Deletion of RhoGDI Protects Against Hepatic Steatosis via Improved Mitochondrial Metabolism in Mice
Yongzhi Wang, Yuanqi Zhou, Yifan Xu, Chen Wang, Shuo Meng, Honglin Li, Huifang Tang, Jian Zhang

TL;DR
Deleting RhoGDI in mice protects against fatty liver disease by improving mitochondrial function and reducing liver fat accumulation.
Contribution
This study identifies RhoGDI as a key regulator of metabolic liver disease and shows that its deletion improves mitochondrial metabolism and reduces hepatic steatosis.
Findings
Hepatocyte-specific deletion of Arhgdia (RhoGDI) reduced hepatic lipid accumulation and fibrosis in mice.
RhoGDI deficiency suppressed the steroid hormone biosynthesis pathway and improved mitochondrial β-oxidation.
A natural compound targeting RhoGDI alleviated liver steatosis and inflammation in a MASLD model.
Abstract
The global incidence of metabolic dysfunction-associated steatotic liver disease (MASLD) is rising alongside epidemics of diabetes and obesity. Rho GDP-dissociation inhibitor (RhoGDI) is now recognized to play dual regulatory roles in disease. A deeper understanding of its mechanistic contributions in MASLD could offer critical insights for developing novel therapies