Genome-Wide Identification and Expression Analysis of the WOX Family Reveals Potential Roles in Stem Development of Euphorbia hirta
Qianyi Lyu, Shutong Chen, Xin Wang, Yuan Yuan, Hongrui Zhang, Wanqi Liang, Han Cheng, Zhi Deng

TL;DR
This study identifies and analyzes 14 WOX genes in Euphorbia hirta, revealing their potential roles in stem and laticifer development.
Contribution
The first comprehensive characterization of the WOX gene family in Euphorbia hirta, including expression and localization data.
Findings
14 EhWOX genes were identified and classified into three evolutionary clades.
EhWOX4-6 and EhWOX14 show high stem-specific expression.
Promoter analysis reveals light, hormone, and stress-responsive elements in EhWOX genes.
Abstract
The homeobox transcription factor (TF) superfamily includes the WUSCHEL-RELATED HOMEOBOX (WOX) family, which plays a critical role in adaptive plant growth. Specifically, WOX regulates stem growth in plants, with stems serving as the structural framework for laticifers in Euphorbia hirta. However, the number of WOX gene family members in the E. hirta genome has not been reported. In this study, we identified 14 EhWOX genes in E. hirta and characterized their physicochemical properties, chromosomal locations, phylogenetic relationships, conserved motifs, gene structures, promoter cis elements, gene ontology (GO) enrichment, tissue-specific expression patterns, and subcellular localization. Chromosomal mapping indicated their distribution across nine chromosomes. Phylogenetic analysis classified these genes into three evolutionary clades. Promoter cis-element analysis identified abundant…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
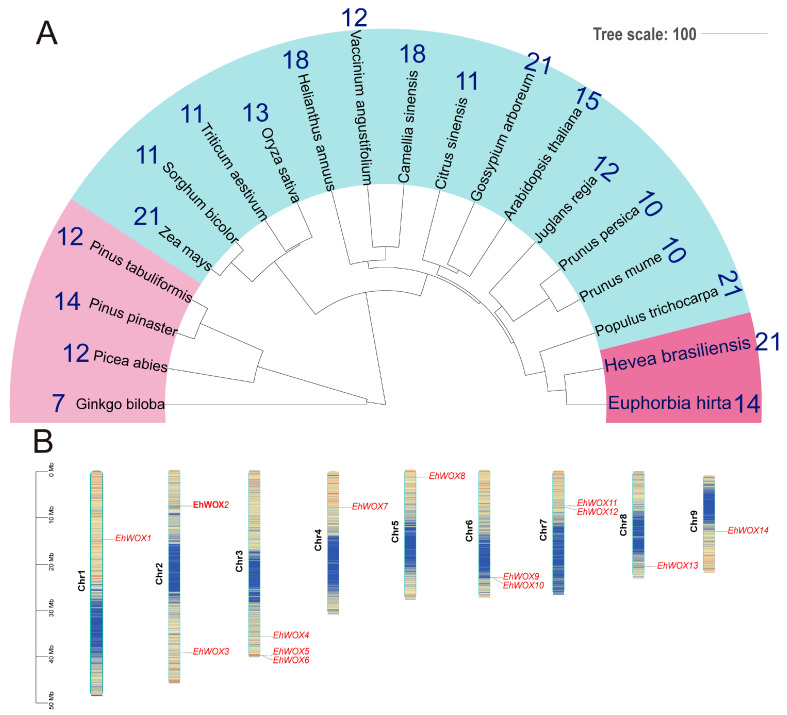 Figure 1
Figure 1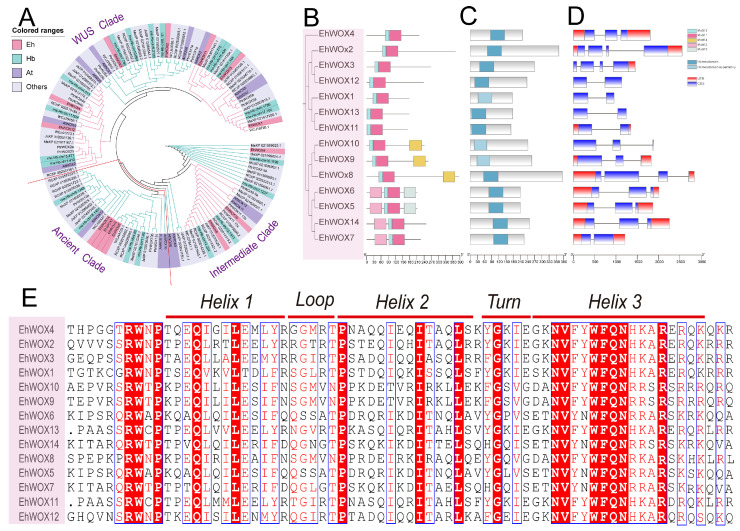 Figure 2
Figure 2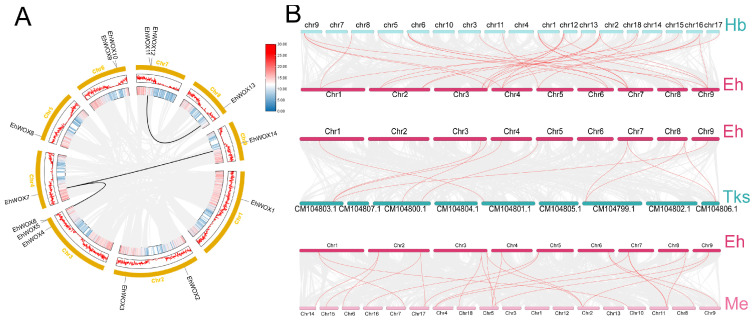 Figure 3
Figure 3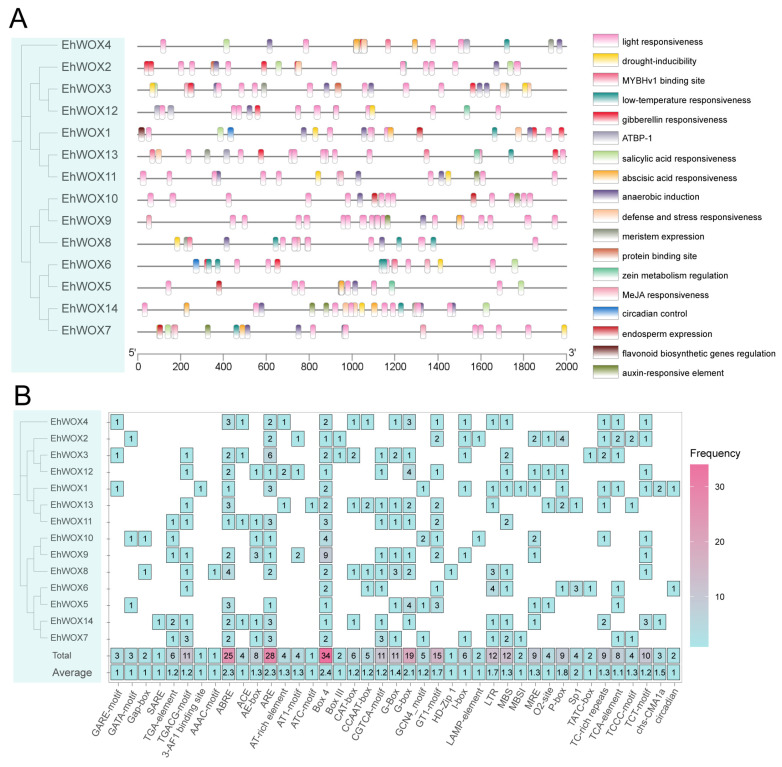 Figure 4
Figure 4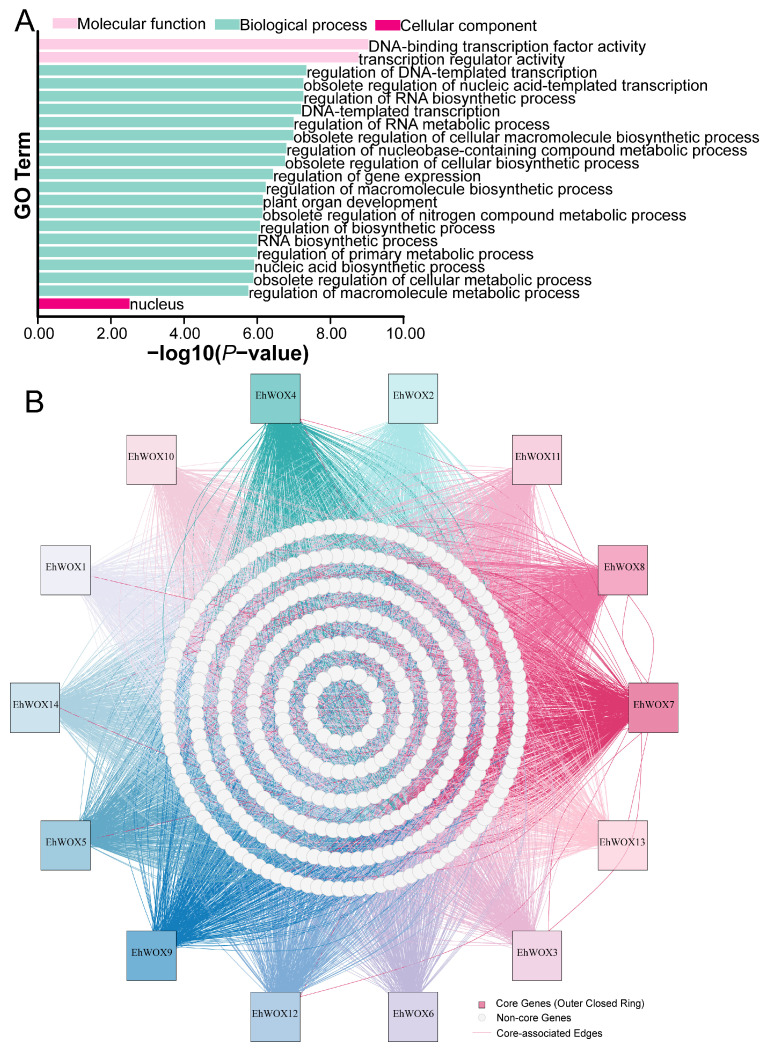 Figure 5
Figure 5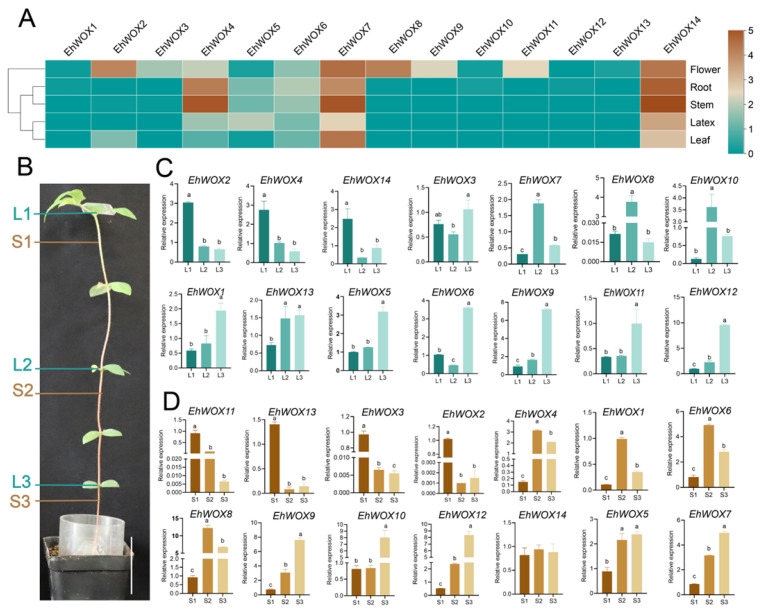 Figure 6
