A Transcriptome Study on Seed Germination of Nitraria roborowskii Kom
Shangfu Ren, Guanghui Lv

TL;DR
This study uses transcriptome sequencing to explore gene expression changes in Nitraria roborowskii seeds during germination, revealing key biological processes and pathways involved in dormancy release.
Contribution
The study provides novel insights into the transcriptomic mechanisms underlying seed dormancy in Nitraria roborowskii through high-throughput sequencing and bioinformatics analysis.
Findings
Transcriptome sequencing identified 215,303 transcripts and 84,450 unigenes in Nitraria roborowskii seeds.
16,130 differentially expressed genes were found, primarily linked to metabolic processes and plant hormone signaling.
Key pathways included starch and sucrose metabolism and plant hormone signal transduction during germination.
Abstract
Nitraria roborowskii Kom. seeds possess pronounced deep dormancy traits. Analyzing changes in gene expression before and after dormancy release is of great significance for elucidating the mechanisms underlying seed dormancy. In this study, transcriptome sequencing and bioinformatics analysis were conducted on N. roborowskii seeds both before and after dormancy release using high-throughput Illumina NovaSeq 6000 sequencing technology. The key findings are as follows: (1) A total of 215,303 transcripts and 84,450 unigenes were obtained through de novo assembly. (2) Comparative analysis revealed 16,130 significantly differentially expressed unigenes during germination, with 10,776 upregulated and 5354 downregulated. Gene Ontology (GO) enrichment analysis indicated that these differentially expressed genes (DEGs) were primarily associated with biological processes and molecular functions,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
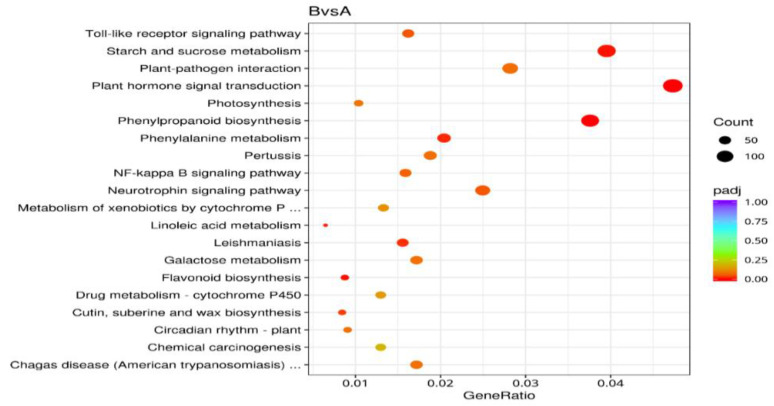 Figure 5
Figure 5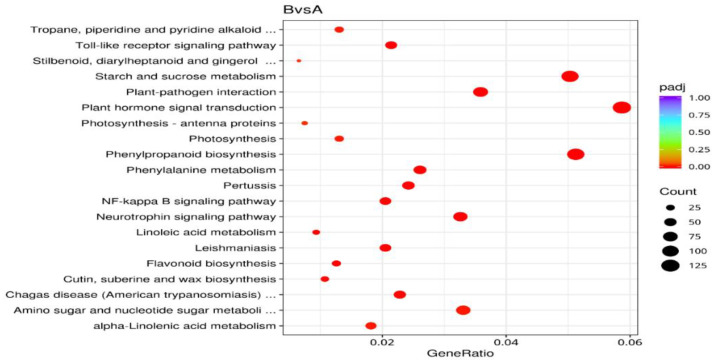 Figure 6
Figure 6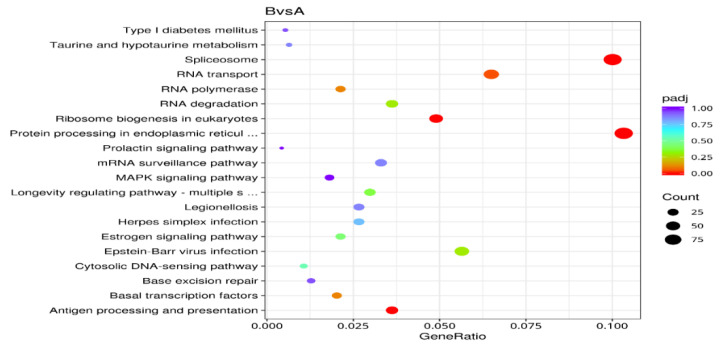 Figure 7
Figure 7- —Kashi University High-Level Talent Project
- —Kashi University Institutional Research Project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced Synthetic Organic Chemistry · Chemical synthesis and alkaloids · Phytochemistry and biological activities of Ficus species
1. Introduction
As an optimal plant for improving saline–alkali conditions, Nitraria roborowskii Kom. belongs to the genus Nitraria of the Nitrariaceae [1,2], and its seeds exhibit deep dormancy characteristics [3,4,5]. N. roborowskii has not only important ecological value but also nutritional value, medicinal value and economic value. Seeds of the Nitraria genus reportedly exhibit physiological dormancy (PD) [6]. The physiological dormancy of seeds is a double inhibition mechanism due to reduced embryo vigor coupled with the restriction of gas exchange caused by the seed coat. Short-term cold stratification, light, dry storage, disruption of the outer coat, and growth promotion can disrupt the physiological dormancy of seeds [7]. According to PIM intensity, physiological dormancy can be divided into three categories: shallow (C1), moderate (C2), and strong (C3) physiological dormancy. PD is the most abundant form and is found in the seeds of gymnosperms and in all major angiosperm clades [8].