against this growing health burden. Immunohistochemical staining was used to examine RhoGDI expression in liver tissues from patients with MASLD. Hepatocyte-specific deletion of Arhgdia (the gene encodes RhoGDI) was generated in mice, and they subjected to NASH diets to induce hepatic steatosis. Transcriptomic sequencing was carried out to identify altered pathways in the Arhgdia-deficient mice, followed by functional investigations of downstream signaling and mitochondrial performance. Finally, the therapeutic potential of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
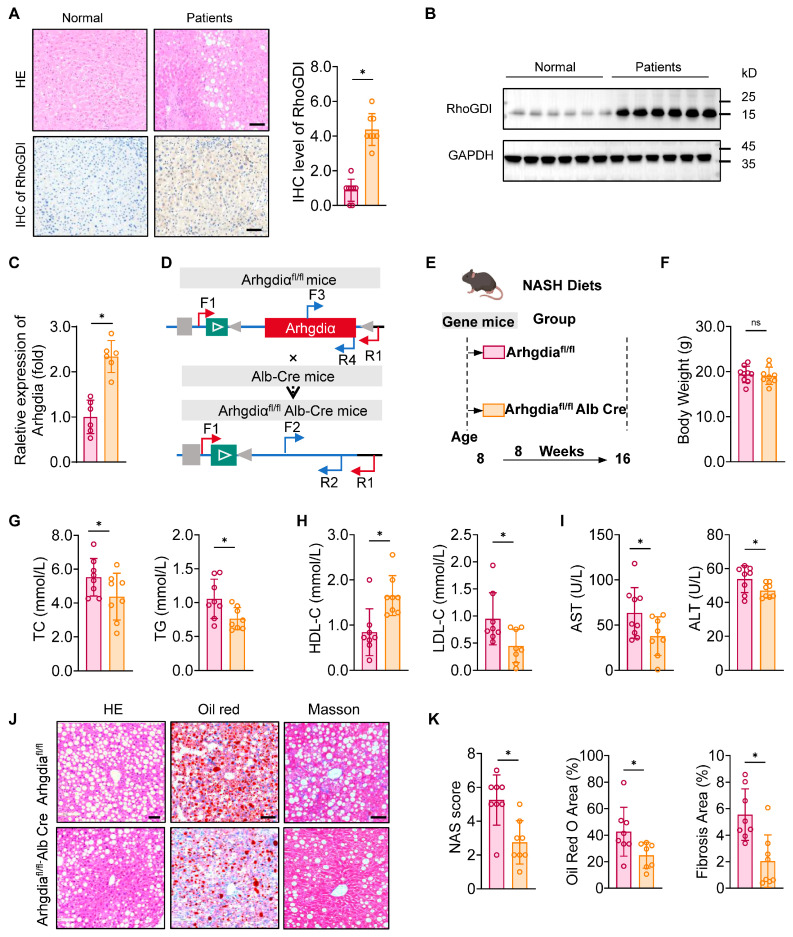 Figure 1
Figure 1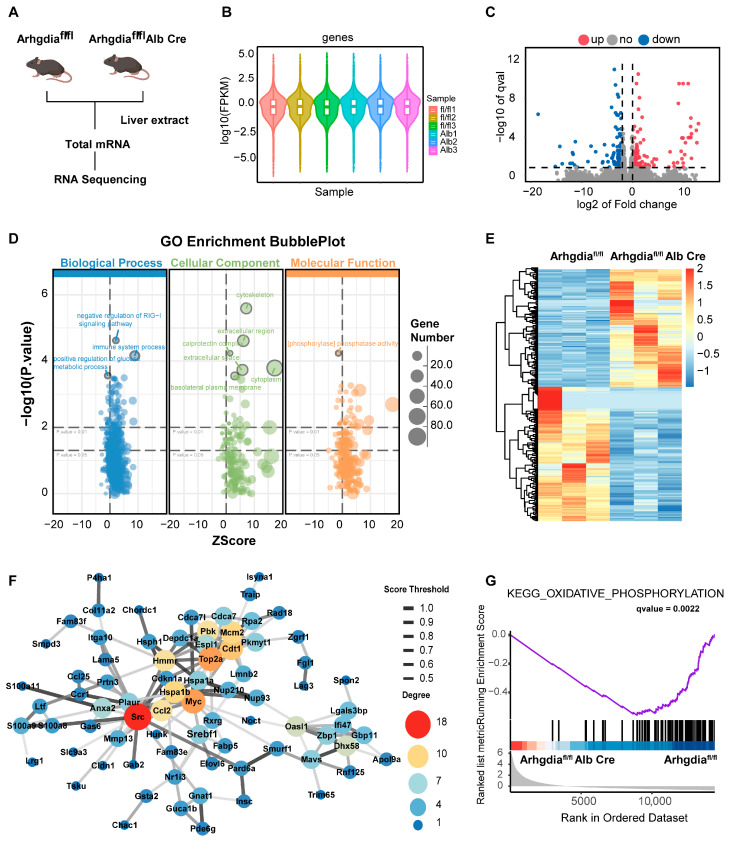 Figure 2
Figure 2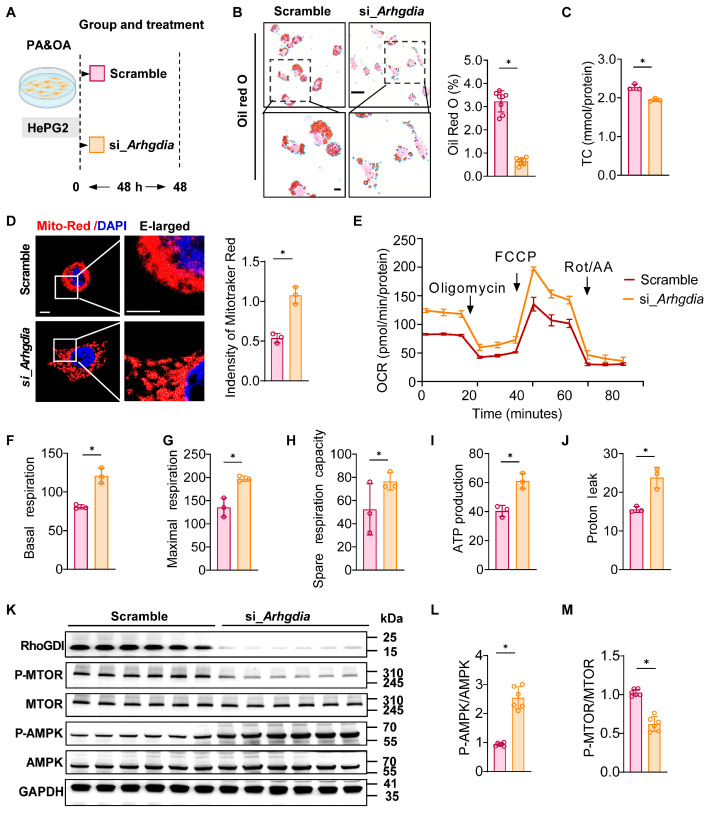 Figure 3
Figure 3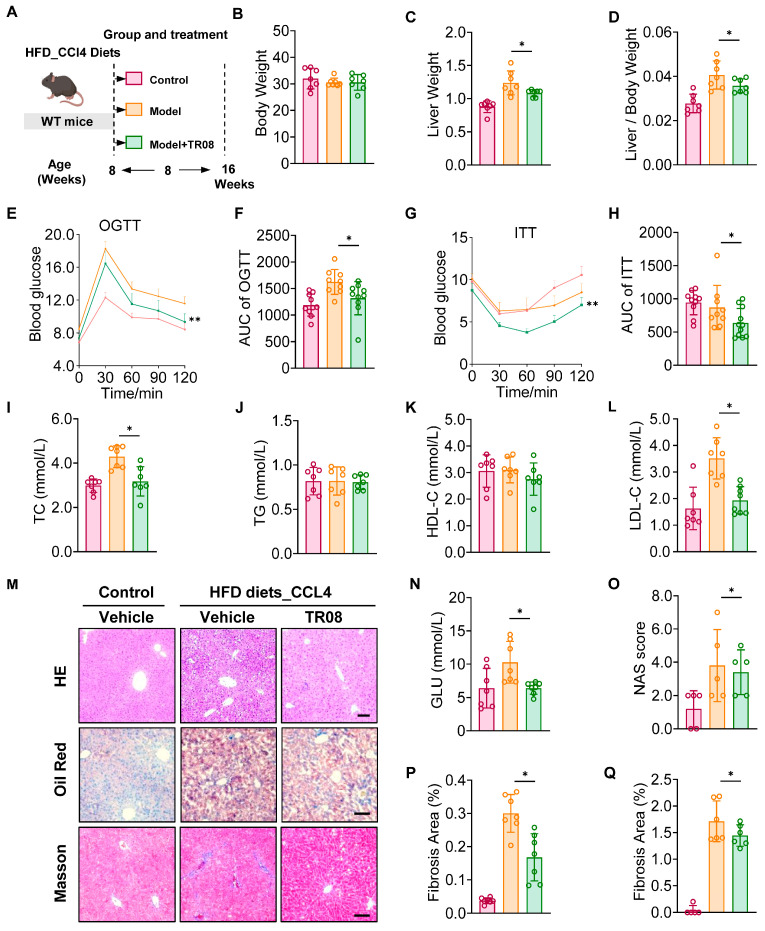 Figure 4
Figure 4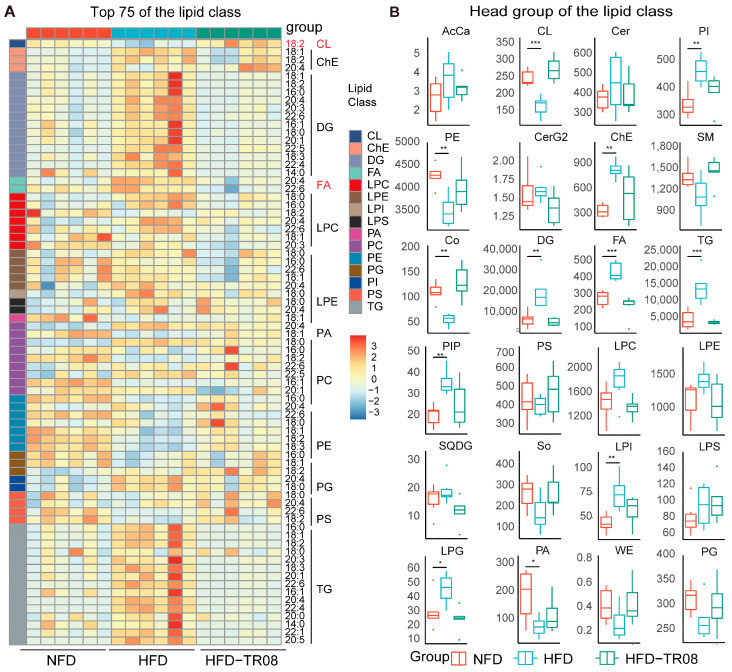 Figure 5
Figure 5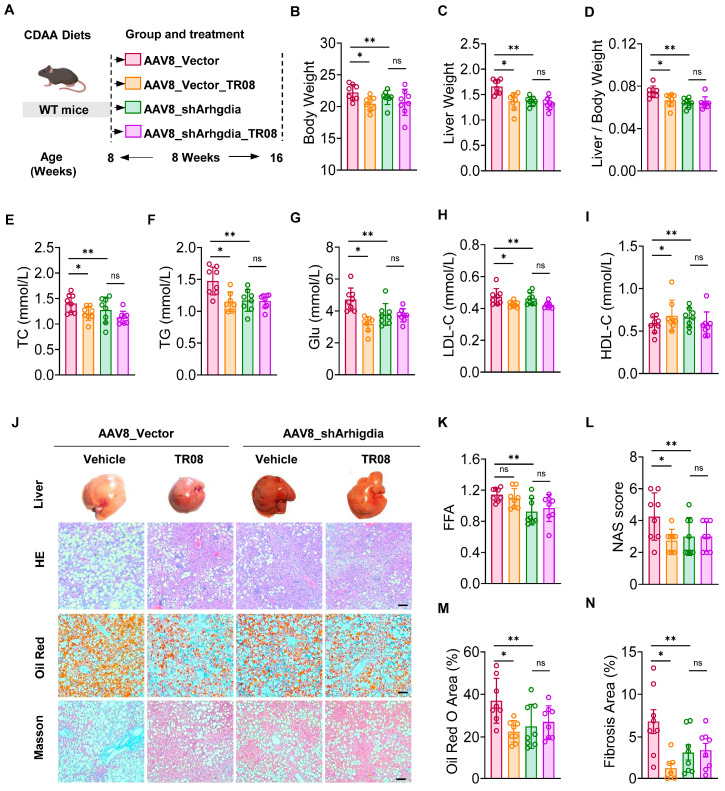 Figure 6
Figure 6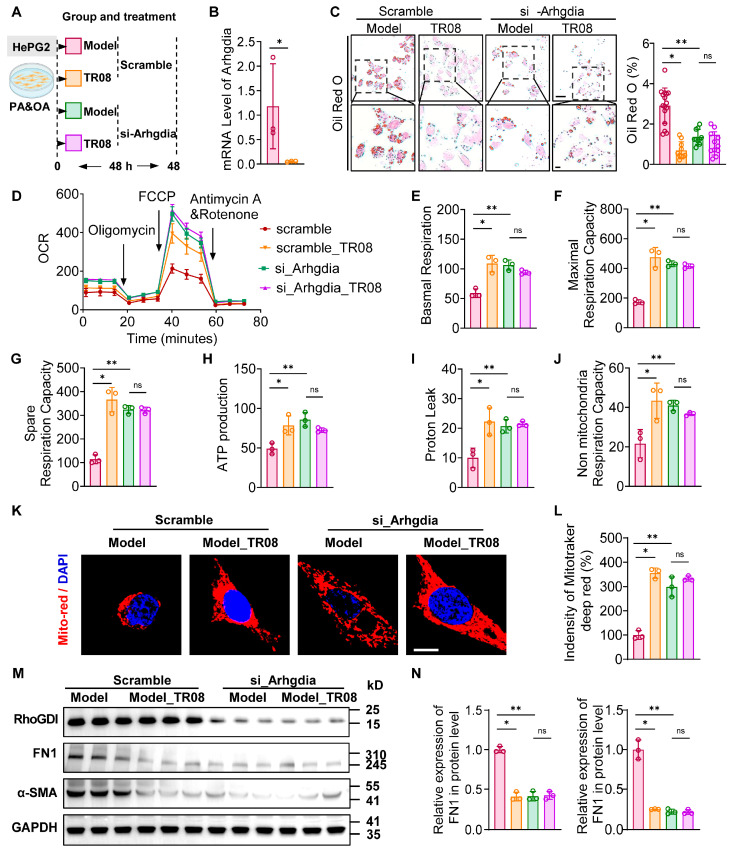 Figure 7
Figure 7- —Noncommunicable Chronic Diseases-National Science and Technology Major Project
- —National Natural Science Foundation of China
- —Innovation Platform and Talent Program