Figure 6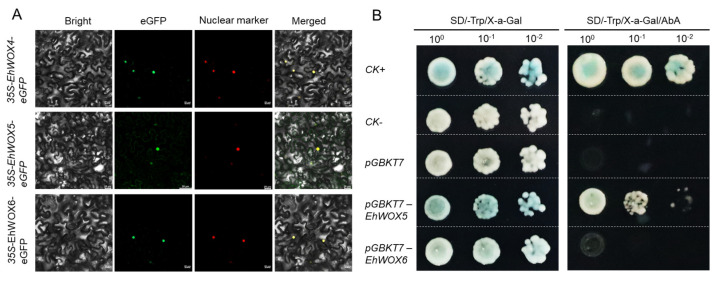 Figure 7
Figure 7- —National Scientific Fund of China
- —Chinese Academy of Tropical Agricultural Sciences for Science and Technology Innovation Team of National Tropical Agricultural Science Center
- —Hainan Province Science and Technology Talent Innovation Project
- —Project of the National Key Laboratory for Tropical Crop Breeding
- —Project of the State Key Laboratory of Tropical Crop Breeding
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Molecular Biology Research · Plant tissue culture and regeneration · Plant Gene Expression Analysis
1. Introduction
Plants possess numerous TF families that play critical roles in regulating diverse cellular metabolic activities and signaling pathways during stress responses and developmental processes. Among TF families, the WOX TF family, first identified in Arabidopsis thaliana in 1996, has been demonstrated to be particularly crucial for shoot meristem maintenance and floral organ development [1]. The WOX genes represent a plant-specific family of TFs belonging to the homeodomain (HD) superfamily, currently classified into 14 distinct subfamilies based on phylogenetic relationships [2,3,4]. Characteristically, WOX proteins contain a highly conserved 60–66 amino acid homeodomain that forms a helix–turn–helix structure, with the N-terminal HD domain enabling high-affinity monomeric binding to specific DNA sequences [5,6]. Phylogenetic analyses have consistently demonstrated that the WOX gene family is evolutionarily classified into three major clades, including the ancient clade (AC), intermediate clade (IC), and modern/WUS clade (WC) [7,8]. Comparative genomic studies reveal that the AC originated in embryophytes and chlorophytes [9], while the IC emerged in lycophytes and seed plants and the WC evolved from the common ancestor shared by spermatophytes and bryophytes [3,10]. This tripartite phylogenetic division reflects the progressive evolutionary trajectory and functional diversification of WOX genes across land plants.
