MELAS Syndrome Presenting with Hypertrophic Cardiomyopathy and Advanced Heart Failure: A Multisystem Diagnostic Challenge
Jozef Dodulík, Marie Lazárová, Eva Kapsová, Jan Václavík

TL;DR
A rare mitochondrial disorder, MELAS, presented as heart failure and kidney disease, highlighting the need for genetic testing in complex cases.
Contribution
Demonstrates MELAS syndrome as a multisystem disorder presenting primarily with heart failure and kidney disease, emphasizing the role of genetic testing.
Findings
MELAS syndrome was diagnosed via whole-exome sequencing in a patient with heart failure and chronic kidney disease.
Cardiac imaging showed non-ischemic cardiomyopathy with diffuse hypertrophy and fibrosis.
The case underscores the importance of considering mitochondrial disorders in unexplained cardiomyopathy with multisystem involvement.
Abstract
Background: Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) is a rare multisystem disorder caused by mitochondrial DNA mutations, most commonly the m.3243A>G variant in the MT-TL1 gene. Although neurological manifestations predominate, cardiac involvement, including hypertrophic cardiomyopathy (HCM), heart failure (HF), and arrhythmias, may be the initial or dominant presentation and often remains underrecognized. Case Presentation: We report a 43-year-old man with chronic kidney disease (CKD) and long-standing bilateral sensorineural hearing loss who presented with progressive dyspnea and acute decompensated HF. Transthoracic echocardiography revealed severe left ventricular (LV) systolic dysfunction with diffuse hypertrophy. Cardiac magnetic resonance showed non-ischemic cardiomyopathy with diffuse late gadolinium enhancement and increased LV wall…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
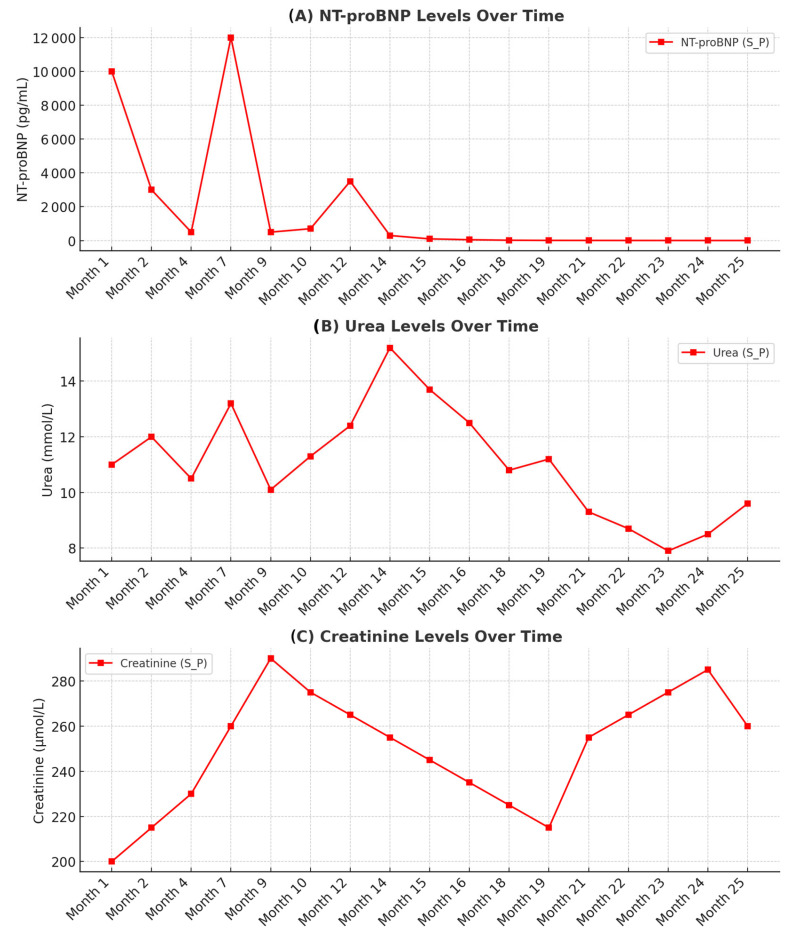 Figure 1
Figure 1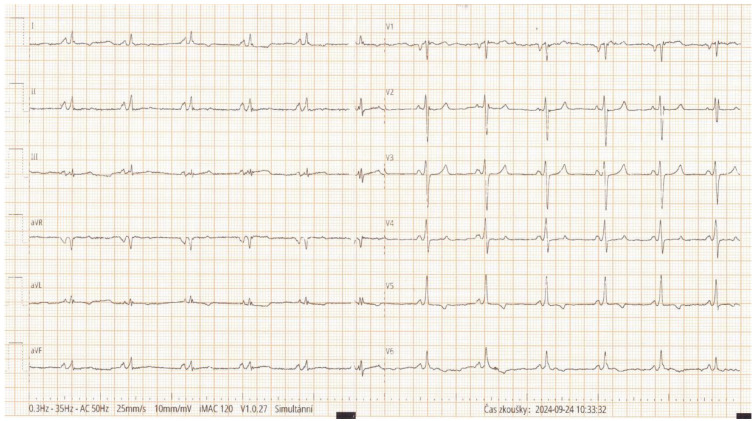 Figure 2
Figure 2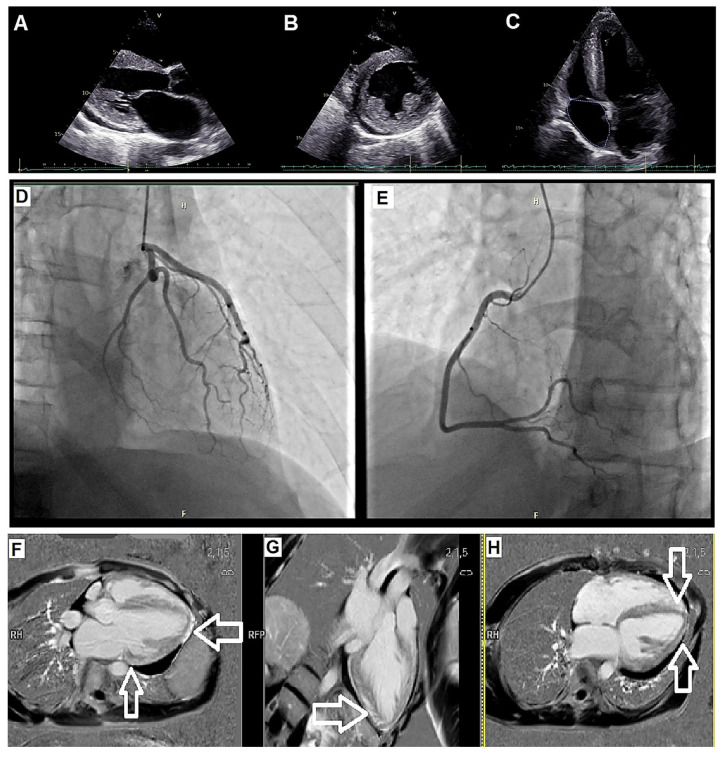 Figure 3
Figure 3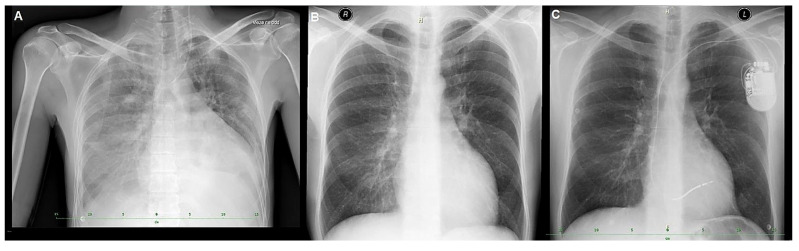 Figure 4
Figure 4- —Ministry of Health
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMitochondrial Function and Pathology · ATP Synthase and ATPases Research · Coenzyme Q10 studies and effects
1. Introduction
Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) syndrome is a rare multisystem disorder caused by pathogenic mutations in mitochondrial DNA (mtDNA), most frequently the m.3243A>G variant in the MT-TL1 gene [1,2]. MT-TL1 encodes mitochondrial transfer RNA for leucine, which is essential for oxidative phosphorylation and normal respiratory chain function.