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLiver Disease Diagnosis and Treatment · Peroxisome Proliferator-Activated Receptors · Diabetes, Cardiovascular Risks, and Lipoproteins
1. Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD) has become one of the most common chronic liver diseases worldwide, affecting 25% of the world’s population as a result of lifestyle changes such as increased consumption of energy-dense diets and sedentary habits [1]. Metabolic steatohepatitis (MASH) is a severe form of MASLD with a potentially progressive course that can lead to liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) [2,3]. The progression of MASLD is difficult to visualize in the early stages of the disease because of uncertainties related to the accuracy of noninvasive markers of liver damage and the lack of effectiveness data related to treatment in patients with MASLD [4]. A growing number of drugs to treat patients with MASLD are being tested in clinical trials [5]. However, despite years of efforts by researchers around the world, only the thyroid hormone receptor β (THR-β) agonist resmetirom and GLP-1 inhibitor semaglutide, which were approved in 2025, have shown great efficiency in patients with MASLD; these were approved for the treatment of MASLD by the Food and Drug Administration (FDA) [6,7]. A major obstacle to the development of drugs for MASLD is the absence of a clear understanding of the mechanisms underlying this metabolic disease [8].
The existing studies in the literature have suggested that MASLD results from a multi-hit pathogenesis [9]. The core driving risk of MASLD progression can be attributed to the accumulation of free fatty acids (FFAs), which either enter the mitochondria to undergo β-oxidation or are esterified and subsequently stored as triglycerides (TG) [10,11]. Previous studies have shown that liver adaptation and mitochondrial flexibility are the main pathological processes in the early stages of MASLD development, which are subsequently lost in MASLD [12]. Thus, alterations in mitochondrial biogenesis and accumulation of damaged mitochondria may occur secondary to defects in the mitophagy pathway observed in liver tissues from patients with MASLD [4,13]. Mitochondrial swelling, cristae disruption, and impaired mitochondrial oxidative phosphorylation may be the markers of MASLD, and the hepatic mitochondrial mass and mitochondrial respiratory capacity are considered related to the transition from MASLD to MASH [14,15].