E. hirta, native to Central America and later introduced to Southeast Asia, is now widely distributed across tropical and subtropical regions worldwide. Commonly known as asthma weed, snakeweed, or ara tanah, it belongs to the Euphorbiaceae family and the Euphorbia genus [11]. As a medicinal herb, the plant is rich in bioactive compounds, including polyphenolic flavonoids, tannins, terpenoids, and triterpenoids [12,13,14], which contribute to its unique pharmacological properties. E. hirta exhibits strong reproductive capacity, thriving in grassy and shrubby hillsides, particularly in sandy soils [15]. Furthermore, this annual herbaceous species typically attains a height of 15–50 cm and exhibits either erect or procumbent stems containing laticifers that produce latex. These morphological characteristics make it an ideal model system for investigating stem and laticifer development.
Indeed, genetic and molecular biological studies on plant WOX genes have demonstrated their crucial roles in various developmental processes [8,16,17]. Since they play vital functions in shoot and floral development [1], research has extensively characterized WOX gene functions in developmental regulation across multiple plant species. During plant growth and development, the plant stem functions as an essential junction connecting roots and leaves, performing multiple pivotal functions, including mechanical support, nutrient transport, storage, reproduction, and photosynthesis. Research has established that in stem development, the WOX4 gene forms a signaling hub with BES1 to regulate the proliferation and differentiation of vascular cambium cells in response to both hormonal and environmental signals, and wox4 mutants exhibit normal xylem differentiation but impaired cambium cell proliferation [18]. In cotton, knockout of GhWOX4 reduces cambium width and division activity, consequently suppressing secondary growth [19]. In hybrid aspen (Populus tremula × tremuloides), WOX4 modulates both cambium cell identity and mitotic activity [20], while in Scots pine (Pinus sylvestris), PsWOX4 expression peaks during active cambium proliferation periods, with the highest cambium cell layers observed in mature trees (63-year-old) [21]. Additionally, further investigations reveal that AtWOX5, AtWOX8/9, and AtWOX14 play crucial roles in stem development [22]. In rubber tree, previous studies have suggested that the CLAVATA-MAPK-WOX signaling pathway may be involved in regulating secondary laticifer differentiation in Hevea brasiliensis [23]. Meanwhile, research has characterized WOX genes in H. brasiliensis, Jatropha curcas, Manihot esculenta, and Ricinus communis [24]. These studies demonstrate that WOX genes have been well studied in across multiple plant species, whereas their functions in the Euphorbiaceae family, which diverged from Arabidopsis approximately 100 million years ago [25], remain largely unexplored and the functions of WOX genes in E. hirta remain unreported.
Despite the established roles of WOX genes in stem development and the economic and medicinal importance of E. hirta, a comprehensive characterization of the WOX family in this species is lacking, and its potential involvement in stem development remains unexplored. In this study, we systematically identified 14 EhWOX genes in E. hirta. These genes were subjected to comprehensive characterization through multiple approaches, such as bioinformatics analyses, including phylogenetic tree construction, chromosomal localization, conserved motif and gene structure analysis, synteny analysis, cis-acting element prediction, GO enrichment, and regulatory network prediction. The expression patterns of EhWOX genes in various tissues were analyzed by RNA-seq; then, expression in stems and leaves was further validated by RT-qPCR. Additionally, functional validation was achieved through subcellular localization in Nicotiana Benthamian and transcriptional autoactivation in yeast. This study presents the first systematic characterization and expression profiling of WOX genes and provides a foundation for functional validation of candidate WOX proteins in E. hirta.
2. Results
2.1. Identification of EhWOXs and Prediction of Physicochemical Properties
The hidden Markov model (HMM) was employed to identify EhWOX homeodomains, followed by local BLASTP searches using A. thaliana WOX genes as a reference. A total of 14 candidate EhWOX genes were identified based on the genome of E. hirta (Supplementary Table S1). Bioinformatic analysis predicted that their amino acid (AA) lengths ranged from 170 to 385 residues, with molecular weights (MW) between 19.9 and 42.36 kDa. Further predicted characterization showed that their theoretical isoelectric points (pI) varied from 5.15 to 9.83. All EhWOX proteins exhibited instability indices exceeding 40 (from 43.09 to 74.23), suggesting their inherent instability. The aliphatic index ranged from 51.75 to 73.86, while the values of the grand average of hydropathicity (GRAVY) were all negative (−0.475 to −1.057) (Table 1), indicating strong hydrophilicity. Additionally, subcellular localization predictions placed EhWOX proteins in the nucleus.
2.2. WOX Numbers Across Species and Chromosomal Locations of EhWOXs
To compare WOX numbers across species, a phylogenetic tree of 20 plant species was constructed, and the number of WOX genes in each species was quantified (Figure 1A) [8]. The divergence time tree revealed that E. hirta and H. brasiliensis clustered within the same clade, indicating their close phylogenetic relationship. However, differences were observed among the WOX gene family members, with 14 EhWOX genes identified in E. hirta and 21 in H. brasiliensis, which may result from gene evolution driven by functional demands [26,27]. Notably, this higher number of WOX genes in H. brasiliensis was comparable to that in Zea mays and Gossypium arboreum. Additionally, chromosomal localization analysis demonstrated that the 14 EhWOX genes were randomly distributed across nine chromosomes in E. hirta (Figure 1B). Chromosome three contained the highest number (three genes), while chromosomes two, six, and seven each harbored two EhWOX genes and the remaining chromosomes contained one EhWOX gene, respectively.
