‐2 exhibited elongated filamentous structures (Figure 1a,d). Cryo‐ET was employed to determine the 3D structure and length of the amphiphilic elongated fibers. To eliminate human bias, the fiber length distribution was semi‐automatically analyzed with Fiji^[^ 41 ^]^ using 3D skeleton analysis^[^ 42 ^]^ of pre‐processed cryo‐ET volume stacks (illustrated for MIACP‐1 in Figure 1b,c and in the supporting movie SM‐01). Using this calculation, the average length for MIACP‐1 and MIACP‐2 were 83.1 ± 29.2 nM and 85.3 ± 19.0 nM, which is aligned well with the theoretical average stretched length of ≈150 nM of MIACPs (average number of repeat units = 500), calculated based on the atomic distance in the polymer chain. The observed length distribution of fibers is plausible considering the polydispersity (Đ = 1.5) in MIACPs.
a) Cryo‐EM image and b) cryo‐electron tomogram of MIACP‐1 in aqueous solution (C = 1.0 mg mL−1); the backbone structures determined with Fiji are highlighted in pink in the enlarged section. c) The 3D volume reconstructed from the cryo‐ET tilt image series (± 64°, 2° increments): Unlike 2D cryo‐EM images (= projections), this allows the determination of the actual 3D length of the amphiphilic elongated fibers. The average calculated length was 83.1 ± 29.2 nM. d), Cryo EM images of MIACP‐2 and e) PGM, each in aqueous solution (C = 1.0 mg mL−1). All scale bars correspond to 100 nM. f) Small angle neutron scattering (SANS) spectra of MIACPs and BSM (C = 20 mg mL−1). A fiber structure should lead to a q−1 power law in mid‐q, which is more pronounced for MIACP‐1. The lower cut‐off of the q−1 power law region, q, is inversely related to the persistence length of the fiber.*
Interestingly, ≈30% of the hydrophobic functional groups in the case of MIACPs did not affect the overall filamentous morphology of the copolymers. Since aliphatic C11 carbon chains are randomly distributed throughout the polymer backbone, the possibility of aggregation is reduced and filamentous structures are formed.
For comparison, the morphology of commercially available porcine gastric mucin (PGM), was also evaluated. It displayed thread‐like elongated fiber structures (Figure 1e), suggesting that MIACPs mimic the mucin‐like elongated morphology.^[^ 43 ^]^
Small angle neutron scattering (SANS) was further applied to evaluate the morphology of MIACPs. The SANS spectra of the MIACPs with subtracted background (cell and incoherent scattering) are shown in Figure 1f. The spectra can be divided into three regimes. At low q, where large structures are visible, the intensity roughly follows a q ^−3^ power law, which indicates the presence of large‐scale inhomogeneities with sizes > 10^2^ nM, i.e., clusters of polymers, as observed frequently in cross‐linked polymer gels^[^ 44, 45 ^]^ or in entangled polymer solutions.^[^ 46, 47 ^]^ The fact that such domains are seen in SANS, when there were none in cryo‐EM, can be explained by the much higher concentration (20 mg mL^−1^ for SANS as opposed to 1.0 mg mL^−1^ for cryo‐EM). The increased concentrations of the SANS measurements were chosen to ensure a sufficiently high scattering intensity. At such high concentrations, the polymers overlap and entangle, giving rise to large‐scale inhomogeneities visible in low‐q. At intermediate q, meaning on a more local scale, the intensity scales roughly as q ^−1^, as expected for a filamentous structure. Notably, the q ^−1^ region is significantly more pronounced for MIACP‐1 and extends to lower q values. This suggests that the carboxylate moiety at the end of the hydrophobic chain promotes the formation of longer and stiffer fibers. In contrast, for MIACP‐2, one observes a markedly different scattering pattern at mid‐ and low‐q, which indicates the formation of more compacted structures on the length scale of 3‐5 nM. This may be attributed to the presence of the hydrophobic C_11_‐chains, whereas the MIACP‐1 has a charged group at the end of this chain. At high q, the spectra of MIACP‐1 and MIACP‐2 look identical. On such small length scales, the local structure of the polymer chain is visible. The q ^−1.8^ scaling suggests that the polymer chains are slightly swollen, as expected for a polymer in a good solvent. For a comparison to mucin, we investigated BSM as a representative of commercially available natural mucin, using small‐angle neutron scattering (SANS). The resulting scattering pattern closely resembles that observed for MIACP‐1. Therefore, SANS data confirmed mucin‐like filamentous morphology for MIACPs.