From a cardiology perspective, mitochondrial cardiomyopathies represent a diagnostic challenge because their phenotypic spectrum overlaps with more prevalent conditions such as sarcomeric hypertrophic cardiomyopathy (HCM), infiltrative cardiomyopathies, and storage diseases. In many patients, cardiac involvement may dominate the clinical presentation for years before the development of classical neurological manifestations, leading to delayed or missed diagnosis. Importantly, early identification of a mitochondrial etiology has implications not only for patient management and prognosis but also for family screening due to maternal inheritance patterns.
Advances in multimodality imaging and genetic testing have substantially improved the recognition of rare metabolic cardiomyopathies. Cardiac magnetic resonance (CMR), in particular, allows for detailed tissue characterization and may reveal patterns of hypertrophy and fibrosis suggestive of mitochondrial disease. Nevertheless, awareness of these entities remains limited in routine clinical practice. This case therefore aims to highlight key diagnostic features and a practical diagnostic approach to unexplained HCM with multisystem involvement.
MELAS typically affects the central nervous system, skeletal muscle, kidneys, and auditory pathways; however, cardiac involvement is increasingly recognized [1,2,3]. Up to 20–40% of patients develop HCM, heart failure (HF), arrhythmias, or conduction abnormalities, often preceding neurological manifestations [3,4].
The diagnosis may be difficult due to the heterogeneous clinical presentation and overlap with more common cardiomyopathies. The concurrent presence of unexplained left ventricular (LV) hypertrophy, renal dysfunction, and sensorineural hearing loss should raise suspicion for an underlying mitochondrial disorder [1,4]. In such cases, genetic testing plays a key role in establishing a unifying diagnosis and guiding multidisciplinary management.
We present the case of a 43-year-old man with progressive HF, chronic kidney disease (CKD), and bilateral sensorineural hearing loss, in whom MELAS syndrome was diagnosed after an extensive diagnostic workup, highlighting the importance of considering rare multisystemic diseases in the differential diagnosis of unexplained HCM and HF.
2. Case Presentation
A 43-year-old man with a history of CKD (stage 3a) and long-standing bilateral sensorineural hearing loss requiring hearing aids since childhood presented with progressive dyspnea, orthopnea, and lower-limb edema. He had no history of diabetes, hypertension, or known cardiovascular disease. There were no prior neurological symptoms suggestive of stroke-like episodes, seizures, or encephalopathy.
On admission, he was hypotensive (90/60 mmHg), tachycardic (120 bpm), and hypoxemic (89% on room air). Physical examination revealed bilateral basal crackles and signs of volume overload. Initial laboratory testing demonstrated markedly elevated NT-proBNP (5430 pg/mL), mild metabolic acidosis, and worsening renal function (creatinine 248 μmol/L; urea 12 mmol/L). Serum lactate measured at admission was mildly elevated (3.1 mmol/L). The longitudinal dynamics of key laboratory parameters during follow-up are summarized in Figure 1.
Electrocardiography showed sinus tachycardia without overt voltage criteria for left ventricular hypertrophy (Figure 2). Transthoracic echocardiography (TTE) revealed severe global LV systolic dysfunction with an ejection fraction (EF) of approximately 20%, diffuse hypokinesis, and concentric LV hypertrophy with a maximal wall thickness of 15 mm measured at the interventricular septum (Figure 3A–C). A small circumferential pericardial effusion was also noted.