The transition of seeds from the dormant stage to the germination stage involves very complex biological processes [8]. There are often more differentially expressed genes (DEGs) between the two phases [9]. Therefore, exploring the dormancy mechanism of N. roborowskii seeds is valuable for their development and utilization. Transcriptome research has become an effective means for discovering key genes involved in seed germination [10]. It can comprehensively reveal differences in gene expression under different physiological conditions and has been widely used for screening genes related to the low-temperature stress response and participating in the seed germination process [11]. In recent years, differential gene studies related to seed dormancy enhancement have been carried out on Polygonatum sibiricum Red [12] and Paris polyphylla [13].
The lack of Nitraria sibirica Pall. Information on its genome limits further exploration of the molecular mechanisms involved in salt stress [14]. To date, no transcriptome studies have been conducted on the dormancy release of large white thorn fruit seeds. Therefore, in this study, transcriptome sequencing was performed on samples before and after dormancy release from N. roborowskii seeds to address the following scientific questions: (1) Which key genes were significantly differentially expressed after dormancy release from N. roborowskii seeds? (2) Which metabolic pathways underwent critical changes?
2. Results
2.1. Transcriptome Sequencing and De Novo Assembly
Since there is no corresponding reference gene sequence for N. roborowskii, this study used the de novo assembly method and Trinity software to assemble the clean reads. More than 20,529,105 raw reads were obtained for each sample, and after filtration, more than 5.8 Gb of sequencing data were obtained. Q20 ≥ 97.20%, Q30 ≥ 91.80%, and the GC content ranged from 44.69~50.19% (Table 1). A total of 215,303 transcripts and 84,450 unigenes were obtained, with the maximum lengths of the transcripts and unigenes being 13,381 bp and 1614 bp, respectively, and the N50 length was 1461 bp. A total of 84,450 unigenes were annotated in the NT, NR, KOG/COG, Swiss-Prot, PFAM, KEGG, and GO databases, and the annotation rates of the unigenes in each database are shown in Table 2.
2.2. Expression Levels of Genes Before and After Germination
The percentages of unigenes with an FPKM [15] > 0.3 were 34.36%, 28.41%, 38.79%, 82.37%, 51.96% and 53.34%, respectively, indicating that the gene expression level of the seeds of N. roborowskii in the dormant period was relatively low, with an average value of 33.85%, of which A3 was the highest at 38.79%, and after the seeds of N. roborowskii were lifted during the dormant period, their gene expression level increased greatly, with an average value of 62.56%, of which B1 was the highest at 82.37%.
According to the experimental sequencing data, the transcriptomes of dormant seeds (A) and dormant-released seeds (B) of N. roborowskii were compared and analyzed, and a total of 16,130 DEGs were identified, of which 10,776 genes were upregulated and 5354 genes were downregulated (Figure 1).
Note: The green dots represent significantly downregulated genes, the red dots represent significantly upregulated genes, and the blue dots represent genes whose expression did not significantly change. The X-axis indicates the base-2 logarithms of the fold changes of the DEGs, and the Y-axis indicates the negative base-10 logarithms of the p values of the DEGs.
