ADD domain added new binding partners for the nuclear hub protein ATRX: ADD domain added new binding partners for ATRX
Yan Chen, Yang Luo, Jielin Sun, Shouhua Wang, Bingbing Wan

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
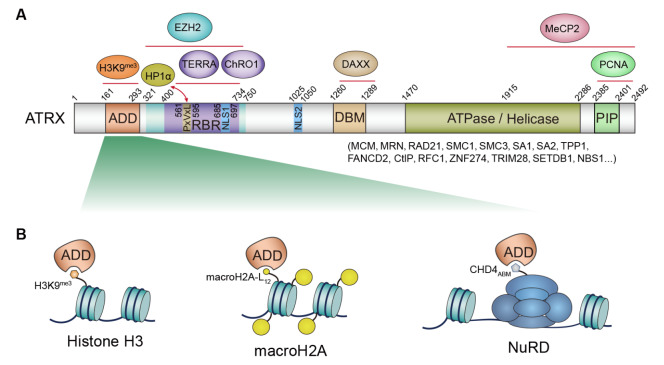 Figure 1
Figure 1- —the grants from Ningbo Science Innovation Yongjiang 2035 Key R&D Program
- —the National Natural Science Foundation of China
- —the Fundamental Research Funds for the Central Universities and the Interdisciplinary Program of Shanghai Jiao Tong University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeuroblastoma Research and Treatments · Nuclear Structure and Function · Retinoids in leukemia and cellular processes
ATRX is a large, multi-domain nuclear protein that functions as a crucial ATP-dependent chromatin remodeler, transcriptional regulator, and guardian of telomeric and genomic integrity [1]. As a member of the SWI/SNF family of chromatin remodeling proteins, a primary and well-defined function of ATRX is to facilitate the replication-independent deposition of the histone variant H3.3 at specific genomic loci, predominantly repetitive sequences such as telomeres, pericentromeric heterochromatin, and ribosomal DNA (rDNA). Mutations in the ATRX gene are associated with a severe X-linked neurodevelopmental disorder and alpha-thalassemia. Moreover, ATRX dysfunction can lead to genomic instability, contributing to the development and progression of various cancers, including gliomas and pancreatic neuroendocrine tumors (PanNETs) [2].
ATRX orchestrates chromatin dynamics through its modular domains. Its N-terminal ADD domain and a PxVxL-like motif recognize histone H3K9me3 and interact with the heterochromatin-binding protein HP1α, respectively. Collaborating with the histone chaperone DAXX, ATRX then utilizes its C-terminal ATPase/Helicase domain to provide the energy needed to remodel chromatin and deposit the histone variant H3.3 into repetitive DNA regions ( Figure 1A) [3]. The Chen lab and other two groups previously elucidated the minimal elements for DAXX interaction, demonstrating that a short 1260‒1289 residues motif (DAXX-binding motif, DBM) of ATRX is solely responsible for ATRX-DAXX heterodimer formation [ 4– 6] . Beyond its role in chromatin remodeling, ATRX employs its RBR (RNA-binding region) to engage the telomeric long non-coding RNA (lncRNA) TERRA, thereby regulating TERRA-mediated R-loops and telomeric G-quadruplex (G4) structures. Interestingly, this same RBR also binds the muscle-specific lncRNA ChRO1 to coordinate constitutive heterochromatin reorganization and regulate cell differentiation. Notably, nearly half of the disease-causing mutations in ATRX [7], leading to a severe neurodevelopmental disorder, are clustered within the ADD domain, highlighting this relatively small domain’s critical functional importance and warranting more intense investigation.
Figure 1 The multifaceted protein ATRX orchestrates a complex regulatory network to modulate epigenetic landscapes and safeguard genomic stability(A) Schematic diagram of full-length ATRX (282 kDa) depicting key protein interaction domains. The ADD domain serves as a binding site for H3K9me3, and the PxVxL motif facilitates interaction with HP1α. The RBR (RNA-binding region) engages with TERRA and ChRO1 lncRNAs, with an overlapping area also binding EZH2. The DBM (DAXX-binding motif) interacts with DAXX, and the C-terminal conserved hydrophobic PIP (PCNA-interacting protein) motif likely mediates PCNA interaction. Additionally, a substantial C-terminal segment (residues 1915‒2492), encompassing a portion of the ATPase/Helicase domain, interacts with MeCP2. (B) The versatile ADD domain coordinate diverse protein interactions. Beyond its established role in binding histone H3K9me3, new research by Yan et al. [12] in this issue significantly broadens the ADD domain’s functional interaction network. Their structural and AP-MS analyses identify the histone variant macroH2A and CHD4, a component of the chromatin remodeling NuRD complex, as new binding partners.