Cardiac magnetic resonance (CMR) imaging demonstrated increased LV wall thickness (up to 15 mm), elevated LV mass index, and diffuse late gadolinium enhancement (LGE) involving the basal and mid-ventricular anteroseptal and inferolateral segments, consistent with non-ischemic cardiomyopathy with replacement fibrosis. Quantitative analysis confirmed the absence of regional ischemia and demonstrated a non-ischemic pattern of LGE distribution (Figure 3F–H).
Selective coronary angiography excluded obstructive coronary artery disease (Figure 3D,E). An endomyocardial biopsy (EMB) was performed at a referring center early during the diagnostic evaluation. Histology revealed nonspecific myocyte hypertrophy and interstitial fibrosis without diagnostic features; detailed micrographic documentation was not available for external review. Given the nondiagnostic biopsy findings and the coexistence of cardiac, renal, and auditory involvement, a multisystem disorder was suspected.
Given the combination of unexplained left ventricular hypertrophy, severe systolic dysfunction, CKD, and long-standing sensorineural hearing loss, an extensive differential diagnostic work-up was performed. Particular attention was paid to conditions associated with hypertrophic or infiltrative cardiomyopathy and systemic involvement.
Fabry disease was excluded by normal α-galactosidase A activity and the absence of characteristic findings on cardiac imaging. Transthyretin amyloidosis was considered unlikely based on negative bone scintigraphy and the absence of typical amyloid patterns on CMR imaging. Other lysosomal storage disorders, including Pompe disease and Alström syndrome, were also considered unlikely based on clinical phenotype and targeted testing.
Despite EMB demonstrating only nonspecific myocyte hypertrophy and interstitial fibrosis, the overall clinical picture raised strong suspicion of a metabolic or genetic disorder. Whole-exome sequencing was performed on peripheral blood DNA and identified the pathogenic m.3243A>G mutation in the MT-TL1 gene and confirming the diagnosis of MELAS syndrome. Genetic testing of myocardial tissue was not performed.
The patient was stabilized with intravenous diuretics and vasodilators, and guideline-directed medical therapy for HF was initiated, including beta-blockers (BB), mineralocorticoid receptor antagonists (MRA), and sodium-glucose co-transporter 2 (SGLT2) inhibitors. Sacubitril/valsartan was introduced after improvement in blood pressure. Coenzyme Q10 and L-arginine supplementation were added as supportive therapy for mitochondrial dysfunction based on current practice recommendations.
NT-proBNP was markedly elevated at baseline (5.430 pg/mL) and increased to 10.000 pg/mL within the first month. A further rise with a peak between months 4 and 9 coincided with a clinically documented HF decompensation requiring treatment intensification (Figure 1). Following stabilization, NT-proBNP gradually decreased in parallel with clinical improvement and optimization of HF therapy.
Due to persistent LV dysfunction, the patient received a single-chamber implantable cardioverter-defibrillator (ICD) for primary prevention of sudden cardiac death (SCD) (Figure 4). A single-chamber ICD was selected because the patient had a narrow QRS (Figure 1) and no indication for atrial pacing, and to minimize additional hardware in the context of CKD and advanced HF management. He was subsequently evaluated by a multidisciplinary HF team and assessed for heart transplantation (HTx). Over 25 months of follow-up, he remained clinically stable without further hospitalizations. Genetic counseling was provided; however, further cascade testing was not feasible due to the absence of siblings and unavailable paternal information.
A timeline summarizing the key diagnostic and therapeutic milestones during the patient’s work-up and management is provided in Table 1.
3. Discussion
This case is noteworthy because MELAS syndrome presented with a predominantly cardiac phenotype and rapid progression to advanced HF, while overt neurological manifestations were absent at presentation. The diagnosis was established only after integrating extracardiac red flags with CMR tissue characterization and genetic testing, despite an initially nondiagnostic EMB.