2.3. Analysis of Gene Function Annotation Differences Before and After Germination
The DEGs were annotated into 3 categories [16], BP (16,549), CC (10,583), and MF (10,377), accounting for 44.12%, 28.21%, and 27.67%, respectively. Among them, 6639 differentially expressed genes in BP were annotated to 66 functions (Figure 2), including mainly cellular nitrogen compound metabolic processes, biosynthetic processes, and transport. There were 4141 DEGs in CC, annotated to 32 functions, and the 3 most important functions were intracellular, protein-containing complex, and organelle. There were 7528 DEGs in the MF category, which were annotated to 37 functions, and the most annotated genes were involved in ion binding, oxidoreductase activity, and DNA binding. The functions of these genes are related to metabolic activities.
During the process of seed dormancy release, the 3 most significantly upregulated annotated genes in the 3 most important biological processes were “cellular protein modification processes”, “cell wall organization or biogenesis”, and “lipid metabolic processes”; the 3 most important cellular components were “cell wall”, “thylakoid”, and “external encapsulation structure”; and the 3 most important molecular functions of the annotated genes were “hydrolase activity acting on glycosyl bonds”, “kinase activity”, and “transferase activity of glycosyl groups” (Figure 3). The 3 most important biological processes with significantly downregulated annotated genes were ribosome biogenesis, the cellular nitrogen compound metabolic process, and mature protein; the 3 most important cellular components were ribosome, lipid droplet, and organelle; and the 3 most important molecular functions were RNA binding protein, ribosome structural constituent, and structural molecule activity (Figure 4).
2.4. Analysis of Gene Expression Differences Before and After Germination
The top 20 pathways with the largest number of genes were included [17] (Figure 5). The five pathways with the greatest enrichment were plant hormone signal transduction, starch and sucrose metabolism, phenylpropanoid metabolism, plant–pathogen interaction, and neurotrophic protein signaling pathways. For the upregulated genes, the top 20 pathways with the most significant enrichment were selected (Figure 6), and the five pathways with the greatest enrichment were plant hormone signal transduction, phenylpropanoid metabolism, starch and sucrose metabolism, plant–pathogen interaction, and the neurotrophic protein signaling pathway. For the downregulated genes, the top 20 pathways with the most significant enrichment were selected (Figure 7), and the five pathways with the greatest enrichment were splicing, protein processing in the endoplasmic reticulum, RNA transport, Epstein–Barr virus infection, and ribosome biosynthesis in eukaryotes. In this study, a total of 10 metabolic pathways related to the release of dormancy in N. roborowskii were identified (Table 3).
Statistics of pathway enrichment of DEGs.
Note: The X-axis represents the enrichment factors of different KEGG pathways, and the Y-axis indicates different KEGG pathways. The different colors of the dots indicate different q-values. The different sizes of the dots indicate different numbers of genes.
Statistics of pathway enrichment of upregulated genes.
Statistics of pathway enrichment of downregulated genes.
2.5. Differentially Expressed Genes in the Hormone Signal Transduction Pathway Before and After Germination
Studies have shown that genes involved in plant hormone signal transduction play a key role in the process of seed dormancy and germination. The DEGs of N. roborowskii seeds before and after germination were associated with the plant hormone signal transduction pathway (ko04075). The Q value also revealed that the hormone signal transduction pathway presented the greatest difference in gene expression. The results revealed 146 annotated DEGs in the hormone signal transduction pathway (Table 4), of which 126 genes were upregulated and 20 genes were downregulated. The numbers of genes associated with GA, ABA, IAA, CTK, ETH, BR and JA were 15, 5, 4, 3, 3, 6, 7 and 5, respectively (Table 5).
