Iron Deficiency Inhibits the Proliferation of Intestinal Stem Cells and Induces Their Differentiation to Enterocytes
Yecheng Xu, Jing Zhao, Shouchuan Jiang, Yu Han, Yi Zheng, Xi Qiao, Xin Wen, Yuanyuan Zhang, Yunqin Li, Jingxia Kong, Huahua Du

TL;DR
Iron deficiency reduces intestinal stem cell proliferation and pushes them to become enterocytes, affecting gut health and repair.
Contribution
This study reveals a novel mechanism linking iron deficiency to altered intestinal stem cell behavior via the Notch signaling pathway.
Findings
Iron deficiency reduces Lgr5-positive intestinal stem cells and Ki67-positive proliferating cells.
Iron deficiency increases Vil1 expression, indicating a shift toward enterocyte differentiation.
Modulation of the Notch signaling pathway is associated with iron deficiency-induced differentiation of intestinal stem cells.
Abstract
Objectives: Iron deficiency impairs intestinal mucosal structure and function, yet its impact on intestinal stem cells (ISCs) remains unclear. This study was therefore designed to examine how iron deficiency affects the proliferation and differentiation of ISCs. Methods: Iron-deficient mouse and enteroid models were established. Expression of key cell markers was analyzed using Western blot, qPCR, and immunofluorescence. Results: Iron deficiency led to structural