The function of the Rho-specific guanine nucleotide dissociation inhibitor (RhoGDI), encoded by the gene of Arhgdia, has been previously reported to be critically modulated by cholesterol-enriched membrane domains like lipid rafts, which serve as targeted platforms for GTPase delivery and facilitate localized release of active GTPases by integrating biophysical cues and extracellular signals such as integrin engagement [16,17]. Therefore, RhoGDI is ubiquitously expressed and interacts with several Rho GTPases. RhoGDI acts as a master regulator, orchestrating the spatiotemporal activity of Rho kinases. Rho-kinase has been suggested to ameliorate metabolic disorders through activation of the adenosine monophosphate (AMP)-activated protein kinase (AMPK) pathway, suggesting that Rho-kinase is a novel therapeutic target of metabolic disorders [18]. However, previous studies have not clarified whether the Rho GTPase companion RhoGDI plays a similar role in MASLD. Moreover, the mechanism underlying the effects of RhoGDI and its regulatory role in lipids and mitochondrial function are not clear.
In this study, we investigated the role of RhoGDI in metabolic regulation by generating liver-specific knockout of its encoding gene Arhgdia in mice. Our results demonstrate that hepatic RhoGDI deficiency attenuates lipid accumulation, inflammation, steatosis, and fibrosis in mice fed with MASLD-inducing diets. Furthermore, we revealed that knockdown of RhoGDI decreased liver lipid accumulation by enhancing mitochondrial function and promoting the phosphorylation of AMPK. In addition, we observed that a natural compound, TR08, which has been reported to be a drug candidate in antihypertension [19], attenuated hepatic lipid accumulation, inflammation, and hepatic steatosis and fibrosis in mice. Mechanistically, targeting RhoGDI with TR08 yielded protective effects by enhancing mitochondrial function and decreasing liver lipid accumulation and fibrosis, indicating a mechanism promoting the phosphorylation of AMPK and mitochondrial function. These data identified RhoGDI as a potential target of MASLD treatment and demonstrated that TR08 is a promising drug candidate that can ameliorate MASLD by promoting fatty acid oxidation and lipid clearance.
2. Results
2.1. Hepatocyte Arhgdia Knockout Attenuated MASLD Progression by Improving Lipid Metabolism and Reducing Fibrosis
To investigate the role of Arhgdia in fatty liver development, we first analyzed RhoGDI expression in human hepatic tissues with MASLD. Notably, RhoGDI exhibited much higher levels of expression in fatty livers than in normal tissues (Figure 1A), indicating a potential association with hepatic lipid metabolism. Furthermore, Both the mRNA and protein expression levels of RhoGDI were significantly upregulated in fatty livers than in normal tissues (Figure 1B,C). Hepatocyte-specific Arhgdia-knockout mice were obtained by crossing Arhgdia^fl/fl^ mice with Alb-Cre transgenic mice, which were then fed high-fat and high-cholesterol diets to induce MASLD (Figure 1D,E). The mice with Arhgdia deletion did not show significant changes in body weight in MASLD (Figure 1F). However, Arhgdia^fl/f,Alb-Cre^ mice showed marked improvements in serum lipid profiles, with reduced total cholesterol (TC) and TG levels (Figure 1G). Furthermore, these mice showed increased high-density lipoprotein cholesterol (HDL-C) levels and decreased low-density lipoprotein cholesterol (LDL-C) levels (Figure 1H). These findings suggest that Arhgdia deficiency mitigates hepatic lipid accumulation. Additionally, the levels of serum markers of liver injury, including aminotransferase (AST) and alanine transaminase (ALT), were significantly lower in Arhgdia^fl/fl-Alb-Cre^ mice (Figure 1I). Histological analyses revealed reduced lipid deposition and fibrosis in Arhgdia^fl/fl-Alb-Cre^ mice (Figure 1J,K). Collectively, our data highlight Arhgdia as a critical regulator of hepatic lipid metabolism and fibrosis, with its ablation conferring protection against MASLD progression.
2.2. Transcriptome Analysis Revealed That Arhgdia Ameliorates Hepatic Steatosis and Reduces Lipid Accumulation
To investigate the mechanistic role of Arhgdia in hepatic lipid metabolism, we conducted hepatocyte-specific RNA-seq analysis of liver tissues from Arhgdia^fl/f,Alb-Cre^ mice and WT littermate controls (Figure 2A). Quality assessments revealed high reproducibility among biological replicates, as demonstrated by uniform distribution patterns in violin plots of gene expression profiles (Figure 2B). Differential expression analysis revealed 867 significantly upregulated genes and 521 downregulated genes in Arhgdia-deficient livers in comparison with WT controls (|log_2_FC| > 1, q Value < 0.05), and the results were visualized using a volcano plot (Figure 2C). Gene ontology (GO) enrichment analysis demonstrated significant enrichment of differentially expressed genes (DEGs) in biological processes related to glucose metabolism, extracellular matrix organization, and protein phosphorylation (Figure 2D). Hierarchical clustering of DEGs revealed distinct expression patterns between genotypes, which were visualized by heatmap analysis (Figure 2E). Protein–protein interaction (PPI) network analysis of these genes revealed key interacting proteins of the differential genes in the liver. Interestingly, Srebf1 was shown to be related to lipid metabolism, especially cholesterol metabolism (Figure 2F). Moreover, GSEA further identified differential changes in Arhgdia-associated genes involved in mitochondrial oxidative phosphorylation pathway (Figure 2G). These findings collectively indicate that Arhgdia directly regulates genes involved in cholesterol metabolism and the mitochondrial oxidative phosphorylation in hepatocytes.