3. Discussion
Seed dormancy is regulated by a variety of genes, and genes encoding phytohormones and key enzymes of the respiratory pathway play important roles in the regulation of seed dormancy release. A total of 44 genes encoding three key enzymes, namely, phosphoglucose isomerase (PGI), which is involved in glycolysis (EMP); malate dehydrogenase (MDH), which is involved in the tricarboxylic acid cycle (TCA); and glucose-6-phosphate dehydrogenase (G6PDH), which is involved in the pentose phosphate pathway (PPP), were found in the transcriptomes of N. roborowskii seeds. Among the dormancy lifting and dormant N. roborowskii seeds with key respiratory pathway metabolism enzyme-related DEGs, 11 DEGs were upregulated, while a total of 8 PGI genes and 7 genes were upregulated. A total of 31 MDH genes and 26 genes were upregulated, and a total of 5 G6PDH genes were upregulated; i.e., three key enzymes were upregulated, and the three key enzymes were significantly upregulated in the process of increasing seed dormancy in N. roborowskii. High α-amylase gene expression is an important indicator of increased seed dormancy. In this study, we determined that there were 11 differentially expressed α-amylase genes in the transcriptome of the seeds of N. roborowskii, of which 8 were significantly upregulated and 3 were significantly downregulated. The elevated content of α-amylase promoted the conversion of starch to soluble sugars in the seeds, and the high and low contents were related to the low-temperature resistance of the plants, which is in line with the preliminary findings. This finding is consistent with the conclusion of the study that the soluble sugar content increased after the increase in seed dormancy in N. roborowskii.
In this study, GO enrichment analysis of DEGs in N. roborowskii seeds before and after germination revealed that the DEGs were involved mainly in metabolic processes, catalytic activity, single-organism metabolic processes, oxidoreductase activity, and ribosomes. KEGG and weighted gene co-expression network analyses indicated that DEGs responsive to saline-alkaline stress were predominantly enriched in pathways including plant hormone signal transduction, MAPK signaling, starch and sucrose metabolism, glutathione metabolism, and phenylpropanoid biosynthesis [18]. KEGG pathway enrichment analysis revealed that the genes that were significantly differentially expressed after seed germination were enriched mainly in pathways such as ribosome, carbon metabolism, amino acid biosynthesis, and protein processing. Among them, 146 genes were involved in the hormone signal transduction pathway, 116 genes were involved in the phenylpropanoid metabolism pathway, 27 genes were involved in the flavonoid biosynthesis pathway, 122 genes were involved in the starch and sucrose metabolism pathways, and many DEGs were significantly expressed in the MAPK signaling pathway, which is consistent with previous studies.
The dormancy of Ginkgo biloba seeds is regulated not only by the balance of ABA/GA but also by other hormones related to embryo morphological development and genes related to embryo differentiation and development [19]. KEGG pathway analysis revealed that plant hormone signal transduction is related to seed germination, and identifying hormone metabolic genes and analyzing gene expression patterns are the keys to understanding the regulation of dormancy and germination [7,20].
The GA signaling pathway consists mainly of target factors regulated by the receptor GID1 [21]. GID1 is a soluble GA receptor that is localized in the nucleus and cytoplasm of rice and Arabidopsis cells [22]. The expression of genes related to gibberellic acid (GA) catabolism and signal transduction (CYP707As, GA2ox, and DELLAs) was consistent with endogenous hormone changes [23]. This study compared the transcriptome data of dormant and dormant-release N. roborowskii seeds and identified a total of 80 genes related to plant hormone (ABA, GA, and IAA) metabolism, among which the GA receptor (GID1A) was significantly upregulated by 1.47-fold. After the dormancy of N. roborowskii seeds was released, the expression of the SLY1 gene increased significantly. A sleepy1 (sly1) mutant was identified in Arabidopsis seeds, and SLY1 was hypothesized to be a key factor in seed sensory gibberellin signaling [24]. GA promotes the generation of growth potential by stimulating cell elongation and inducing the expression of genes encoding cell wall-modifying enzymes, such as the expansin gene (EXP), which can regulate cell wall loosening and is important for cell elongation in dormant-released N. roborowskii seeds. During the process of releasing the dormancy of N. roborowskii seeds, the expression of genes related to GA synthesis (GA2OX1, GA2OX2, GA2OX6 and GA2OX8) and gibberellin regulatory proteins (GASA4, GASA9, GASA13 and GASA14) increased significantly. Five C19-GA2ox genes have been identified in Arabidopsis thaliana [25].
The expression of the GA-positive regulatory gene PP2A significantly differed before and after the release of dormancy in N. roborowskii seeds. Several studies have shown that the molecular marker of the germination period phosphatase (PP2A), which is involved in the release of dormancy in Arabidopsis seeds, is upregulated in GCR1 plants [26].