impairment of the intestinal mucosa, characterized by decreased small intestinal villus height. In iron-deficient mice, expression of ChrA (enteroendocrine cell marker), Lyz (Paneth cell marker), and Muc2 (goblet cell marker) was significantly downregulated across duodenum, jejunum and ileum, whereas Vil1 (enterocyte marker) expression increased. Moreover, both Lgr5 (an ISC marker) expression and the number of Ki67-positive…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
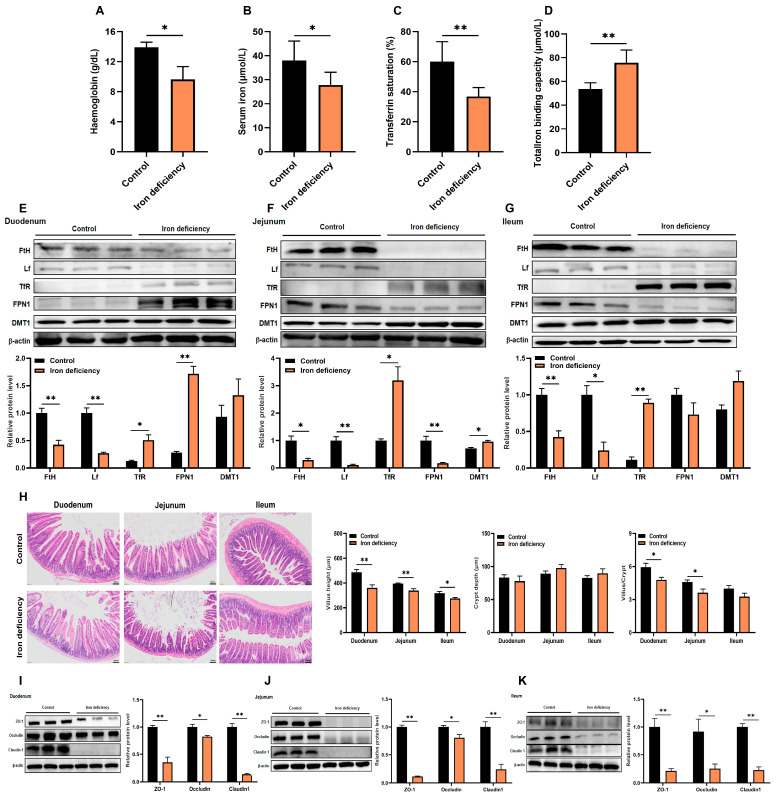 Figure 1
Figure 1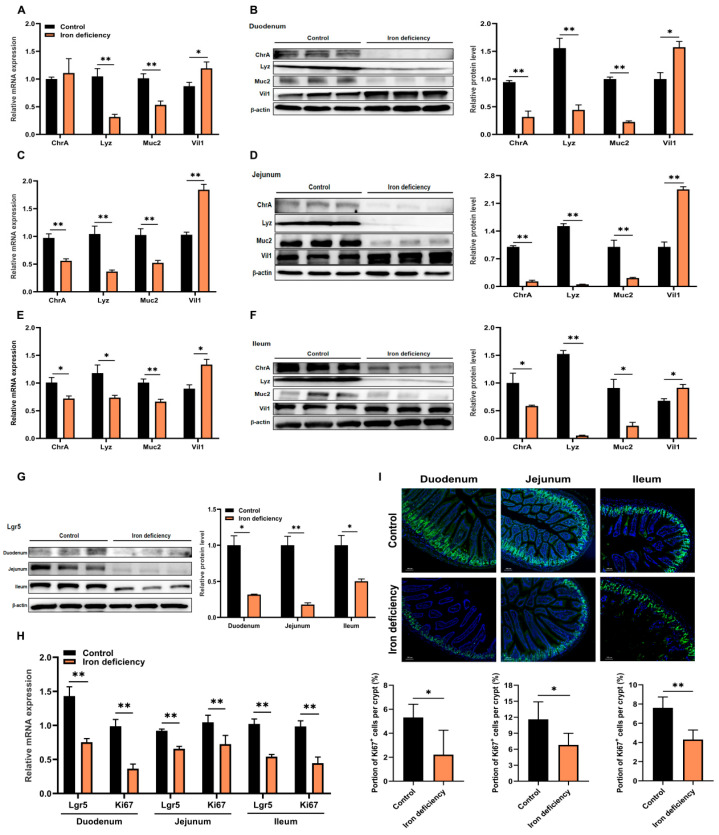 Figure 2
Figure 2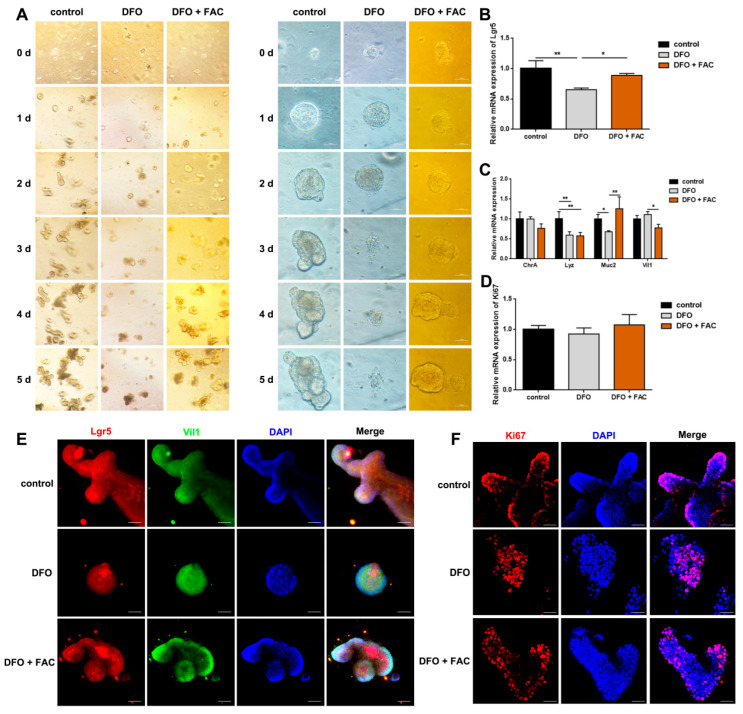 Figure 3
Figure 3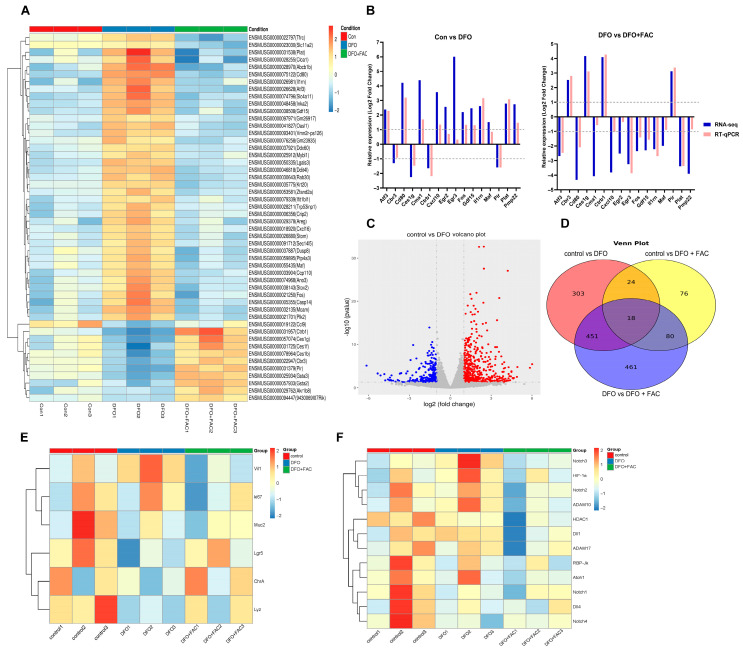 Figure 4
Figure 4- —Natural Science Foundation of Zhejiang province of China
- —National Natural Science Foundation of China
- —National Key Research and Development Program of China
- —Zhejiang Shuren University Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDigestive system and related health · Iron Metabolism and Disorders · Nutrition, Genetics, and Disease
1. Introduction
Iron deficiency represents a significant global public health challenge, affecting approximately one-third of the world’s population [1]. Its consequences extend far beyond the classic hematological manifestations of anemia, impacting multiple organ systems [2].