MELAS syndrome is a rare mitochondrial disorder most commonly caused by the m.3243A>G mutation in the MT-TL1 gene, which encodes mitochondrial tRNA^Leu and is essential for mitochondrial protein synthesis and oxidative phosphorylation [1]. Although neurological involvement is classical, cardiac manifestations, including HCM, HF, arrhythmias, and conduction abnormalities, occur in up to 20–40% of patients and may precede neurologic symptoms [2,3,4,5]. In the present case, the patient exhibited no stroke-like episodes or seizures, and his dominant presentation was rapidly progressive HF, emphasizing the highly variable phenotype of MELAS.
The differential diagnosis of a hypertrophic phenotype is broad and includes numerous ‘phenocopies’ (e.g., infiltrative, storage, and mitochondrial diseases), for which a stepwise approach integrating clinical red flags, ECG, echocardiography, CMR tissue characterization, and genetics is recommended [6].
Diagnosing mitochondrial cardiomyopathy remains challenging due to its overlap with more common etiologies. The coexistence of unexplained LV hypertrophy, renal dysfunction, sensorineural hearing loss, and mildly elevated serum lactate in our patient prompted consideration of a multisystem disorder. In such settings, multimodality imaging plays a central role. CMR findings of diffuse LGE with a non-ischemic distribution pattern and increased LV mass strongly suggested a metabolic or mitochondrial etiology [7,8]. These features have been repeatedly described in mitochondrial cardiomyopathies and may provide diagnostic clues even in the absence of specific histological confirmation.
EMB may support the diagnosis of specific cardiomyopathy etiologies; however, in mitochondrial disease, the diagnostic yield can be limited and may depend on the availability of specialized analyses (e.g., mitochondrial-focused histochemistry and/or ultrastructural assessment). In the present case, biopsy performed at a tertiary center showed nonspecific findings and the detailed biopsy protocol (including any mitochondrial-specific staining or electron microscopy) was not available to the authors. Given the strong multisystem clinical suspicion, the nondiagnostic biopsy reinforced the need to proceed to genetic testing to establish a unifying diagnosis.
Whole-exome sequencing confirmed the m.3243A>G mutation in MT-TL1, providing definitive evidence of MELAS syndrome. Genetic testing was performed on peripheral blood DNA. In mtDNA disorders, heteroplasmy may vary substantially across tissues and may not directly reflect the degrees of organ involvement particularly between blood and post-mitotic tissues such as myocardium. Quantitative heteroplasmy data were not provided by the external laboratory in this case; therefore, we interpreted the confirmed pathogenic variant in the context of the patient’s multisystem phenotype and characteristic imaging findings. The presence of hearing loss since childhood and progressive renal impairment further supported a unifying mitochondrial etiology [9,10,11].
Management of MELAS-related cardiomyopathy is largely supportive. Our patient was treated with guideline-directed medical therapy for HF, including BB, MRA, SGLT2 inhibitors, and sacubitril/valsartan [12]. Coenzyme Q10 and L-arginine supplementation were added as adjunctive therapies, reflecting current practice in mitochondrial disorders, although robust clinical evidence remains limited [13,14,15,16]. Given persistent LV dysfunction, an ICD was implanted for primary prevention [17], in accordance with current recommendations. Ultimately, the patient was evaluated for HTx, which remains a consideration in advanced mitochondrial cardiomyopathy.
From a clinical standpoint, this case underscores several important lessons. First, mitochondrial disease should be considered in patients with unexplained cardiomyopathy when cardiac findings coexist with extracardiac features such as renal dysfunction or sensorineural hearing loss, even in the absence of neurological symptoms. Second, CMR plays a pivotal role in raising suspicion of a metabolic etiology through identification of diffuse, non-ischemic late gadolinium enhancement patterns.
Third, this case highlights the limitations of EMB in mitochondrial cardiomyopathies, particularly when specialized histochemical or ultrastructural analyses are not available. In such scenarios, genetic testing may provide the most definitive diagnostic information and should be considered early in the diagnostic pathway. Finally, establishing a unifying diagnosis enables appropriate genetic counseling, cascade testing, and long-term planning, including timely referral to advanced HF programs.