ADD Domain Functions in Chromatin-Binding Protein ATRX and DNMTs
The ADD domain is a compact, globular domain, typically around 140 residues in length. Its most well-understood function is its ability to recognize and bind to specific post-translational modifications on the N-terminal tail of histone H3. This domain’s unique three-dimensional structure is characterized by the intricate packing of three distinct modules: an N-terminal GATA-like C2C2-type zinc finger, a central C4C4-type PHD zinc finger, and a C-terminal single α-helix [8]. Beyond histone recognition, the DNMT3A ADD domain is known to bind to unmodified or post-translationally modified peptides from proteins such as HDAC1, SETDB1, histone H4R3me2, and EZH2 [9]. This multifaceted binding property resembles the more evolutionarily conserved chromatin-binding BRCT (BRCA1 C-terminal) domain [ 10, 11] , which recognizes phosphorylated histone H2A and various unmodified DNA repair proteins from yeast to humans. Intriguingly, whether ATRX also utilizes its ADD domain to engage a distinct set of proteins, similar to DNMT3A, is still enigmatic until Yan et al. [12] reported in this issue, through structural analysis, that the histone variant macroH2A binds the ATRX ADD domain.
To investigate the macroH2A-binding region of ATRX, Yan et al. first utilized a GST-pulldown assay with purified macroH2A-H2B heterodimer and truncated ATRX proteins. This approach successfully narrowed the interaction site from the previously reported 1‒841 amino acid region [13] to the more precise N-terminal ADD domain (161‒292 residues). Their analysis further demonstrated that the compact N-terminal three-helix bundle-shaped histone fold (HF) domain of macroH2A-H2B, rather than its macro domain, mediates this interaction. Notably, while confirming the previously reported interaction between macroH2A1-H2B and ATRX [13], the study extended these findings by identifying a direct interaction between ATRX and its structural homolog macroH2A2-H2B ( Figure 1A).
Molecular Basis of ATRX
ADD Domain Recognition of macroH2A-H2B
To delineate the precise interaction, Yan et al. [12] leveraged AlphaFold2-multimer to predict the macroH2A1-H2B-ATRX complex structure. The high prediction local distance difference test (pLDDT) values and low predicted aligned error (PAE) rates indicated high reliability of the predicted model. Their analysis revealed that the interaction between ATRX and macroH2A1-H2B is primarily driven by electrostatic forces. Specifically, a negatively charged loop (amino acids 207‒225) connecting the GATA-like and PHD zinc finger of ATRX, rich in aspartic (D) and glutamic (E) residues, docks onto a positively charged surface of macroH2A1 HF L 12 (linking α-helix-1 and α-helix-2 of macroH2A1 HF). They pinpointed that ATRX residues Y203, D207, D208, E218 and macroH2A1 residue R40 play the most critical roles in this engagement. Furthermore, the guanidinium group of macroH2A1 HF L 12 R40 forms a cation–π interaction with the phenol ring of ATRX Y203, additionally strengthening the overall electrostatic and hydrogen bonding network between ATRX and macroH2A.
To dissect the molecular basis by which ATRX distinguishes between canonical histone H2A and macroH2A, Yan et al. [12] performed a comparative analysis of their respective nucleosome core particle (NCP) structures. They identified that the preceding residues _36_HPKY 39 of macroH2A1 L 12 residue R40 adopts a configuration distinct from the corresponding _39_NYAE 42 segment of canonical H2A (containing R43). This unique conformation of the macroH2A segment facilitates closer contacts with the ATRX ADD domain. Strikingly, swapping the _39_NYAE 42 of H2A segment into _36_HPKY 39 of macroH2A1 L 12 abrogated ATRX binding, whereas the reverse swap in H2A awarded ATRX binding capacity.
Does the ATRX ADD domain employ a similar mechanism to bind both macroH2A and H3K9me3? Yan and colleagues addressed this question through competitive GST pull-down assays and isothermal titration calorimetry (ITC). Their experiments demonstrated that increasing concentrations of H3K9me3 peptide progressively reduced macroH2A/H2B-ATRX binding. ITC measurements revealed the wild-type H3K9me3 peptide binds ATRX with a dissociation constant (K d) of around 4 μM, while Y203A and D/E mutations in ATRX significantly weakened this interaction. Moving forward, it will be crucial to further explore whether macroH2A or H3K9me3 binds ATRX more strongly under both in vitro and in vivo conditions.