Within the gastrointestinal tract, iron deficiency is clinically associated with a spectrum of structural and functional impairments, most notably villous atrophy, malabsorption, and intestinal barrier dysfunction [3]. These alterations not only reduce quality of life but also create a pernicious cycle in which impaired gut function further impedes nutrient absorption, aggravating the underlying deficiency [4]. Although these clinical outcomes are well established, the fundamental cellular and molecular mechanisms driving this intestinal pathology remain inadequately defined, representing a critical obstacle to the development of targeted therapeutic strategies.
At the heart of intestinal resilience lies the dynamic population of intestinal stem cells (ISCs), residing within the crypts of Lieberkühn. These cells serve as the primary architects of epithelial renewal, responsible for the continuous and high-fidelity regeneration of the entire intestinal lining [5]. Through precisely orchestrated divisions, ISCs give rise to transient amplifying cells, which subsequently differentiate into the diverse, functional epithelial lineages of the epithelium: nutrient-absorbing enterocytes and various secretory cells, including mucus-producing goblet cells, antimicrobial Paneth cells, and hormone-secreting enteroendocrine cells. This exquisite balance between ISC self-renewal and lineage commitment is the cornerstone of mucosal homeostasis [6,7]. Crucially, the maintenance of this stem cell compartment is both energetically and biochemically demanding.
Iron serves as an essential cofactor for numerous proteins involved in critical biological processes, including mitochondrial electron transport, oxidative phosphorylation, and DNA synthesis, making it a fundamental metabolic currency [8]. The emerging recognition that a majority of functional ISCs operate in an iron-sensitive metabolic state underscores their potential susceptibility to fluctuations in iron bioavailability [9]. This intrinsic dependency positions iron availability as a likely pivotal regulator of ISC behavior, influencing their fate decisions between proliferation, quiescence, and differentiation [10]. However, a direct, mechanistic link between systemic iron deficiency and the autonomous reprogramming of ISC biology has not been conclusively established.
To address this critical knowledge gap, our study aims to systematically investigate the hypothesis that iron deficiency directly impairs intestinal homeostasis by reprogramming the metabolism of ISCs and biasing their differentiation commitment. We will employ an integrated experimental approach that combines a physiologically relevant in vivo mouse model of dietary iron deficiency with a controlled in vitro intestinal organoid system. This strategy will allow us to dissect the cell-autonomous effects of iron scarcity on ISCs, independent of systemic secondary influences. Our primary objectives are to (1) quantitatively evaluate the impact of iron deficiency on ISC proliferation and pool maintenance; (2) define alterations in lineage specification under iron-restricted conditions; and (3) identify key signaling pathways and metabolic shifts that mediate the ISC response to iron deficiency.
2. Materials and Methods
2.1. The Animals
Three-week-old male C57BL/6 mice were purchased from Charles River Laboratories (Beijing, China). All mice were housed under specific pathogen-free (SPF) conditions at Zhejiang University and had free access to water and appropriate diets. A chronic model of dietary iron deficiency was generated by feeding 3-week-old mice either a low-iron diet (5 mg/kg Fe, n = 8) or a standard diet (150 mg/kg Fe, n = 8) for 60 days. Both the low-iron diet and standard diet, formulated to be identical in all nutritional components except for iron content, were purchased from Xietong Biotechnology (Nanjing, China). At the conclusion of the experiment, blood samples were collected by retro-orbital puncture. The mice were then euthanized via cervical dislocation, after which intestinal tissues were rapidly frozen in liquid nitrogen and stored at −80 °C for subsequent analysis. Additionally, enteroids derived from intestinal crypts of 8-week-old mice were treated for 48 h under three conditions: control (culture medium), 500 μM deferoxamine (DFO, iron chelation group, n = 6), and 500 μM DFO + 500 μM ferric ammonium citrate (FAC, iron repletion group, n = 6), establishing an in vitro intervention model to resolve cellular iron compensation mechanisms. The procedures were approved by Zhejiang University Animal Care Committee (ZJU20240918).