2.3. Phylogenetic Analysis, Conserved Domains and Gene Structural Analysis of EhWOXs
Phylogenetic reconstruction was conducted using the neighbor-joining method with amino acid sequences of WOXs from E. hirta, H. brasiliensis, A. thaliana, and other species. Consistent with the phylogenetic clade of A. thaliana AtWOXs [28], the EhWOX proteins were classified into three distinct subfamilies (Figure 2A). Each subfamily contained WOX members from both A. thaliana and H. brasiliensis. Notably, the EhWOX proteins formed distinct branches that were phylogenetically distant from their orthologs in H. brasiliensis and A. thaliana, suggesting potential functional divergence during the evolution of E. hirta. Conserved motif and gene structure analyses were performed to investigate the evolutionary relationships among the 14 EhWOX genes. MEME analysis identified five conserved motifs in the EhWOX family (Figure 2B). Notably, motif 1 and motif 3 were present in all 14 EhWOX genes, with motif 1 consistently positioned to the right of motif 3, and seven genes (EhWOX4, EhWOX2, EhWOX3, EhWOX12, EhWOX1, EhWOX13, and EhWOX11) only contained motifs 1 and 3 of five motifs. Additional motif analysis revealed that four genes (EhWOX6, EhWOX5, EhWOX14, and EhWOX7) possessed motif 2, while three genes (EhWOX10, EhWOX9, and EhWOX8) contained motif 4. Moreover, only two genes (EhWOX6 and EhWOX5) exhibited motif 5, suggesting potential functional diversification among EhWOX members. NCBI-CDD domain prediction showed that all 14 genes belong to the Homeodomain superfamily, with 11 possessing canonical homeodomains (Homeodomain family) and 3 (EhWOX10, EhWOX9, and EhWOX1) showing structural variations (Figure 2C). Gene structure analysis revealed that EhWOX genes contained one to three introns (Figure 2D), while protein sequence alignment revealed that the Helices, Loops, and Turns within the conserved homeodomain of the 14 EhWOX proteins exhibited highly similar amino acid distributions (Figure 2E). These findings suggest that this gene family is highly conserved during evolution and may play a crucial role in plant growth and development.
2.4. Duplication Events and Evolutionary Characterization of EhWOXs
Gene duplication events include whole-genome duplication (WGD), tandem duplication, segmental duplication, and dispersed duplication, which contribute to the generation of homologous genes with sequence similarity. Tandem duplication events are defined as chromosomal regions containing two or more genes within a 200 kb genomic interval; they are particularly important for evolutionary adaptation, gene regulation, and genome stability [29]. Using the MCScanX algorithm, we identified diverse duplication types among EhWOX genes (Supplementary Table S2). Specifically, EhWOX6 originated from proximal duplication, while EhWOX10 and EhWOX9 resulted from tandem duplication. Five genes (EhWOX13, EhWOX11, EhWOX5, EhWOX14, and EhWOX7) were derived from WGD or segmental duplication events, with the remaining six genes classified as dispersed duplications, demonstrating the heterogeneous origins of EhWOX genes. Collinearity analysis revealed three syntenic gene pairs distributed across five chromosomes (Figure 3A). Notably, EhWOX7 formed two syntenic pairs and clustered with EhWOX14 in the phylogenetic tree, despite their localization to different chromosomes (Chr4 and Chr9). Additionally, comparative synteny analysis with H. brasiliensis, Taraxacum kok-saghyz, and M. esculenta demonstrated distinct evolutionary patterns (Figure 3B). E. hirta exhibited 28, 10, and 23 syntenic gene pairs with these species. The higher number of syntenic pairs with H. brasiliensis and M. esculenta compared to T. kok-saghyz suggests stronger evolutionary conservation between these species. These findings indicate that WOX genes have undergone multiple duplication events during speciation while maintaining functional conservation, underscoring their essential roles in plant growth and development.
2.5. Characterization of Cis-Acting Elements in Promoters of EhWOXs
To elucidate the potential regulatory relationship of EhWOX genes, we analyzed cis-acting elements within the 2000 bp upstream regions of all 14 EhWOX genes. As a result, a total of 18 distinct cis-regulatory elements were predicted (Figure 4). The promoter regions were particularly enriched with light-responsive, hormone-related, and stress-responsive elements. Light-responsive elements were the most abundant category, with each EhWOX gene containing 5–15 Light-responsive elements (Box 4, G-Box, and GT1 motif) and EhWOX9 showing the maximum number (15 elements). Hormone-responsive elements were also widely distributed, including abscisic acid-responsive ABRE elements and JA-responsive CGTCA/TGACG motifs. Notably, five genes (EhWOX4, EhWOX2, EhWOX1, EhWOX10, and EhWOX5) lacked JA-responsive elements, while the remaining genes contained varying numbers of JA-responsive motifs. Only two genes (EhWOX13 and EhWOX6) lacked ARE elements. The predicted cis-element profile suggests that EhWOX genes are likely involved in diverse physiological processes, including light signaling, hormonal regulation, and stress responses, highlighting their potential roles in adaptive plant growth and development.
2.6. GO and Regulatory Network Predictions of EhWOXs
Studies across plant species have demonstrated the involvement of WOX gene family members in diverse biological processes. In this study, the predicted result revealed significant enrichment in the GO terms of 45 biological process (Supplementary Table S3). Figure 5A displays the top 18 most significantly enriched biological process terms, 2 molecular function terms, and 1 cellular component term. The five most significantly enriched terms comprised regulation of DNA-templated transcription (GO:0006355), obsolete regulation of nucleic acid-templated transcription (GO:1903506), regulation of RNA biosynthetic process (GO:2001141), DNA-templated transcription (GO:0006351), and regulation of RNA metabolic process (GO:0051252).These predicted analyses support the canonical role of WOX family members as TFs that regulate plant growth and development by binding to promoter regions of target genes and directly modulating DNA/RNA-related activities. In addition, based on the complete sequences of E. hirta, we identified transcription factor binding motifs across all sequences, obtaining a total of 10,155 potential binding motif pairs involving 14 EhWOX TFs through the FIMO tool, encompassing 630 genes, including these 14 EhWOX genes (Supplementary Table S4). A total of 362 potential target genes with prediction scores of “Excellent” or “Fair” were displayed (Figure 5B). Among these, EhWOX4, EhWOX14, and EhWOX12 exhibited the highest predicted motif binding counts (465, 461, and 413 pairs, respectively), while EhWOX6 showed the lowest (259 pairs). These predicted results demonstrate that EhWOX TFs exhibit differential DNA-binding properties in E. hirta, suggesting their potential involvement in plant physiological processes through varying regulatory intensities on target genes. The construction of the EhWOX transcriptional regulatory network provides foundational data for subsequent research.