New ATRX Binding Partners Are Added Through Its ADD Domain
The DNMT3A ADD domain is well-known as a versatile module involved in diverse protein interactions [9]. Intriguingly, the ATRX ADD domain utilizes a similar surface region to bind both H3K9me3 and macroH2A. This similarity raised the possibility that the ATRX ADD domain might interact with other proteins beyond histones. Indeed, Yan and colleagues employed conventional AP-MS to identify numerous new ATRX-interacting proteins. These included several components of the Nucleosome Remodeling and Deacetylase (NuRD) complex, such as MTA1/2, HDAC1/2, RBBP4/7, CHD4, and MBD2/3. Subsequent experiments confirmed these interactions, as Flag-tagged ATRX successfully co-purified endogenous NuRD complex proteins including CHD4, MTA2, and HDAC2. Leveraging AlphaFold3, a short fragment within the N-terminal disordered region of CHD4 (termed CHD4 ADD-binding motif, or CHD4 ABM) was predicted to pair with the ATRX ADD domain by forming a β-strand. Interestingly, CHD4 ABM and H3K9me3 share structural commonalities: both adopt very similar β-strand configurations and possess key AR/KTK residues at the N-terminus of their respective peptides. Notably, ITC revealed a K d of approximately 46 μM for the binding of CHD4 ABM to ATRX. This interaction was further validated by ITC experiments using ATRX Y203A and D/E mutations, known to impair its binding.
While the structural and biochemical data presented by Yan et al. [12] have substantially advanced our comprehension of the ATRX ADD domain, several critical questions remain to be addressed.
First, despite the strong association between ATRX ADD mutations and human diseases, the key residues identified for H3K9me3, macroH2A, and CHD4 interactions do not align with the most prevalent disease-causing point mutations within the ADD domain [ 7, 14] . This discrepancy strongly suggests that these disease-associated residues may serve as docking sites for other crucial, yet-to-be-discovered, ATRX ADD-interacting proteins. Identifying these unknown binding partners could directly illuminate the etiology of ATR-X syndrome and other diseases.
Second, a pertinent question arises regarding the DNMT3A ADD domain: does it, like its ATRX counterpart, utilize a similar competitive binding strategy to interact with its diverse set of functionally distinct partners, including HDAC1, SETDB1, histone H4R3me2, and EZH2?
Finally, the physiological and pathological roles of the newly identified ATRX ADD-CHD4 interaction in cell and mouse models warrant further investigation. Given that the ADD domain generally functions as a molecular “GPS” for ATRX, it is crucial to ascertain whether CHD4 ABM actively recruits ATRX to NuRD-associated chromatin regions, thereby enabling ATR’s ATPase/helicase to remodel local chromatin structure. To fully elucidate these aspects, comprehensive functional assays in various cell and mouse models are essential to evaluate this interaction’s potential involvement in transcriptional regulation, cellular differentiation and development, genomic instability, telomere malfunction, or tumorigenesis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Aguilera P López-Contreras AJ ATRX, a guardian of chromatin Trends Genet 20233950551910.1016/j.tig.2023.02.00936894374 · doi ↗ · pubmed ↗
- 2Jiao Y Shi C Edil BH de Wilde RF Klimstra DS Maitra A Schulick RD et al. DAXX/ ATRX, MEN 1, and m TOR pathway genes are frequently altered in pancreatic neuroendocrine tumors Science 20113311199120310.1126/science.120060921252315 PMC 3144496 · doi ↗ · pubmed ↗
- 3Dyer MA Qadeer ZA Valle-Garcia D Bernstein E ATRX and DAXX: mechanisms and mutations Cold Spring Harb Perspect Med 20177 a 02656710.1101/cshperspect.a 02656728062559 PMC 5334245 · doi ↗ · pubmed ↗
- 4Wang X Zhao Y Zhang J Chen Y Structural basis for DAXX interaction with ATRX Protein Cell 2017876777110.1007/s 13238-017-0462-y 28875424 PMC 5636753 · doi ↗ · pubmed ↗
- 5Hoelper D Huang H Jain AY Patel DJ Lewis PW Structural and mechanistic insights into ATRX-dependent and -independent functions of the histone chaperone DAXX Nat Commun 20178119310.1038/s 41467-017-01206-y 29084956 PMC 5662737 · doi ↗ · pubmed ↗
- 6Li Z Zhao D Xiang B Li H Structural and biochemical characterization of DAXX-ATRX interaction Protein Cell 2017876276610.1007/s 13238-017-0463-x 28875283 PMC 5636754 · doi ↗ · pubmed ↗
- 7Argentaro A Yang JC Chapman L Kowalczyk MS Gibbons RJ Higgs DR Neuhaus D et al. Structural consequences of disease-causing mutations in the ATRX-DNMT 3-DNMT 3L (ADD) domain of the chromatin-associated protein ATRX Proc Natl Acad Sci USA 2007104119391194410.1073/pnas.070405710417609377 PMC 1924575 · doi ↗ · pubmed ↗
- 8Iwase S Xiang B Ghosh S Ren T Lewis PW Cochrane JC Allis CD et al. ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome Nat Struct Mol Biol 20111876977610.1038/nsmb.206221666679 PMC 3130887 · doi ↗ · pubmed ↗