2.2. Measurement of Serum Indicators
The serum iron concentration was measured using a commercial assay kit (Iron Assay Kit, Elabscience Biotechnology, Wuhan, China; Catalog No. E-BC-K945-M) based on the principle of ferrous iron reacting with ferrozine to form a purple complex, with absorbance detected at 562 nm. Total iron binding capacity was measured using a specialized test kit (Total Iron Binding Capacity Assay Kit, Jiancheng, Nanjing, China; Catalog No. A040-1-1) following the chromogenic principle of iron-binding protein combining with iron ions. Absorbance readings were taken at 520 nm with a microplate reader (Molecular Devices, Sunnyvale, CA, USA). Transferrin saturation was calculated as follows: (serum iron [mg/L]/total iron binding capacity [mg/L]) 100. Hemoglobin concentration in peripheral blood was measured using an automated hematology analyzer (XN-1000V, Sysmex, Hamburg, Germany) following the manufacturer’s standard procedures for erythrocyte parameter detection.
2.3. RNA Sequencing and Analysis
We used total RNA with a minimum amount of 800 ng to create strand-specific libraries. This process involved several steps: poly-A selection, cDNA fragmentation, and adapter ligation. We quantified the resulting libraries using a Qubit 3.0 fluorometer and validated their size with a Qsep100 system. Then, we sequenced them on the NovaSeq 6000 platform using a 150 bp paired-end approach. We rigorously filtered the raw sequencing data, applying Q20 and Q30 thresholds and performing adapter trimming. Next, we used featureCounts to count reads and then quantified gene expression as FPKM. Differential expression analysis was conducted using DESeq2. Using biological triplicates, we set the criteria for differential expression as |log_2_FC| greater than 1 and a p-value less than 0.05. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed with the cluster Profiler package. The significance threshold was set at p less than 0.05. We effectively visualized the key findings using various graphical representations such as heatmaps, volcano plots, and Venn diagrams.
2.4. Histological Examination
Tissue specimens were fixed overnight in 4% paraformaldehyde, rinsed in phosphate-buffered saline (PBS), dehydrated through an ethanol series, and embedded in paraffin. Subsequently, 5 μm sections were deparaffinized, rehydrated, and stained with hematoxylin and eosin (H&E). Images were taken by Olympus APX100 and BX63 (Beijing, China). The villus height (from the tip of the villi to the villus-crypt junction) and crypt depth (depth of the invagination between adjacent villi) were measured and recorded using Image-Pro Plus 7.0 (Media Cybernetics, Rockville, MD, USA). For each intestinal cross-section, measurements were obtained from at least three structurally intact and properly oriented crypt–villus units to ensure accurate morphological assessment.
2.5. Analysis of mRNA Expression
Total RNA was extracted using the SteadyPure Universal RNA Extraction Kit (Accurate Biotechnology, Changsha, China). Subsequently, 1 μg of the isolated RNA was reverse-transcribed into cDNA with the Evo M-MLV Mix Kit with gDNA Clean for qPCR (Accurate Biotechnology, Changsha, China). Quantitative real-time PCR analysis was performed using SYBR Green Pro Taq HS Premix (Accurate Biotechnology, Changsha, China). The PCR protocol consisted of an initial denaturation at 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s. Relative gene expression was calculated using the 2^−ΔΔCT^ method, with β-actin as the endogenous control. All primer sequences are listed in Table 1.
2.6. Western Blot Analysis
Protein extraction was performed by homogenizing cellular or murine tissue samples in RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China). The buffer contained a cocktail of protease and phosphatase inhibitors (Lablead, Beijing, China). The homogenate was then clarified by centrifugation. Protein concentration was quantified with a BCA assay kit (Beyotime Biotechnology, Shanghai, China). Following SDS-PAGE separation, proteins were transferred onto PVDF membranes, which were then blocked using a rapid protein-free blocking buffer (Epizyme Biotech, Shanghai, China) and incubated overnight at 4 °C with the respective primary antibodies. Primer antibodies are listed in Table 2. Subsequently, membranes were probed with species-matched secondary antibodies for 60 min at room temperature. HRP-conjugated anti-rabbit and anti-mouse IgG (Biosharp, Hefei, China) were used as secondary antibodies. Immunoreactive bands were then visualized with Immobilon Western Chemiluminescent HRP Substrate and quantified by densitometric analysis using ImageJ software 1.54p (National Institutes of Health, Bethesda, MD, USA).