Biocompatibility and Anticoagulant Assay
2.3
The cytocompatibility of the MIACPs and MIP was evaluated on A549, HBE, and Vero E6 cell lines at polymer concentrations of up to 1.0 mg mL^−1^. The results suggest that MIACPs are biocompatible (Figure 2a–c) in different cell lines and the introduction of C11 aliphatic functionality did not result in significant toxicity compared to the control polymer, MIP. The inherent anticoagulant activity of sulfated polymers generally limits their direct application.^[^ 48 ^]^ In this context, the MIACPs exhibited negligible anticoagulant activity up to 10 µg mL^−1^ compared to heparin (Figure 2d). Our studies further revealed that anticoagulant activity increased with the degree of sulfation, although it remained substantially lower than that of heparin.
Cell viability of MIACPs and MIP in a) A549, b) HBE and c) Vero E6 cell line up to 1.0 mg mL−1 after 24 h of incubation. d) Activated partial thromboplastin time (aPTT) of plasma treated with MIACP‐1 (dark blue), MIACP‐2 (light blue), MIP (light green) at different concentrations. Heparin (dark green) and non‐sulfated MIACP, MIACP‐0 (black) were used as controls. Values are expressed as mean ±SD, n = 3.
Respiratory Syncytial Virus Inhibition and Virucidal Experiments
2.4
We investigated the antiviral activity of MIACPs and MIP against hRSV using an hRSV subtype A long strain containing a GFP reporter gene (hRSV‐GFP).^[^ 49 ^]^ A549 lung epithelial cells were used to evaluate the anti‐hRSV efficacy (Figure 3). In an infection inhibition assay, the dose‐response curves demonstrated that both MIACPs and MIP inhibited hRSV‐GFP, whereas the non‐sulfated copolymer (MIACP‐0) showed no inhibitory activity (Figure 3b), confirming the charge‐dependent mechanism of inhibition.^[^ 22 ^]^
Activity of MIACPs against hRSV infection. hRSV‐GFP was mixed with compounds at indicated concentrations for 45 min at 37 °C before incubating with A549 cells for 2 h. Viruses mixed with double‐distilled water (ddH2O, the solvent of compounds) were used as the control group. The inoculum was then replaced with fresh medium. 24 h later, cells were imaged, and the number of infected cells (with GFP signal) was detected by Incucyte. a) Fluorescent images of cells infected by hRSV‐GFP treated with given compounds (1.0 mg mL−1). The green fluorescence observed in the infected cells is attributable to the expression of GFP; scale bar: 500 µm. b) Dose‐response curves of MIACPs and MIP against hRSV‐GFP (n = 3). Relative inhibition rates were expressed relative to the number of infected cells detected in the control group. The error bars indicate mean ± SEM. c) Table with the IC50 values for the hRSV inhibition of the investigated copolymers. Commercially available natural mucins (BSM and PGM) and non‐sulfated MIACP, MIACP‐0 were used as controls.
The similar viral inhibition activities of MIP (IC_50_ = 1.57 nM) and MIACP‐1 (IC_50_ = 0.83 nM) confirm that ≈70% sulfation is optimal for maximum inhibition, aligning with previously reported degrees of sulfation required for maximal inhibitory activity. In fact, the relatively better inhibition observed for MIACP‐1 compared to MIP is likely due to the presence of ≈30% carboxylate functionality, which may contribute additional inhibitory effects owing to its electronegative nature. A remarkably low half‐maximal inhibitory concentration (IC_50_ = 0.51 nM) highlights the excellent antiviral efficacy of these mucin‐inspired polymers against hRSV. Such low IC_50_ values make them among the most potent electronegative inhibitors reported for hRSV.MIACP‐2 also exhibited a low IC_50_ value for hRSV‐GFP inhibition (IC_50_ = 1.82 nM), reinforcing their potential as effective antiviral agents.