2.3. Arhgdia Knockdown Reduces Lipid Accumulation and Enhances Mitochondrial Function in Hepatocytes
To investigate the role of Arhgdia in lipid metabolism, we first established a PA/oleic acid (OA)-induced cellular model in HepG2 cells (Figure 3A). We found that Arhgdia knockdown significantly reduced lipid accumulation in hepatocytes under PA/OA-induced steatotic conditions (Figure 3B). Additionally, TC levels were markedly decreased in these hepatocytes (Figure 3C). Mito tracker red staining revealed an increased mitochondrial number in the cells treated with small interfering RNA (siRNA) targeting Arhgdia (Figure 3D). Given the critical role of mitochondria in lipid metabolism, we assessed the OCR using Seahorse XFe96 Analyzer. Cells transfected with Arhgdia siRNA exhibited stronger mitochondrial respiratory capacity, particularly in basal and maximal respiration (Figure 3E–G). Moreover, these cells also showed elevated spare respiratory capacity and ATP production (Figure 3H–J). Concurrently, we measured the phosphorylation levels of AMPK and mammalian target of rapamycin (mTOR). Arhgdia knockdown led to a significant increase in AMPK phosphorylation, while the levels of phosphorylated mTOR remained were markedly reduced (Figure 3K–M).
2.4. TR08 Ameliorates HFD-CCl4-Induced Metabolic Steatohepatitis in Mice
To evaluate the therapeutic potential of the natural compounds in MASLD, we established a murine model by feeding male mice an HFD for 12 weeks, combined with intraperitoneal CCl_4_ injections followed by the last 4 weeks of HFD feeding, the control group was on a normal diet, while the administration group received TR08 via oral gavage under modeled conditions (Figure 4A). We found that TR08 (10 mg/kg/d) administration did not significantly alter body weight (Figure 4B). However, the mice showed significant reductions in liver weight (Figure 4C) and liver-to-body weight ratio (Figure 4D). Oral glucose tolerance tests (OGTTs) revealed improved glucose metabolism in TR08-treated mice after 16 weeks (Figure 4E,F). Meanwhile, the ITT curve for the condition with insulin injection and the area under the curve of the ITT curve showed an improvement in insulin sensitivity in response to TR08 treatment (Figure 4G,H). TR08 administration significantly reduced the TC and LDL-C levels in the serum of mice. No marked changes were observed in the TG or HDL-C levels of mice (Figure 4I–L). Hematoxylin and eosin (H&E) staining and Oil Red O staining of liver sections showed a reduction in lipid accumulation in the TR08-treated group in comparison with the model group. Masson staining showed a markedly reduced fibrosis area in liver tissues (Figure 4M). In parallel with the decrease in blood glucose levels post-administration (Figure 4N) in serum, the NAS (Figure 4N), the Oil Red O area (Figure 4P), and the fibrosis area (Figure 4Q) were simultaneously reduced. Collectively, these results demonstrated the therapeutic efficacy of TR08 in ameliorating MASLD progression in various mouse models.
Lipidomics, based on high-throughput analytical technologies, enables systematic characterization of the composition and expression profiles of lipids in biological systems. To investigate the effects of TR08 on fatty acid metabolism, we performed untargeted lipidomic analysis of liver tissues. As shown in the heatmap in Figure 5A, lipids in the livers of TR08-treated mice were significantly downregulated compared with those in the control group, with a notable reduction in lipids containing fatty acid chains longer than 18 carbons, indicating impaired elongation of C18 and longer-chain fatty acids. In particular, significant downregulation of free fatty acids, triglycerides (TGs), and long-chain fatty acids was observed, while a marked upregulation of cardiolipin (CL) was also noted which reveals the mitochondrial relationships (Figure 5B). These findings suggest that TR08 treatment significantly downregulates hepatic lipid metabolism and exerts its ameliorative effects on MASLD by disrupting lipid metabolic homeostasis.