2.7. Expression Patterns of WOX Genes in E. hirta Tissues
To investigate the potential roles of EhWOX genes in E. hirta, we analyzed their expression patterns across five tissues (flower, latex, leaf, root, and stem) using RNA-seq data. Heatmap analysis revealed distinct tissue-specific expression profiles among the 14 EhWOX genes (Figure 6A, Supplementary Table S5). Notably, EhWOX7 and EhWOX14 showed consistently high expression across all examined tissues, with particularly high expression in stems, roots, and flowers. The EhWOX4, EhWOX5, and EhWOX6 genes also exhibited stem-predominant expression (S2 and S3), with these genes predicted to play crucial roles in stem development and maintenance. Furthermore, EhWOX14 and EhWOX7 displayed relatively high expression in latex, implying their potential involvement in latex biosynthesis. To validate these expression patterns, RT-qPCR was performed on three distinct segments of leaves (L1–L3) and stems (S1–S3) from seedlings (Figure 6B–D). Leaf RT-qPCR showed that three genes were highly expressed in L1, with 7 genes highly expressed in L2, and 9 genes highly expressed in L3 (Figure 6C). Additionally, the result revealed four genes with high expression in S1, eight genes with high expression in S2, and eight genes with high expression in S3(Figure 6D). Importantly, the stem-specific expression patterns of EhWOX4, EhWOX7, EhWOX14, EhWOX5 and EhWOX6 observed in RNA-seq data were confirmed by RT-qPCR. The RNA-seq results demonstrate that EhWOX genes exhibit tissue-specific expression patterns, and further RT-qPCR analysis of WOX gene expression revealed that several genes may be involved in stem development.
2.8. Subcellular Localization and Autoactivation of EhWOXs
Bioinformatic analysis predicted nuclear localization for 14 EhWOX TFs (Table 1). To validate these predictions, GFP fusion vectors (pCAMBIA2300-EhWOX4-eGFP, pCAMBIA2300-EhWOX5-eGFP, and pCAMBIA2300-EhWOX6-eGFP) were constructed and transiently expressed in Nicotiana benthamiana. Microscopic detection demonstrated that three EhWOX-GFP fusion proteins exhibited nuclear localization, which colocalized with the OsWRKY70-RFP nuclear marker (Figure 7A). These results confirm the computational predictions and provide experimental evidence supporting the TF function of EhWOX proteins. Furthermore, sequence alignment of EhWOX5 and EhWOX6 revealed only one amino acid difference in the homeodomain (EhWOX5: Leu; EhWOX6: Pro) and a total of 11 amino acid differences across their full-length sequences (Figure 2E, Supplementary Table S1). Therefore, to further investigate their functional differences, autoactivation validation was first performed. The CDSs of EhWOX5 and EhWOX6 were cloned into the pGBKT7 vector to generate recombinant pGBKT7-EhWOX vectors. Then, the yeast colonies were dropped onto SD/-Trp/-X-α-gal medium to assess their autoactivation capability (Figure 7B). Additionally, the addition of AbA (200 ng/mL) as an inhibitor, combined with X-α-gal staining analysis, revealed that 200 ng/mL AbA significantly suppressed the growth of yeast colonies (Figure 7B). Collectively, the results confirm that these two proteins are autoactivating.
3. Discussion
The regulation of plant growth and development involves numerous TFs, among which the WOX gene family represents one of the most conserved TF families in plants. With advances in genome sequencing technologies, WOX gene families have been identified and characterized in various plant species (Figure 1A), including H. brasiliensis, J. curcas, M. esculenta, and R. communis within the Euphorbiaceae family [24]. However, systematic characterization of the EhWOX family in E. hirta has not been reported. In this study, through a combination of local BLASTP, HMMER, CDD, and conserved motif analyses, we identified 14 EhWOX genes distributed across nine chromosomes of E. hirta (Table 1, Figure 1B, Supplementary Table S1). Subsequently, a series of bioinformatics analyses, including assessments of protein physicochemical properties, phylogenetic tree construction, gene structure analysis, synteny analysis, cis-regulatory element identification, and GO and transcriptional regulatory network analysis, was performed. Additionally, tissues expression profiling was conducted, followed by validation of stem and leaf expression patterns through RT-qPCR. Moreover, subcellular localization and autoactivation assays were performed. These comprehensive analyses establish a foundational framework for further functional characterization of the EhWOX family.
Studies have demonstrated that the evolution of the WOX family is closely associated with morphological innovations during plant speciation [28,30,31]. In this study, we conducted temporal evolutionary analysis of E. hirta, H. brasiliensis, A. thaliana, and 17 other species and quantified WOX gene family members in each species [8] (Figure 1A). Notably, H. brasiliensis contains 21 WOX genes, while E. hirta has 14 WOX genes. In E. hirta, only three syntenic gene pairs were identified, whereas 28 homologous pairs were found between E. hirta and rubber tree (Figure 3). Studies have shown that H. brasiliensis underwent a recent WGD event, resulting in more retained duplicate gene copies in the rubber tree genome [32]. Moreover, as a perennial latex-producing plant, the demands of secondary metabolism in the rubber tree may have driven the expansion of the WOX gene family, while the life history strategy of annual herb E. hirta, which favors rapid growth and reproduction, may reduce dependence on certain developmental regulatory genes. Previous phylogenetic classifications divided WOX proteins into three major clades [28,30,31,33]. The phylogenetic tree revealed conserved evolutionary patterns, with both H. brasiliensis and E. hirta containing four members in the ancient clade. Previous studies have shown that some members participate in lateral root development and floral organ formation (e.g., AtWOX13) in the ancient clade, while the Intermediate Clade plays a conserved role in root organogenesis. For example, IC-WOX proteins in ferns are expressed in root founder cells of adventitious and lateral roots, while seed plant IC-WOX subgroups (e.g., WOX11 and WOX8/9) are involved in root or shoot formation [8]. In this analysis, H. brasiliensis and E. hirta displayed differential retention in the Intermediate Clade (four in H. brasiliensis and three in E. hirta). Strikingly, the WUS clade showed the most dramatic expansion, containing 13 and seven members in H. brasiliensis and E. hirta, respectively. Previous studies have demonstrated that the core members of the WUS clade (e.g., WUS and WOX5) maintain stem-cell homeostasis in apical shoot meristems and apical root meristems, which are critical for post-embryonic plant development [8]. Additionally, a phylogenetic analysis of the 14 EhWOX proteins consistently resolved them into three characteristic clades (Figure 2B). These findings suggest functional divergence among WOX subfamilies, with WUS-clade genes potentially playing enhanced roles in species-specific secondary metabolism (e.g., latex biosynthesis), while ancient-clade members likely maintain conserved functions in fundamental developmental processes.