In this study, the expression of ABA biosynthetic 9-cis-epoxycarotenoid dioxygenase (NCED6), which is involved in the ABA metabolic signal transduction pathway, was significantly downregulated 8.14-fold. The NCED6 gene is expressed specifically in seeds, plays a major role in ABA synthesis during seed development and germination and can induce seed dormancy [27]. Under low-temperature conditions, the expression of genes involved in ABA synthesis (NCED) decreases, whereas the expression of genes involved in gibberellin and ethylene synthesis (GA3oxl and ACS1) increases [28]. In this study, the expression of NCED decreased, and the expression of ACS1 increased. The ABA content in seeds is related not only to the synthetic pathway but also to its inactivation. The CYP707A2 gene encoding ABA-8′-hydroxylase is a key protein that regulates ABA metabolism. In this study, the expression of CYP707A2, which is a key regulatory enzyme involved in the release of dormancy in N. roborowskii seeds, was significantly upregulated by 4.13-fold. Studies have shown that the DELLA gene family is the main factor regulating GA signaling [29]. The expression of DELLA genes (GAI and GAIP) in N. roborowskii seeds was upregulated, which proved that the GA content increased and that ABA synthesis decreased or decreased in the seeds released from dormancy, which jointly promoted the germination of N. roborowskii seeds. The results of this experiment revealed that 40 BHLH genes in the seeds of N. roborowskii released from dormancy were significantly upregulated. BHLH transcription factors indirectly regulate the ABA synthesis gene NCED2, which can regulate ABA metabolism and synthesis and achieve accurate regulation of ABA in seeds, thereby affecting the process of seed dormancy or germination [30]. The relative expression levels of ABA synthesis- and metabolism-related genes change [31]. ABI5 shares several target genes that negatively regulate embryo germination together with the bHLH transcription factor PIF1 [32].
4. Materials and Methods
4.1. Experimental Materials
The seeds of N. roborowskii required for this experiment were purchased from the planting base of Minqin County Linquan Ecological Seed Industry Co., Ltd. (Wuwei, China). According to the seed collection requirements specified in the “Technical Code for Sowing and Seedling Cultivation of Nitraria (LY/T 2295-2014),” the following procedures were followed during fruit harvesting: healthy, tall, and high-yielding mother plants were selected. Fruits showing signs of pests or diseases were excluded. The fruits were manually picked or shaken from the branches and collected separately to prevent mixing. After natural air-drying, the dried N. roborowskii fruits were soaked in water until fully expanded. They were then placed in an iron sieve with a mesh size smaller than the seed diameter and repeatedly rubbed to break down the pulp. The mixture was rinsed under running water to remove floating peels, empty grains, rotten grains, and pest-damaged grains. After multiple rounds of rubbing and rinsing, clean seeds were obtained. These seeds were air-dried naturally, packaged in sealed bags, and stored in a refrigeration chamber.
4.2. Material Treatment
The seeds of N. roborowskii for this study were sourced from the Minqin County Linquan Ecological Seed Industry Co., Ltd. (Wuwei, China) planting base. Mature, healthy seeds (viability: 83.00% by TTC method) were selected, surface-sterilized with 0.1% KMnO_4_ for 30 min, and then soaked in 500 mg/L GA_3_ for 24 h, with distilled water as control. The GA_3_-treated seeds were mixed with moist river sand (1:3, mass ratio; ~75% moisture—assessed as “hand-squeezed into a ball, crumbles when released”) and subjected to outdoor sand stratification for 90 days. Radicle emergence was taken as the criterion for dormancy break. Most GA_3_-treated seeds showed radicle and plumule exposure, indicating dormancy release, whereas control seeds displayed no germination signs and remained dormant [33]. Seed embryos in dormant (A1, A2, A3) and dormancy-released (B1, B2, B3) states were separately used for RNA extraction and transcriptome sequencing. For each sample, approximately 0.1 g material was collected with three biological replicates.
4.3. Total RNA Extraction, Library Construction and Sequencing
RNA was extracted via the cetyltrimethylammonium bromide (CTAB) method (QIAGEN, Germany). The Panomics default sequencing platform used was an Illumina NovaSeq 6000 (Illumina, San Diego, CA, USA) with S4 suite components. After the library was constructed, it was first quantified via a Qubit 2.0 fluorometer, and then the insert size of the library was detected via an Agilent 2100 bioanalyzer. After the insert size met the expectations, the effective concentration of the library was accurately quantified via qRT–PCR (the effective concentration of the library was greater than 2 nM) to ensure the quality of the library.