2.5. TR08 Attenuates MASLD Progression by Enhancing Lipid Disposion Through Arhgdia
To investigate whether TR08 attenuates MASLD progression through Arhgdia, mice were fed MASLD diets, followed by TR08 administration for an additional 8 weeks while they continued to receive MASLD diets (Figure 6A). This model was expected to show substantially higher hepatic steatosis and more inflammation. We observed that the reductions in body weight, liver weight, and the liver weight-to-body weight ratio of HFD-fed mice were significantly affected by infection with sh-Arhgdia compared to the AAV8_vector group. Administration of TR08 significantly reduced the body weight, liver weight, and liver weight-to-body weight ratio compared to those of the AAV8_vector group, but these changes were not observed when mice underwent both TR08 administration and Arhgdia transfection compared to the AAV8_sh-Arhgdia group (Figure 6B–D). Meanwhile, the TC, TG, and Glu levels in serum, as well as the LDL-C levels and HDL-C levels in serum, reduced after infection with sh-Arhgdia compared to those of the AAV8_vector group (Figure 6E–I). These changes showed similar trends in response to TR08 treatment, but these changes were not observed when mice underwent both TR08 administration and Arhgdia transfection compared to the AAV8_sh-Arhgdia group. In addition, H&E staining showed that hepatocyte ballooning was reduced in mice treated with TR08 in comparison with those that received the AAV8 vector. However, hepatocyte ballooning was similar in mice treated with TR08 and those injected with AAV8-sh-Arhgdia (Figure 6J). Additionally, the liver FFA level decreased in response to TR08 treatment in comparison with that in the AAV8-vector group. The Arhgdia and Arhgdia + TR08 groups underwent identical injections, with TR08 was added only later. However, the two groups did not show significant differences in liver FFA levels (Figure 6K). Pathological assessment, including the NAS, Oil Red O staining, and fibrosis analysis, consistently indicated that TR08 treatment ameliorated liver steatosis, inflammation, and injury compared to the AAV8-vector control group. Notably, the Arhgdia and Arhgdia + TR08 groups received identical viral injection, with TR08 administration commencing only afterward. Despite this treatment difference, the two groups did not exhibit statistically significant differences in pathological assessment (Figure 6L–N). These results suggest that TR08 regulates lipid metabolism and liver injury through RhoGDI to enhance mitochondria function and reduce lipid accumulation in the liver, thereby delaying the progression of MASLD.
2.6. TR08 Ameliorates Lipid Accumulation by Enhancing Mitochondria Function Through Arhgdia
To investigate the mechanism of TR08, we employed a lipid accumulation model in vitro via the administration of PA/OA in HepG2 cells (Figure 7A). The Arhgdia knockdown mice were observed and revealed a decrease in Arhgdia in HepG2 cells (Figure 7B). We found that lipid accumulation in HepG2 cells was significantly reduced by TR08 treatment. Notably, the extent of this reduction varied across Arhgdia knockdown conditions. Oil Red O staining of PA/OA-treated cells confirmed that TR08 intervention effectively decreased lipid deposition (Figure 7C). Given the central role of mitochondria in lipid metabolism, we measured the OCR using Seahorse XFe96 Analyzer. HepG2 cells transfected with Arhgdia siRNA exhibited enhanced mitochondrial respiratory capacity, particularly in basal and maximal respiration compared to the scramble group, while the TR08 treatment reduced mitochondrial function in response to Arhgdia siRNA (Figure 7D–G). Additionally, spare respiratory capacity and ATP production revealed TR08 enhanced mitochondria function (Figure 7H–J). These findings indicate that si-Arhgdia enhanced mitochondrial function, while TR08 administration caused a marked increase in mitochondrial function, suggesting an increase in oxidative phosphorylation during FFA metabolism. Subsequent mitochondrial functional assays demonstrated that Arhgdia knockdown significantly improved mitochondrial activity (Figure 7K). Although TR08 alone also enhanced mitochondrial function, its effects on liver lipid levels were lower than those observed in response to Arhgdia knockdown. In fact, early intervention targeting mitochondria showed potential in preventing or slowing fibrosis progression. We also evaluated fibrosis markers and found that Arhgdia knockdown significantly reduced the levels of fibronectin 1 (FN1) and α-smooth muscle actin (SMA) (Figure 7M,N). These results indicate that Arhgdia inhibits liver fibrosis by enhancing the mitochondrial function of hepatocytes.
3. Discussion
In this study, we found that RhoGDI is upregulated in the liver of patients and that hepatocyte-specific knockout of Arhgdia improves liver lipid accumulation and fibrosis in mice by enhancing AMPK phosphorylation and mitochondrial function and reducing lipid accumulation. Additionally, we identified a natural compound, TR08, that targeted RhoGDI to protect against the progression of MASLD in mice. Subsequently, infection with Arhgdia shRNA AAV8 ameliorated lipid accumulation and fibrosis in the liver. Meanwhile, TR08 also ameliorated lipid accumulation and fibrosis, providing further evidence for the efficacy of targeting RhoGDI with TR08. We further demonstrated that TR08 improves mitochondrial function through RhoGDI by administering TR08 to cells and knocking down RhoGDI.