This case highlights the importance of considering mitochondrial disorders in patients with unexplained cardiomyopathy and multisystem involvement. Early genetic testing is particularly valuable when standard diagnostic evaluations are nondiagnostic. Family counseling is essential due to maternal inheritance, and cascade testing was recommended to the patient’s maternal relatives. The case underscores the diagnostic value of integrating clinical features, CMR findings, and genetic testing to establish a unifying diagnosis in complex multisystem presentations.
From an advanced HF perspective, mitochondrial disease raises additional considerations for HTx evaluation. Candidate assessment should explicitly address extracardiac involvement, particularly neurological status, renal function, and the overall trajectory of multisystem disease, because these factors may influence perioperative risk and post-transplant outcomes. In our patient, the severity of cardiac involvement prompted transplantation evaluation, while the extent and progression of extracardiac manifestations required careful multidisciplinary consideration.
Future research should focus on defining imaging and genetic markers that allow for the earlier identification of mitochondrial cardiomyopathies in cardiology practice. Prospective registries and collaborative studies are needed to better characterize the natural history, optimal heart failure management strategies, and transplant outcomes in this patient population. Increased awareness among cardiologists may ultimately lead to earlier diagnosis, improved patient selection for advanced therapies, and better integration of multidisciplinary care.
4. Conclusions
This case illustrates that MELAS syndrome may present predominantly with rapidly progressive cardiomyopathy and advanced HF, even in the absence of overt neurological manifestations. Integrating extracardiac ’red flags’ with CMR tissue characterization and early genetic testing can enable a unifying diagnosis and guide advanced HF management.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hirano M. Pavlakis S.G. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): Current concepts J. Child. Neurol.1994941310.1177/0883073894009001028151079 · doi ↗ · pubmed ↗
- 2Chinnery P.F. Elliott H.R. Hudson G. Samuels D.C. Relton C.L. Epigenetics, epidemiology and mitochondrial DNA diseases Int. J. Epidemiol.20124117718710.1093/ije/dyr 23222287136 PMC 3304530 · doi ↗ · pubmed ↗
- 3Sproule D.M. Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: Basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome Ann. N. Y. Acad. Sci.20081142133158 Erratum in Ann. N. Y. Acad. Sci. 2009, 1161, 601. PMID: 1899012510.1196/annals.1444.01118990125 · doi ↗ · pubmed ↗
- 4Arbelo E. Protonotarios A. Gimeno J.R. Arbustini E. Barriales-Villa R. Basso C. Bezzina C.R. Biagini E. A Blom N. A de Boer R. ESC Scientific Document Group. 2023 ESC Guidelines for the management of cardiomyopathies Eur. Heart J.2023443503362610.1093/eurheartj/ehad 19437622657 · doi ↗ · pubmed ↗
- 5Gorman G.S. Chinnery P.F. Di Mauro S. Hirano M. Koga Y. Mc Farland R. Suomalainen A. Thorburn D.R. Zeviani M. Turnbull D.M. Mitochondrial diseases Nat. Rev. Dis. Primers 201621608010.1038/nrdp.2016.8027775730 · doi ↗ · pubmed ↗
- 6Teresi L. Trimarchi G. Licordari R. Restelli D. Taverna G. Liotta P. Micari A. Smecca I. Dendramis G. Turturiello D. Hypertrophic Cardiomyopathy Phenocopies: Classification, Key Features, and Differential Diagnosis Biomedicines 202513306210.3390/biomedicines 1312306241463072 PMC 12730691 · doi ↗ · pubmed ↗
- 7Finsterer J. Scorza F.A. Renal manifestations of primary mitochondrial disorders Biomed. Rep.2017648749410.3892/br.2017.89228515908 PMC 5431253 · doi ↗ · pubmed ↗
- 8Parikh S. Goldstein A. Koenig M.K. Scaglia F. Enns G.M. Saneto R. Anselm I. Cohen B.H. Falk M.J. Greene C. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society Genet. Med.20151768970110.1038/gim.2014.17725503498 PMC 5000852 · doi ↗ · pubmed ↗