The lower IC_50_ of MIACP‐1 compared to MIACP‐2 is likely due to the additional ≈30% carboxylate content in MIACP‐1. Commercially available porcine gastric mucin (PGM) and bovine submaxillary mucin (BSM) also showed hRSV‐GFP inhibition, but with significantly higher IC_50_ values (Figure 3c). This suggests that the synthetic inhibitors are more effective than commercially available natural mucins in terms of antiviral activity.
MIACPs exhibit strong charge‐dependent interactions with viral glycoproteins, as previously reported for polyglycerol sulfate groups, thereby inhibiting viral infection.^[^ 13 ^]^ Additionally, the long‐chain polysulfate amphiphilic elongated fibers of MIACPs likely engage in polyvalent interactions, providing sufficient steric shielding that contributes to their excellent inhibitory activity against hRSV. These features mark MIACPs as outstanding viral inhibitors.
We next wanted to investigate the virucidal activity of copolymers MIACPs through a dilution experiment.^[^ 24 ^]^ In this experiment, hRSV‐GFP (≈2 × 10^4^ PFU mL^−1^) was incubated with the desired amount of MIACPs, achieving a final concentration of 100 µg mL^−1^ (≈IC_90_ concentration), for 45 min at 37 °C. The virus‐MIACPs mixtures were then diluted 1:1000 fold into fresh medium, yielding a MIACPs concentration of 0.1 µg mL^−1^ (Figure 4a). Then the diluted mixtures were applied to A549 cells to quantify the viral titer. At 24 h post‐infection, the viral titre was calculated by counting the number of infected cells per well. If the interaction between virus and MIACPs were reversible (virustatic), the dilution would be expected to promote the disassociation of MIACPs from the virus surface and restore the infectivity towards the control (≈90–100%, Figure 3b). Conversely, a virucidal activity would have prevented recovery after dilution. No notable recovery of viruses was observed after serial dilution in MIACPs treated groups, indicating that MIACP‐1 and MIACP‐2 are virucidal, confirming their strong binding to hRSV (Figure 4b,c). On the other hand, MIP, which lacks the C11 aliphatic group, did not exhibit any virucidal activity (Figure 4d). This further confirms the importance of the C11 hydrophobic functional groups in conferring virucidal efficacy.
*Serial dilution experiments for the evaluation of the virucidal efficacy of MIACPs: a) Schematic diagram of the workflow. Virucidal activity detection of b) MIACP‐1, c) MIACP‐2 d) MIP against hRSV (n = 3). Viruses mixed with ddH2O were used as the control group. The error bars indicate mean ± SD. *p < 0.01; ns, p > 0.5.
Representation of the ECD Domain of hRSV and Its Possible Interaction with Inhibitors
2.5
Both the attachment protein G and the homotrimeric fusion protein F mediate viral binding to negatively charged heparan sulfate residues on the host cell surface,^[^ 6, 50 ^]^ providing a rationale for the inhibitory effect of the negatively charged polymer structures used in this study. It can be hypothesized that these structures interact with positively charged motifs of the ectodomain (ECD). It has been concluded that the binding of the G protein occurs via a stretch of positively charged amino acids (aa 187‐198: KRIPNKKPGKK; Uniprot P03423|GLYC_HRSVA) located between two mucin‐like motifs.^[^ 7, 51, 52 ^]^ To date, no motif has yet been identified for the ECD of the F protein. However, as we^[^ 53, 54 ^]^ and other groups^[^ 55, 56, 57 ^]^ have shown, analyzing the surface potential of the ECD of viral proteins can provide clues for binding sites. Nevertheless, this requires knowledge of the 3D structure. The ECD of the G protein essentially corresponds to an intrinsically disordered structure,^[^ 58 ^]^ as we have confirmed by an analysis using Alphafold3 (not shown). This makes it impossible to analyze the surface potential. However, it can be assumed that the flexibility of the structurally disordered ECD enables efficient interaction with negatively charged groups.