2.7. Isolation of Intestinal Crypts and Enteroids Culture
Intestinal organoids were generated from freshly isolated crypts taken from the small intestine as previously described [11]. The intestinal tissue was cut longitudinally and thoroughly washed before processing. Tissue segments of 5 mm length were treated with 2 mM ethylenediaminetetraacetic acid (EDTA) in PBS at 4 °C for 30 min. After EDTA treatment, the fragments were washed with ice-cold PBS. We aspirated the villus-rich supernatant and resuspended the crypt-containing pellet in fresh PBS. Mechanical dissociation was performed sequentially, followed by centrifugation, to enrich crypt structures in the supernatant. Contaminating villus debris was eliminated by 40 μm filtration (Corning, NY, USA). Purified crypts were pelleted at 200× g for 3 min and embedded in 50 μL Matrigel^®^ (Corning, NY, USA) in 24-well plates. Following Matrigel polymerization (37 °C, 10 min), cultures were maintained in IntestiCult™ Organoid Growth Medium (Stemcell Technologies, Vancouver, BC, Canada), with the medium replaced every 48 h. Organoids were passaged every 4–6 days.
2.8. Immunofluorescence (IF) Microscopy
Small intestine tissues were fixed overnight in 4% paraformaldehyde, subsequently paraffin-embedded, and sectioned at 5 µm. The sections were then deparaffinized in xylene (Google Biotech, Beijing, China) and rehydrated through a graded ethanol series. After antigen retrieval, the sections were then blocked, permeabilized, and incubated with primary antibodies and fluorescent secondary antibodies (Table 2). For immunostaining in organoids, the organoids in Matrigel were fixed in 4% PFA, then permeabilized, blocked, and incubated overnight with anti-rabbit Ki67 (1:100, Abcam, Shanghai, China), followed by incubation with anti-rabbit Alexa Fluor 594 (1:200, Abcam, Shanghai, China). Nuclei were counterstained with DAPI (1:5000, Sangon Biotech, Shanghai, China). Fluorescence images were captured using a confocal microscope (Zeiss, Jena, Germany) and an Olympus APX100 system (Beijing, China), followed by cell counting with Image-Pro Plus software IPP7.0 (Media Cybernetics, MD, USA).
2.9. Statistical Analysis
Statistical analysis was performed with GraphPad Prism 8.0 (GraphPad Software, San Diego, CA, USA). Data are expressed as mean ± standard error of the mean (SEM). Differences between two groups were assessed by unpaired two-tailed Student’s t-tests, while comparisons across multiple groups were analyzed by one-way ANOVA with Tukey’s post hoc test. A p-value < 0.05 was considered statistically significant, denoted as * p < 0.05 and ** p < 0.01.
3. Results
3.1. Iron Deficiency Induces Intestinal Villus Shortening and Impairs Tight Junction Proteins
In order to explore the role of iron deficiency in the intestine, we established an iron deficient murine model by feeding a low-iron diet (5 mg/kg Fe) for 60 days. Successful induction of iron deficiency was confirmed by characteristic hematological alterations: a 30.1% reduction (p < 0.05) in hemoglobin levels, 27.0% decrease (p < 0.05) in serum iron concentration, 30.1% decline (p < 0.01) in transferrin saturation, and a 1.4-fold increase (p < 0.01) in total iron binding capacity (Figure 1A–D). Western blot analysis revealed consistent iron regulatory responses across intestinal segments. In the duodenum, jejunum, and ileum of iron-deficient mice, protein expressions of ferritin heavy chain (FtH) and lactoferrin (Lf) were significantly (p < 0.01) downregulated, while transferrin receptor (TfR) and divalent metal transporter 1 (DMT1) were markedly (p < 0.05) upregulated (Figure 1E–G). Notably, ferroportin 1 (FPN1) exhibited segment-specific regulation, with significant (p < 0.01) upregulation in the duodenum but downregulation in the jejunum. These findings demonstrated that iron deficiency triggered adaptive increases in intestinal iron transporters (DMT1 and TfR) while suppressing iron storage proteins (FtH and Lf), collectively impairing erythropoiesis [12]. Histopathological analysis further confirmed intestinal damage. H&E staining showed significant villus shortening in iron-deficient mice: 29.9% reduction (p < 0.01) in the duodenum, 14.4% (p < 0.01) in the jejunum, and 13.4% (p < 0.05) in the ileum (Figure 1H). The villus height/crypt depth ratio was significantly diminished in both duodenum and jejunum (p < 0.05). The atrophic villi displayed structural disorganization, suggesting compromised nutrient absorption capacity and impaired mucosal barrier function. Tight junctions (TJs) are essential for epithelial barrier integrity and regulate paracellular permeability [13]. To determine whether iron deficiency affects TJ integrity, we measured the protein expression of key TJ markers (ZO-1, occludin, and claudin 1) in intestinal segments. Western blot analysis revealed that their levels were significantly reduced (p < 0.05) in the duodenum, jejunum, and ileum of iron-deficient mice compared with controls (Figure 1I–K), indicating that iron is critical for maintaining junctional complex integrity.