4.4. Quality Control and Assembly
Since there is no reference genome for N. roborowskii seeds, Trinity [34] was used to assemble the clean reads to obtain the unigene library during the process of seed dormancy release. In this study, the de novo assembly method was used, and Trinity software (v2.4.0) was used to assemble the clean reads. With Corest software (v4.6), the transcripts were clustered and found to be redundant, and each cluster was ultimately defined as a unigene. KEGG annotations were performed on the unigenes via KAAS, and GO annotations were performed via Blast2 GO.
4.5. Gene Functional Annotation
To obtain the functional information of the sequence genes, all the unigenes after assembly were subjected to BLAST (v2.0.6) [35] searches against seven functional databases, namely, the NR, NT, PFAM, COG, Swiss-Prot, KEGG, and GO databases, via BLASTX (v2.0.6).
4.6. Data Analysis
The transcript sequences obtained in this study were used as reference sequences, and RSEM [36] was used to map the clean reads to the reference sequence with Bowtie2 via default parameters. Through a BLASTx search, the key genes involved in the dormancy and germination of N. roborowskii seeds were identified.
5. Conclusions
In this study, using N. roborowskii seeds before and after dormancy release as the research subjects, a total of 16,130 differentially expressed genes (DEGs) were identified, of which 10,776 were upregulated and 5354 were downregulated. Among these DEGs, 165,450 were functionally annotated by GO classification into three main categories and 43 subcategories, covering biological processes, molecular functions, and cellular components. In KEGG pathway analysis, 36,540 DEGs were mapped to 303 metabolic pathways, with notable involvement in pathways such as the ribosome, carbon metabolism, biosynthesis of amino acids, protein processing in the endoplasmic reticulum, and spliceosome. Based on the annotation results from the KEGG database, ten metabolic pathways closely related to the dormancy release of N. roborowskii seeds were identified. Among these, the plant hormone signal transduction pathway and the starch and sucrose metabolism pathway play key regulatory roles during seed germination. Additionally, this study identified key genes (GID1A, GA2OX, PP2A, NCED, CYP707A2, SLY1, EXP, GASA, and BHLH) that exhibited significant expression changes before and after seed germination.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Liu Y. Nitraria X. Xu L. Huang C. The Flora Reipublicae Popularis Sinicae (FRPS)Beijing Science Press Beijing, China 1998 Volume 43117123
- 2Liu Y.X. Zhou L.H. Nitariaceae, Peganaceae, Zygophyllaceae Flora of China Wu Z.Y. Raven P.H. Science Press Beijing, China Missouri Botanical Garden Press St. Louis, MO, USA 2008 Volume 114150
- 3Commander L.E. Merritt D.J. Rokich D.P. Dixon K.W. Seed biology of Australian arid zone species: Germination of 18 species used for rehabilitation J. Arid Environ.20097361762510.1016/j.jaridenv.2009.01.007 · doi ↗
- 4Zeng Y.J. Wang Y.R. Zhang J. Li Z.B. Germination responses to temperature and dormancy breaking treatments in Nitraria tangutorum Bobr. and Nitraria sibirica Pall Seed Sci. Technol.20103853755010.15258/sst.2010.38.3.02 · doi ↗
- 5Zeng Y.J. Wei J.P. Yu L. Wang Y.R. Seed viability, germination and dormancy of Nitraria roborowskii (Nitrariaceae)Seed Sci. Technol.20164464765310.15258/sst.2016.44.3.17 · doi ↗
- 6Baskin C.C. Baskin J.M. Seeds: Ecology, Biogeography, and Evolution of Dormancy and Germination 2nd ed.Elsevier Academic Press San Diego, CA, USA 2014
- 7Khan A.A. The Physiology and Biochemistry of Seed Dormancy and Germination North-Holland Publishing Company Amsterdam, The Netherlands New York, NY, USA Oxford, UK 1977
- 8Finch-Savage W.E. Leubner-Metzger G. Seed donnancy and the control of gemination New Phytol.2006175052310.1111/j.1469-8137.2006.01787.x 16866955 · doi ↗ · pubmed ↗