In contrast, the 3D structure of the ECD of the F protein has been experimentally elucidated (Figure 5a). Recently, we have analyzed the surface potential of the crystal structure of the F ECD (PDB 5UDE).^[^ 54 ^]^ However, the latter is missing an essential domain (aa 75‐111) of the ECD which contains two stretches of positively charged amino acids. To take this domain into account, we modeled the 3D structure of the complete ECD sequence (Uniprot P03420|FUS_HRSVA) using Alphafold3. As illustrated in Figure 5b, the 3D structure aligns with the experimentally determined structure. The domain aa 75‐111 is depicted in magenta which could be described as intrinsically disordered structure. The positively charged amino acids of two stretches are shown as blue spheres. The sequence RARR (aa 81‐84) is exposed on the ECD surface, while the second sequence KKRKRR (aa 106‐111) is localized inside the trimer.
The surface potential of the ECD of human RSV. a) Crystal structure of the trimeric ECD of hRSV (pdb 5UDE). b–d) Upper part: 3D structure of the trimeric ECD of hRSV determined by Alphafold3. The sequence was taken from Uniprot P03423|GLYC_HRSVA. c) The structure depicted in (b) has been rotated by 90°. d) Top view. The sequence aa 75‐111, absent from the crystal structure (a), is shown in magenta (back thin arrow). Stretches of positively charged amino acids are shown as blue spheres (yellow arrow: RARR (aa 81‐84); blue arrow: KKRKRR (aa 106‐111)). Lower part: Surface potential of the ECD. The solvent accessible surface is shown. The calculation of the surface potential was performed using the ABPS plugin of PyMOL, with an ionic strength of 0.15 m and a pH of 7.0.
Figure 5d shows the surface potential of the ECD. It is evident that there are multiple regions of positive surface potential that have the potential to function as binding sites for negatively charged groups. Notably, the exposed stretch RRAR contributes to a distinct region of positive potential. This sequence exhibits a striking similarity to a Cardin‐Weintraub motif, which has been implicated as a binding motif for heparan sulfate residues.^[^ 59, 60 ^]^
Conclusion
3
In this work, using RAFT polymerization, we developed a series of high molecular weight amphiphilic copolymers (MIACPs) composed of ≈70% sulfated dendronized oligoglycerol methacrylate repeat units and ≈30% C11 aliphatic chain repeat units bearing terminal alkyl and carboxylate functionality. We also developed a homopolymer (MIP) of similar molecular weight, composed solely of sulfated dendronized methacrylate repeat units, without any C11 aliphatic functionality. The synthesized polymers were well characterized by ^1^H NMR, SEC, zeta potential, and elemental analysis. Cryo‐EM of the MIACPs confirmed a single‐chain filamentous structure. SANS corroborated the filamentous structure. The strategic statistical distribution of ≈30% aliphatic chains led to the formation of a single‐chain filamentous structure instead of an aggregated morphology.
MIACPs demonstrated good biocompatibility and less anticoagulant activity, as confirmed by cell viability assays across various cell lines and by anti‐complementary assays, respectively. A plaque‐reduction assay confirmed that MIACPs inhibit hRSV infectivity in a sulfate‐dependent manner, with activity significantly superior to commercially available natural mucins such as PGM and BSM. The excellent inhibitory activity is further supported by the possible interaction of the ECD domain of hRSV with the polymers. The inhibitory activity of MIACPs is comparable to that of MIP, confirming that ≈70% sulfation is optimal for