3.2. Iron Deficiency Inhibits the Proliferation of ISCs and Regulates Their Differentiation
To further explore the impact of iron deficiency on intestinal cell fate, we examined key cell lineage markers in iron-deficient mice [14,15]. Compared with control mice, the duodenum of iron deficient mice exhibited significantly (p < 0.01) reduced mRNA and protein expression levels of Lyz (marker of Paneth cells) and Muc2 (marker of goblet cells), whereas Vil1 (marker of enterocytes) was significantly (p < 0.05) increased (Figure 2A,B). In the jejunum, iron deficiency led to a marked (p < 0.01) decrease in ChrA (marker of enteroendocrine cells), Lyz and Muc2 expression, while inducing Vil1 levels (Figure 2C,D). A similar expression profile was observed in the ileum (Figure 2E,F). These findings suggest that iron deficiency did influence the differentiation pattern of ISCs. Besides differentiation, ISCs also self-renew and proliferate to maintain the intestinal epithelium. Iron deficiency significantly (p < 0.05) suppressed both mRNA and protein levels of Lgr5 (marker of ISCs) across the duodenum, jejunum, and ileum (Figure 2G,H). Consistent with this, Ki67 transcriptional level and the number of Ki67-positive proliferating cells were significantly (p < 0.05) lower in the iron-deficient group than those in the control group (Figure 2H,I). Taken together, our results indicate that iron deficiency not only biases ISCs differentiation toward enterocytes but also impairs their proliferative capacity.
3.3. Iron Is Essential for the Proliferation and Differentiation of ISCs in Enteroids
In order to verify the effect of iron deficiency on intestinal cells in vivo, we established an iron-deficient enteroid model by treating mouse enteroids with 500 μM deferoxamine (DFO). Compared with the control group, DFO-treated enteroids showed collapsed structure and cell death after 3 days, which was effectively rescued by supplementation with 500 μM ferric ammonium citrate (FAC) (Figure 3A). Based on these observations, we selected a 2-day treatment period for establishing the iron-deficient mouse enteroid model. DFO treatment significantly (p < 0.01) reduced the relative mRNA expression of Lgr5, which was reversed by FAC supplementation (Figure 3B). In addition, the mRNA levels of Lyz and Muc2 was significantly (p < 0.05) decreased in DFO-treated enteroids compared with controls, and the reduction in Muc2 transcript was restored by FAC treatment (Figure 3C). In contrast, the mRNA expression of Ki67 remained unaffected by either DFO or FAC treatment (Figure 3D). Immunofluorescence analysis further confirmed these findings, showing consistent changes in Lgr5-positive ISCs, Vil1-positive cells and Ki67-positive proliferative cells (Figure 3E,F). These results align with the in vivo data, supporting the conclusion that iron deficiency inhibits the proliferation and differentiation of ISCs without affecting their proliferative capacity.
3.4. Iron Deficiency Disrupts ISC Self-Renewal and Enhances Enterocyte Differentiation by Activating Notch Signaling
To investigate how iron deficiency affects the proliferation and differentiation of ISCs, we performed RNA sequencing on mouse enteroids from control, DFO and DFO + FAC groups (Figure 4A). Following data filtration, quality control of sequencing error rates and GC content distribution, high-quality reads were obtained for downstream analysis. Differentially expressed genes (DEGs) were identified using the thresholds |log_2_FoldChange| > 0 and P_adj_ < 0.05. The reliability of the RNA-seq data was validated by RT-qPCR on 16 randomly selected DEGs, which showed consistent expression trends (Figure 4B). Detailed primer sequences and validation data are provided in Supplementary Figure S1. Volcano plot analysis revealed 796 DEGs between the control and DFO groups, with 528 upregulated and 286 downregulated genes (Figure 4C). Venn analysis indicated that 469 of these genes were reversibly regulated upon iron supplementation (Figure 4D). GO enrichment analysis showed that DEGs were significantly associated with biological processes (BP) such as cellular movement and motility; cellular components (CC) including the apical membrane and adherens junctions; and molecular functions (MF) related to receptor regulation and ligand activity (Figure S2A). These results suggest that iron deficiency disrupts epithelial organization and promotes cell migration, potentially impairing intestinal barrier integrity. KEGG pathway analysis further identified significant enrichment in apoptosis and endocrine resistance-related pathways (Figure S2B). Clustering of ISC-related markers demonstrated that DFO treatment significantly upregulated the enterocyte marker Vil1, while downregulating the ISC marker Lgr5 and secretory lineage markers (Muc2, ChrA, Lyz) (Figure 4E). These alterations were rescued by FAC, confirming that iron deficiency skews ISC differentiation toward the absorptive lineage. Notably, key components of the Notch signaling pathway were modulated under iron deficiency: the ligand Dll1, receptors Notch2 and Notch3, and protease ADAM10 were upregulated, whereas the negative regulator Atoh1 was downregulated (Figure 4F). This suggests that Notch pathway modulation may contribute to enterocyte differentiation during iron deprivation, though further functional validation is needed.