The TF motifs serve as core regulatory elements that are crucial for regulating gene expression. Although the homeodomain of WOX proteins is highly conserved, substantial variations exist in other motifs [34]. EhWOX motif prediction revealed that motif1 and motif3 represent two segments of the homeodomain and constitute characteristic signatures of the WOX family, being present in all EhWOX proteins, consistent with reports of motif1 and motif2 in four Euphorbiaceae species [24,35,36,37]. Notably, motif2 was exclusively predicted in the four proteins of the ancient clade, corresponding to motif4, which is uniquely present in the four Euphorbiaceae species [24]. Remarkably, motif5 was only detected in two proteins of the ancient clade, suggesting the significance of motif2 and motif5 in plant development and highlighting the evolutionary conservation of ancient-clade proteins (Figure 2B). These predicted motif distribution patterns demonstrate considerable divergence among different clades, implying functional differentiation across WOX families. WOX proteins belong to the homeobox TF superfamily, characterized by a conserved 60-amino-acid homeodomain that forms a helix–loop–helix–turn–helix structure for specific DNA binding (Figure 2E) [28,38]. In this study, 11 EhWOX proteins were found to belong to the canonical homeodomain family, while 3 (EhWOX10, EhWOX9, and EhWOX1) showing structural variations were found to belong to the Homeodomain superfamily (Figure 2C). In the conserved sequence alignment, compared with other members, EhWOX10, EhWOX9, and EhWOX1 exhibit amino acid variations; for instance, EhWOX1 shows a Glu–to–Thr substitution at position 9 in Helix1. These variations may represent potential markers of functional innovation or Euphorbiaceous-specific subfamily differentiation. Gene structure prediction further revealed that the exon–intron organization of WOX families in E. hirta showed high similarity to those in four Euphorbiaceae species, typically containing two to four CDS regions (Figure 2D) [24], highlighting both the high degree of structural conservation in WOX genes throughout evolution and their critical function in plant growth and development. Through chromosomal localization and synteny analysis in E. hirta, three syntenic gene pairs were identified, whose evolution likely resulted from tandem and segmental duplication events (Figure 3A, Supplementary Table S2). Additionally, comparative genomic analysis was performed to investigate the syntenic relationships of WOX genes among E. hirta, H. brasiliensis, T. kok-saghyz, and M. esculenta, reflecting phylogenetic relationships based on gene-family evolution. The predicted results indicate that orthologous genes in plants have undergone duplication or loss during evolution, leading to differential syntenic conservation of WOX genes among species.
Cis-acting elements act as indispensable molecular switches in gene expression [39,40,41]. In predicted elements, development-related elements were the most abundant, including light-responsive elements (e.g., Box 4, G-box, and GT1 motif). All promoters of EhWOX gene contained Box 4, with the EhWOX10 gene harboring the highest number (nine), indicating extensive involvement of WOX genes in light-regulated development. Furthermore, hormone-related elements (e.g., ABA and JA-responsive elements) were present in EhWOX promoters, as corroborated between hormones and malate in regulating MdWOX11-mediated development in apple [42]. Additionally, stress-related elements (e.g., ARE, O2-site, and CAT-box) were identified in the EhWOX family. Studies have demonstrated that WOX genes widely participate in abiotic stress responses, for example, OsWOX5 and OsWOX10 in rice, along with overexpression of JcWOX5 from J. curcas, enhancing drought tolerance [43,44,45], while OsWOX11 in rice and PagWOX11/12a in poplar respond to salt and nutrient stress. Collectively, these results underscore the crucial roles of the WOX family in plant development and abiotic stress adaptation, with evolutionary conservation further emphasizing their significance. Moreover, we predicted intergenic regulatory relationships based on transcription factor binding motifs (TFBMs). Although prediction indicates low functional overlap between TF binding and gene regulation, alongside the non-linear relationship between motif binding and gene regulation [46], the TFBM-based regulatory network established here provides a foundational framework for subsequent research.
Gene function is closely linked to tissue-specific expression patterns, and different members of the WOX gene family are expressed and functional in diverse plant tissues, including roots, stems, leaves, flowers, and meristems [8,47]. In this study, RNA-seq expression analysis revealed that EhWOX7 and EhWOX14 are highly expressed in roots, stems, leaves, flowers, and latex (Figure 6A), with RT-qPCR validating the accuracy of RNA-seq data in stems and leaves (Figure 6C,D). Furthermore, phylogenetic analysis placed EhWOX14 within the ancient clade (Figure 2A), suggesting functional conservation of this gene in E. hirta. RNA-seq results indicated that seven genes were highly expressed in flowers and four in roots, consistent with expression patterns reported in Jatropha curcas [24]. Two EhWOX genes were expressed in latex and leaves, respectively, implying their functions in these tissues. In stems, EhWOX4, EhWOX5, and EhWOX6 were expressed. Phylogenetically, EhWOX5 and EhWOX6 clustered with A. thaliana AtWOX14 in the ancient clade, while EhWOX4 clustered with AtWOX4 in the WUS clade (Figure 2A). Furthermore, RT-qPCR confirmed EhWOX4, EhWOX5 and EhWOX6 high expression in stems (Figure 6D). Additionally, as TFs, EhWOX4, EhWOX5 and EhWOX6 were predicted and verified to localize to the nucleus (Table 1, Figure 7A), and yeast assays confirmed their transactivation activity (Figure 7B). Previous research has shown that AtWOX14 promotes cell differentiation in the cambium region to regulate stem development in A. thaliana [22] and WOX4 regulates stem development [18,19,20]. The expression profiles and bioinformatics analysis of EhWOX4, EhWOX5, and EhWOX6 suggest their potential involvement in stem development, though further validation is required. Notably, E. hirta possesses advantageous traits for model plant studies, including a compact stature, short life cycle, and adaptability to controlled cultivation (Figure 6B), making it an ideal system for the study of stem development. Consequently, the validation of highly expressed genes in the stems of this species provides significant application value.
This study represents the first systematic identification of 14 EhWOX genes in E. hirta, along with comprehensive characterization of their evolutionary conservation and expression patterns. Our integrative analyses suggest potential roles for EhWOXs in stem development, thereby establishing a foundation for functional studies of EhWOX genes in E. hirta. In the future, investigations employing gene knockout, overexpression, spatial expression analysis of stems, and chromatin immunoprecipitation to identify downstream targets could validate the proposed regulatory mechanisms of EhWOX genes in stem development. Therefore, such a study would provide a systematic roadmap for functional studies of the WOX family and highlight the utility of E. hirta as a model system for investigation of laticifer development.