effective virus inhibition. Additionally, MIACPs exhibited virucidal activity, as demonstrated by dilution experiments. In contrast, MIP did not exhibit any virucidity. Therefore, the strategy to develop MIACPs is unique in that they not only destroy the virus through their virucidal functionality, but also maintain strong inhibitory activity, despite having only 70% electronegative functionality. In addition, surface analysis of hRSV F revealed multiple potential MIACPs engagement sites, suggesting a high genetic barrier to resistance mutation, as single‐point substitutions are unlikely to abrogate inhibition. In addition, RSV F is a highly conserved protein, which makes it an ideal target for antiviral and vaccine‐based interventions.^[^ 61, 62 ^]^ These findings demonstrate that combining high sulfation and virucidal functionality within a filamentous polymer architecture is an effective strategy for developing potent antiviral materials.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Supporting Information
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1H. Nair , D. J. Nokes , B. D. Gessner , M. Dherani , S. A. Madhi , R. Singleton , K. J. O'Brien , A. Roca , P. F. Wright , N. Bruce , A. Chandran , E. Theodoratou , A. Sutanto , E. R. Sedyaningsih , M. Ngama , J. Munywoki , R. Kartasasmita , E. A. Simoes , I. Rudan , M. W. Weber , H. Campbell , Lancet 2010, 375, 1545.20399493 10.1016/S 0140-6736(10)60206-1PMC 2864404 · doi ↗ · pubmed ↗
- 2R. Lozano , M. Naghavi , K. Foreman , S. Lim , K. Shibuya , V. Aboyans , J. Abraham , T. Adair , R. Aggarwal , S. Y. Ahn , M. Alvarado , H. R. Anderson , L. M. Anderson , K. G. Andrews , C. Atkinson , L. M. Baddour , S. Barker‐Collo , D. H. Bell , E. J. Benjamin , K. Bennett , F. Bhalla , A. J. Bikbov , G. Birbeck , M. B. Blyth , I. Bolliger , S. Boufous , C. Bucello , M. Burch , P. Burney , J. Carapetis , et al., Lancet 2012, 380, 2095.23245604
- 3A. R. Falsey , P. A. Hennessey , M. A. Formica , C. Cox , E. E. Walsh , N. Engl. J. Med. 2005, 352, 1749.15858184 10.1056/NEJ Moa 043951 · doi ↗ · pubmed ↗
- 4Q. Zhu , J. S. Mc Lellan , N. L. Kallewaard , N. D. Ulbrandt , S. Palaszynski , J. Zhang , B. Moldt , A. Khan , C. Svabek , J. M. Mc Auliffe , R. S. Wrapp , J. Patel , E. Yang , L. A. Graham , M. Z. Reinhardt , T. W. Nieusma , Z. P. Souza , C. M. Lynch , M. Z. Saunders , L. Sullivan , U. Zydek , T. M. Connolly , H. P. Ko , H. Y. Wang , D. M. Boyington , S. F. Douglas , L. M. Dieseldorff , C. E. Motyka , E. S. Welch , R. G. Foreman , et al., Sci. Transl. Med. 2017, 9, 1 · doi ↗ · pubmed ↗
- 5S. M. Sake , X. Zhang , M. K. Rajak , M. Urbanek‐Quaing , A. Carpentier , A. P. Gunesch , C. Grethe , A. Matthaei , J. Rückert , M. Galloux , T. Larcher , R. Le Goffic , F. Hontonnou , A. K. Chatterjee , K. Johnson , K. Morwood , K. Rox , W. A. M. Elgaher , J. Huang , M. Wetzke , G. Hansen , N. Fischer , J.‐F. Eléouët , M.‐A. Rameix‐Welti , A. K. H. Hirsch , E. Herold , M. Empting , C. Lauber , T. F. Schulz , T. Krey , et al., Nat. Commun. 2024, 15, 1173.38332002 10. · doi ↗ · pubmed ↗
- 6S. A. Feldman , S. Audet , J. A. Beeler , J. Virol. 2000, 74, 6442.10864656 10.1128/jvi.74.14.6442-6447.2000 PMC 112152 · doi ↗ · pubmed ↗
- 7S. A. Feldman , R. M. Hendry , J. A. Beeler , J. Virol. 1999, 73, 6610.10400758 10.1128/jvi.73.8.6610-6617.1999 PMC 112745 · doi ↗ · pubmed ↗
- 8C. D. Griffiths , L. M. Bilawchuk , J. E. Mc Donough , K. C. Jamieson , F. Elawar , M. J. O'Neill , R. F. Diel , J. E. Read , S. J. Orton , S. J. Tanzer , P. J. Hamilton , J. R. Collins , S. J. Shaw , J. M. Hargreaves , S. P. Jones , N. J. Smith , Nature 2020, 583, 615.32494007