4. Discussion
Iron deficiency is a prevalent nutritional disorder that often leads to intestinal malabsorption and mucosal damage [16]. However, current research lacks comprehensive studies on how iron deficiency affects ISC and their differentiation potential, with limited validation using animal and laboratory models [17]. This study elucidated the multifaceted impact of dietary iron deficiency on intestinal homeostasis, primarily through altering the fate determination of ISCs. By integrating in vivo mouse models and in vitro organoid systems, we demonstrated that iron deprivation disrupts the intestinal epithelial barrier via a dual mechanism: suppressing the self-renewal capacity of ISCs and skewing their lineage commitment, ultimately compromising tissue regeneration and integrity.
Our findings revealed a compartmentalized adaptation of the intestine to systemic iron deficiency. The duodenum exhibited a coordinated upregulation of transporters (TfR, FPN1) to enhance absorption, while the jejunum and ileum engage DMT1 and TfR. Paradoxically, this is accompanied by a systemic downregulation of iron storage (FTH), export (FPN1), and regulatory (HAMP, LF) proteins in distal segments, creating a maladaptive cycle that exacerbates iron retention dysfunction [12]. This dysregulated iron dynamics correlates with a significant reduction in ISC numbers and proliferative activity, as evidenced by decreased expression of the canonical stem cell marker Lgr5, coupled with a reduction in Ki67-positive proliferating cells. The association between nutrient deficiency and impaired ISC proliferation has been noted in other models of malnutrition, highlighting a potential common pathway in nutrient stress responses [18,19].
Crucially, beyond merely inhibiting proliferation, iron availability actively shaped differentiation fate. We observed a coordinated downregulation of key secretory lineage markers (ChrA, Lyz, Muc2) alongside a marked upregulation of the absorptive enterocyte marker Vil1. In addition, the iron chelator DFO also effectively recapitulated the phenotypes of dietary deficiency, confirming the central role of iron availability. DFO treatment reduced Lgr5+ ISCs, inhibited organoid growth, and mirrored the in vivo shift in differentiation markers. This “pro-absorptive, anti-secretory” shift was consistent across both experimental models, suggesting it is an intrinsic response to iron scarcity. This adaptive response may aim to maximize the absorptive surface area, albeit at the potential cost of compromising secretory functions critical for mucosal defense (e.g., antimicrobial peptide secretion by Paneth cells, mucus production by goblet cells) [20].
To explore the underlying mechanism, transcriptional profiling of iron-deficient enteroids was performed. RNA-seq analysis demonstrated that iron deficiency not only suppressed the proliferative capacity of ISCs (as indicated by downregulated Lgr5 and Ki67) but, more importantly, biased their differentiation toward the absorptive enterocyte lineage at the expense of secretory lineages (e.g., Paneth cells, goblet cells) by activating the Notch signaling pathway (upregulating Dll1, Notch2/3, and downregulating Atoh1). The Notch pathway is a well-established arbiter of secretory-absorptive fate decisions in the gut [21]. Therefore, our data support a model wherein iron deficiency creates a cellular milieu that favors Notch activation, thereby directing ISCs toward the absorptive lineage while suppressing secretory cell differentiation. Mechanistically, iron chelation is known to stabilize HIF-1α, which may subsequently influence critical stem cell pathways such as Wnt/β-catenin. However, the precise hierarchical relationship and integration between iron-sensing mechanisms (HIF), key developmental pathways (Wnt/Notch), and the observed epigenetic regulators in driving the ISC phenotype remain unclear and represent a crucial area for future research using gene-editing approaches in advanced models.