4. Experimental Procedures
4.1. Experimental Materials
The wild-type E. hirta plants used in this study were provided by the Chinese Academy of Tropical Agricultural Sciences, located in Sanya City, Hainan Province, China (18°15′16.9″ N, 109°30′27.5″ E). For RT-qPCR analysis, E. hirta plants were cultivated under controlled growth-chamber conditions at 28 °C with 16 h of light (100 μmol m^−2^ s^−1^) followed by 8 h of darkness for 40 days. Then, stems and leaves from uniformly grown plants at three distinct developmental positions (S1–3 for stems; L1–L3 for leaves) were collected for RNA extraction and subsequent RT-qPCR analysis. N. benthamiana plants were used for subcellular localization assays. All plant materials were maintained under standardized conditions prior to experimental use to ensure consistency in physiological status.
4.2. Identification and Physicochemical Analysis of EhWOX Genes
The chromosome-level genome assembly of E. hirta and corresponding annotation files (GFF3) were provided by the Chinese Academy of Tropical Agricultural Sciences (unpublished). To identify WOX family members in E. hirta, we employed a dual approach: a Hidden Markov Model (HMM) search against the genome protein sequences using the homeodomain (http://pfam.xfam.org/, Pfam: PF00046, accessed on 15 May 2025) as a query with an E-value cutoff of 0.01 and a local BLASTP (BLAST+ v2.14.0) analysis using A. thaliana WOX protein sequences as queries with a stringent E-value threshold of <1 × 10^−10^. Candidate EhWOX genes were identified and named according to their chromosomal order. Subsequently, their physicochemical properties were analyzed using ExPASy ProtParam (https://web.expasy.org/protparam/, accessed on 17 May 2025). Protein-domain architecture was verified using the NCBI Conserved Domain Database (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi, accessed on 15 May 2025), while subcellular localization was predicted using WoLF PSORT (https://wolfpsort.hgc.jp/, accessed on 20 May 2025).
4.3. Phylogenetic Analysis of WOX Genes
To investigate the evolutionary relationships of WOX genes across species, we first analyzed the divergence times of 20 species, including E. hirta, H. brasiliensis, and A. thaliana, using the TimeTree web resource (TimeTree: The Timescale of Life, https://timetree.org/, accessed on 23 May 2025) while simultaneously counting the number of WOX family members in each species. For phylogenetic reconstruction of WOXs from eight selected species (Eh: Euphorbia hirta; WCJ: Euphorbia lathyrism; Ja: Jatropha curcas; Me: Manihot esculenta; Rc: Ricinus communis; rna-Hb: Hevea brasiliensis; Ptr: Populus tomentosa; At: Arabidopsis thaliana), we performed multiple sequence alignment of full-length protein sequences using MUSCLE (Version 5) with default parameters. The resulting alignment was subsequently refined using trimAL to remove poorly aligned regions. Maximum likelihood (ML) phylogenetic trees were constructed using IQ-TREE with 5000 ultrafast bootstraps replicates (default parameters) to assess node support in TBtools (v2.331) [48], and the appropriate model was selected utilizing the ModelFinder method. The JTTDCMut+G4 model was chosen according to the Bayesian Information Criterion (BIC). The same analytical pipeline was applied to construct the phylogenetic tree of E. hirta EhWOX family members, and the JTT+G4 model was used. All phylogenetic trees were visualized and annotated using the iTOL platform (https://itol.embl.de/help.cgi, accessed on 20 June 2025).
4.4. Structural Analysis of EhWOX Genes
The chromosomal localization and gene density distribution of the EhWOX gene family were visualized using the “Gene distribution Visualize” function in TBtools software with the input GTF/GFF annotation file. For conserved motif analysis, we employed the MEME suite (http://meme-suite.org/tools/meme, accessed on 27 May 2025) with the following parameters: number of motifs set to 5 and site distribution option configured as Zero or One Occurrence Per Sequence (zoops). The Conserved Homeodomain Domain prediction tool is the NCBI Conserved Domain Database tool (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi, accessed on 28 May 2025) with a cutoff of 0.01. The resulting motif patterns were subsequently integrated with phylogenetic relationships, a conserved homeodomain architecture, and exon–intron organization using the “Gene Structure View (Advanced)” plugin in TBtools, which enabled comprehensive visualization of these multi-dimensional structural features in a unified graphical representation.
4.5. Synteny Analysis
Intra-species synteny analysis of EhWOX genes in E. hirta was performed using the “One Step MCScanX” function in TBtools (v2.331) with default parameters (1 × 10^−10^), and the results were visualized, along with gene density and GC content data, using the “Advanced Circos” tool. For inter-species synteny analysis, the H. brasiliensis genome and GFF3 files, the T. kok-saghyz genome and GFF3 files downloaded from NCBI (https://www.ncbi.nlm.nih.gov, accessed on 5 June 2025), and the M. esculenta genome and GFF3 files obtained from Ensembl Plants (Ensembl Plants, accessed on 6 June 2025) were utilized. Comparative synteny analysis between these three species and E. hirta was conducted using the “One Step MCScanX” pipeline in TBtools with default settings, followed by comprehensive visualization of the syntenic relationships [49].
4.6. Analysis of Cis-Acting Elements of EhWOX Promoters
The 2000 bp upstream sequences of all identified EhWOX genes were extracted as putative promoter regions using the Gtf/Gff3 Sequences Extract function in TBtools software. These promoter sequences were subsequently analyzed using the PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/, accessed on 8 June 2025) to identify potential cis-regulatory elements. For downstream analysis, we filtered out common core promoter elements, including TATA-box, CAAT-box, and un-named elements, to focus on functionally relevant regulatory motifs. The distribution patterns of these cis-acting elements were visualized by mapping their positions along promoter sequences using the “Simple Biosequence Viewer” function in TBtools and quantitative analysis of element counts per gene presented as heatmaps generated with R statistical software (version 4.4.3).
4.7. Gene Ontology Enrichment and Regulatory Network Analysis
E. hirta GO annotation was performed using eggNOG-mapper (https://github.com/eggnogdb/eggnog-mapper, accessed on 11 June 2025) with the latest GO-basic.obo file (http://purl.obolibrary.org/obo/go/go-basic.obo, accessed on 11 June 2025) to functionally characterize E. hirta genes using TBtools (default parameters). The resulting GO terms were visualized using TBtools. To analyze the transcriptional regulatory network of EhWOX transcription factors, the “Plant TF Binding Motif Shift” function (default parameters) in TBtools was employed to retrieve binding motifs for all TFs in E. hirta, with data sourced from PlantTFDB (https://planttfdb.gao-lab.org/). Subsequently, the FIMO tool (default parameters) was utilized to predict the regulatory binding sites of 14 EhWOX TFs (Supplementary Table S4) in TBtools, and the potential regulatory-network target genes were visualized using R (version 4.4.3).