The consequences of these cellular changes are profound for intestinal physiology. The depletion of Paneth (reduced Lyz) and goblet (reduced Muc2) cells directly weakens innate immune defense and mucus barrier integrity, respectively [20]. The potential impairment of enteroendocrine cell function (reduced ChrA) may disrupt hormonal signaling. This collective erosion of secretory lineage functions shares pathological similarities with conditions like inflammatory bowel disease (IBD), suggesting a plausible mechanistic link between chronic iron deficiency and increased susceptibility to intestinal inflammation. The disruption likely extends beyond mature cells, as the depletion of the Lgr5+ stem cell pool indicates a fundamental reprogramming of epithelial regeneration, possibly through iron-dependent epigenetic modifications affecting master differentiation regulators (e.g., Atoh1, Neurog3).
A limitation of this study is that the role of Notch signaling in iron deficiency-induced ISC differentiation is supported only by transcriptional data; we have not excluded the possibility that these changes are a compensatory response to other iron deficiency-related cellular events, and functional validation (e.g., using Notch inhibitors such as DAPT) will be necessary to confirm whether Notch signaling directly mediates the observed phenotype. Additionally, the sample size and experimental duration may constrain the detection of long-term adaptive effects of iron deficiency on ISC behavior and intestinal homeostasis. Importantly, our experimental design did not account for the gut microbiota—a well-established modulator of host iron metabolism, intestinal epithelial function, and stem cell niche regulation. The complex interplay between iron deficiency, microbial dysbiosis, and ISC proliferation/differentiation represents a critical frontier for future research, which will require integrated multi-omics approaches and organoid-microbe co-culture systems to dissect the underlying mechanisms.
5. Conclusions
In summary, iron deficiency perturbed intestinal homeostasis by directly targeting ISCs, impairing their self-renewal and biasing differentiation toward enterocytes at the expense of secretory lineages. This is mediated through a complex interplay of localized iron transport adaptations, systemic regulatory imbalances, and activation of Notch signaling pathway. Our findings underscore the essential role of iron in maintaining the intestinal stem cell niche and epithelial barrier function. From a translational perspective, these insights highlight that iron supplementation strategies for conditions like anemia should consider their reparative effects on the ISC compartment and mucosal regeneration, offering potential avenues for improving intestinal health and productivity in both clinical and agricultural settings. Future work should focus on dissecting the precise epigenetic and molecular mechanisms linking iron sensing to stem cell fate determination.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bao H. Wang Y. Xiong H. Xia Y. Cui Z. Liu L. Mechanism of iron ion homeostasis in intestinal immunity and gut microbiota remodeling Int. J. Mol. Sci.20242572710.3390/ijms 2502072738255801 PMC 10815743 · doi ↗ · pubmed ↗
- 2Al-Samkari H. Loren A.W. Lee A.I. The case for classical haematology: The impact of a name and the future of a field Lancet Haematol.20229 e 455e 45910.1016/S 2352-3026(22)00096-535500590 · doi ↗ · pubmed ↗
- 3Montoro-Huguet M.A. Belloc B. Domínguez-Cajal M. Small and large intestine (I): Malabsorption of nutrients Nutrients 202113125410.3390/nu 1304125433920345 PMC 8070135 · doi ↗ · pubmed ↗
- 4Al-Beltagi M. Saeed N.K. Bediwy A.S. Elbeltagi R. Breaking the cycle: Psychological and social dimensions of pediatric functional gastrointestinal disorders World J. Clin. Pediatr.20251410332310.5409/wjcp.v 14.i 2.10332340491742 PMC 11947882 · doi ↗ · pubmed ↗
- 5Häfliger J. Schwarzfischer M. Atrott K. Stanzel C. Morsy Y. Wawrzyniak M. Spalinger M.R. Glycoprotein (GP) 96 is essential for maintaining intestinal epithelial architecture by supporting its self-renewal capacity Cell. Mol. Gastroenterol. Hepatol.20231571773910.1016/j.jcmgh.2022.12.00436516930 PMC 9879791 · doi ↗ · pubmed ↗
- 6Shivdasani R.A. Clevers H. de Sauvage F.J. Tissue regeneration: Reserve or reverse?Science 202137178478610.1126/science.abb 684833602845 · doi ↗ · pubmed ↗
- 7Malagola E. Vasciaveo A. Ochiai Y. Kim W. Zheng B. Zanella L. Wang T.C. Isthmus progenitor cells contribute to homeostatic cellular turnover and support regeneration following intestinal injury Cell 20241873056307110.1016/j.cell.2024.05.00438848678 PMC 11164536 · doi ↗ · pubmed ↗
- 8Nolfi-Donegan D. Braganza A. Shiva S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement Redox Biol.20203710167410.1016/j.redox.2020.10167432811789 PMC 7767752 · doi ↗ · pubmed ↗