4.8. RNA-Seq and RT-qPCR Expression Analysis of EhWOXs
The RNA-Seq raw sequence data reported in this study have been submitted to the Genome Sequence Archive (Genomics, Proteomics and Bioinformatics 2025) at the National Genomics Data Center (Nucleic Acids Research 2025), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (https://ngdc.cncb.ac.cn/gsa, GSA accession number: CRA037671). Expression levels based on FPKM values were quantified and visualized as heatmaps using TBtools. For experimental validation, total RNA was extracted from E. hirta stems and leaves using an RNAprep Pure Plant Kit (Tiangen Biotech) following the manufacturer’s protocol. First-strand cDNA synthesis was performed with 1 μg total RNA as template using the HiScript II Q RT SuperMix reverse transcription kit (Vazyme Biotech). RT-qPCR reactions were carried out on a Light Cycler 480 II Real-Time PCR System (Roche, Basel, Switzerland) in 20 μL reaction volumes (10 μL AceQ qPCR SYBR Green Master Mix, 1 μL cDNA template, and 1 μL each of 10 μm forward and reverse gene-specific primers for 35 cycles) (primers in Supplementary Table S6). The actin gene (ACT1.2373) was used as internal reference for normalization, and relative gene expression levels were calculated using the 2^−ΔΔCt^ quantitative method. All expression data were statistically analyzed and visualized using GraphPad Prism software (version 10).
4.9. Subcellular Localization of EhWOX Proteins
Based on phylogenetic analysis and expression profiling, we selected proteins potentially involved in stem development for subcellular localization. The subcellular localization of EhWOX4, EhWOX5, and EhWOX6 was determined by transiently expressing pCAMBIA2300-EhWOX4-eGFP, pCAMBIA2300-EhWOX5-eGFP, and pCAMBIA2300-EhWOX6-eGFP fusion constructs in N. benthamiana. First, the CDSs of the three EhWOX genes were amplified by PCR and subsequently cloned into the pCAMBIA2300-35S-eGFP vector [15]. The recombinant plasmids were verified by sequencing, then transformed into Agrobacterium tumefaciens strain GV3101. Bacterial suspensions (OD 600 ≥ 0.6) were infiltrated into N. benthamiana leaves using a needleless syringe. After infiltration, the plants were incubated in the dark for 48 h, and GFP fluorescence signals (488 nm) were observed and acquired using a confocal laser scanning microscope (Zeiss LSM800, Göttingen, Germany). Meanwhile, OsWRKY70 was co-injected for concurrent expression as a fluorescence-based localization marker.
4.10. Yeast Autoactivation Assay for EhWOXs
A yeast assay was employed to determine whether the TF EhWOXs possessed autoactivation. The CDSs of EhWOXs were amplified by PCR and subsequently cloned into the pGBKT7 vector to construct recombinant plasmids pGBKT7–EhWOX. These plasmids were then transformed into yeast strain AH109. The transformed yeast cells were plated on SD/-Trp medium and cultured at 30 °C for 2–3 days, after which single colonies were selected for further verification. Positive transformants were inoculated onto SD/-Trp or SD/-Trp/-AbA (AbA final concentration: 200 ng/mL) plates and cultured at 30 °C for 2–3 days to observe growth. Subsequently, X-α-gal (4 μg/mL) was spotted onto the yeast colonies, followed by incubation at 30 °C for 2–12 h to monitor the formation of blue colonies.
4.11. Statistical Analysis
Statistical significance was defined as p < 0.05, and analyses were performed using One-way ANOVA and Tukey’s multiple comparison test. Data visualization was performed using GraphPad Prism (v 9.0.0).
5. Conclusions
In this study, we systematically identified and characterized 14 EhWOX genes in E. hirta. Phylogenetic and structural analyses classified them into three conserved clades and revealed group-specific motifs. Expression profiling revealed tissue-specific patterns, with several genes showing stem-predominant expression that was validated by RT-qPCR. The nuclear localization and transcriptional autoactivation potential of stem-expressed members were confirmed. While RNA-seq data suggest some members may be expressed in latex-producing tissues, their specific cellular roles require future spatial and functional validation. Collectively, this comprehensive genomic and transcriptomic characterization establishes an essential foundation and identifies specific candidate genes for future functional studies on stem development and, potentially, laticifer biology in E. hirta.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Laux T. Mayer K.F. Berger J. Jürgens G. The WUSCHEL Gene Is Required for Shoot and Floral Meristem Integrity in Arabidopsis Development 1996122879610.1242/dev.122.1.878565856 · doi ↗ · pubmed ↗
- 2Gehring W.J. Affolter M. Bürglin T. Homeodomain Proteins Annu. Rev. Biochem.19946348752610.1146/annurev.bi.63.070194.0024157979246 · doi ↗ · pubmed ↗
- 3Lian G. Ding Z. Wang Q. Zhang D. Xu J. Origins and Evolution of WUSCHEL-Related Homeobox Protein Family in Plant Kingdom Sci. World J.2014201453414010.1155/2014/534140 PMC 391339224511289 · doi ↗ · pubmed ↗
- 4Mukherjee K. Brocchieri L. Bürglin T.R. A Comprehensive Classification and Evolutionary Analysis of Plant Homeobox Genes Mol. Biol. Evol.2009262775279410.1093/molbev/msp 20119734295 PMC 2775110 · doi ↗ · pubmed ↗
- 5Khan F.S. Goher F. Hu C.G. Zhang J.Z. WUSCHEL-Related Homeobox (WOX) Transcription Factors: Key Regulators in Combating Abiotic Stresses in Plants Hortic. Adv.20242210.1007/s 44281-023-00023-2 · doi ↗
- 6Sun R. Zhang X. Ma D. Liu C. Identification and Evolutionary Analysis of Cotton (Gossypium hirsutum) WOX Family Genes and Their Potential Function in Somatic Embryogenesis Int. J. Mol. Sci.2023241107710.3390/ijms 24131107737446257 PMC 10342170 · doi ↗ · pubmed ↗
- 7Alvarez J.M. Bueno N. Cañas R.A. Avila C. Cánovas F.M. Ordás R.J. Analysis of the WUSCHEL-RELATED HOMEOBOX Gene Family in Pinus Pinaster: New Insights into the Gene Family Evolution Plant Physiol. Biochem.201812330431810.1016/j.plaphy.2017.12.03129278847 · doi ↗ · pubmed ↗
- 8Rasheed H. Shi L. Winarsih C. Jakada B.H. Chai R. Huang H. Plant Growth Regulators: An Overview of WOX Gene Family Plants 202413310810.3390/plants 1321310839520025 PMC 11548557 · doi ↗ · pubmed ↗