RhoA has been shown to mitigate hepatic lipid accumulation in MASLD, yet its broader pathophysiological roles in inflammation and fibrosis remain unclear [20]. Further investigation into its natural inhibitor, RhoGDI, which is implicated in fibrosis and cardiovascular disease, is necessary [21]. Investigating RhoGDI could reveal deeper mechanistic insights into RhoA’s function in metabolic liver disease. In this study, we found that RhoGDI also plays an important role in preventing MASLD progression. RhoGDI improved liver lipid accumulation and fibrosis in mice by enhancing AMPK phosphorylation and mitochondrial function and reducing lipid accumulation. The compound, TR08, interacted with RhoGDI to significantly alleviate hepatic steatosis, fibrosis, and inflammation in the mouse model used. Mechanistically, TR08 not only decreased hepatic steatosis by enhancing lipid degradation, but also decreased fibrosis, thus preventing MASLD progression. Lipid accumulation is characteristic of liver steatosis, and evidence indicates that increased fatty acid uptake is associated with lipid accumulation [22]. In the present study, treatment with TR08 ameliorated hepatic lipid accumulation and fibrosis. Slowing down the tricarboxylic acid (TCA) cycle impairs fatty acid oxidation, resulting in fat accumulation in the liver. In this regard, elevated plasma levels of TGs and FFAs may contribute to HFD-induced body weight gain, while HFD-triggered glucose intolerance and insulin resistance play key roles in obesity and systemic metabolic dysfunction in mice.
Over the past decade, the discovery of AMPK substrates has improved the understanding of cellular metabolism from anabolism to catabolism, and AMPK has been identified as a central mediator of cellular metabolism that coordinates multiple features of metabolism as well as the mitochondrial response to energetic stress and mitochondrial insults [23]. The genetic and pharmacological disruption of RhoA activity has been shown to effectively mitigate MASLD phenotypes in mice [24]. Researchers have reported that the AMPK-independent pathway may be regulated by Rho GTPase, although the detailed mechanisms underlying this finding remain to be elucidated [25,26,27]. However, our findings for RhoGDI indicate the mechanism of regulation of AMPK in metabolism. We observed that knockdown of RhoGDI promoted the phosphorylation of AMPK, and that TR08 targeted RhoGDI by activating the AMPK pathway. In conclusion, our results highlighted a regulatory mechanism based on RhoGDI and identified a molecule that could facilitate metabolic regulation. TR08 showed protective effects against lipid deposition and fibrosis in a murine MASLD model. Moreover, TR08 activated the AMPK pathway, which decreased FFA synthesis and promoted mitochondrial activation in male mice by targeting Arhgdia. TR08 reduced progressive liver fibrosis, providing a potential therapeutic strategy for MASLD treatment. The RhoGDI-targeting approach and the mechanism underlying AMPK regulation can serve as the foundation for new drug design in MASLD treatment.
4. Materials and Methods
4.1. Animals
All mice were housed in isolated ventilated cages in an animal barrier facility at the Hefei University of Science and Technology (Hefei, China). The mice were maintained on a 12/12 h light/dark cycle at 22 °C ± 2 °C with ad libitum access to pellet food and water. Adult male mice were used at various ages, as indicated for the respective experiments.
4.2. Ethics Statements
Animal studies were conducted in compliance with the recommendations in the Guidelines on the Care and Use of Animals for Scientific Purposes of the National Advisory Committee for Laboratory Animal Research. All experimental procedures were approved by Hefei University of Technology (HFUT20210503001). A trained researcher was responsible for conducting all animal experimental procedures in accordance with the laws governing animal research in China. A total of 16 adults who underwent liver biopsy for suspected MASLD were also enrolled in this analysis. The study was approved by the Research Ethics Committee of Putuo Hospital, Shanghai University of Traditional Chinese Medicine (No. PTEC-A-2025-13(S)-1). The severity of liver steatosis, inflammation, and fibrosis was assessed using the non-alcoholic fatty liver disease (NAFLD) activity score (NAS) [28] and NAFLD fibrosis stage by an experienced senior pathologist who was blinded to the clinical information of the patients.
4.3. Generation of Genetically Modified Mice
Arhgdia^fl/+^ mice were purchased from GemPharmatech Co., Ltd. (Shanghai, China). Arhgdia^fl/fl^ mice were generated using the CRISPR/Cas9 system and Cre-loxP-mediated recombination technology. First, two single-guide RNAs (sgRNA1 and sgRNA2) were used to target a fragment of Arhgdia exon 1–6. Then, the single-stranded oligodeoxynucleotides (ssODNs), sgRNAs, and Cas9 mRNA were injected into zygotes (GenePharmatech, Shanghai, China). We selected one founder mouse and crossed it with a C57BL/6J mouse to generate Arhgdia^fl/fl^ mice. Arhgdia ^fl/fl^ mice were mated with B6.FVB (129)-Tg (Alb-cre) mice, where the Cre was directed to specifically express in the liver, to generate Arhgdia ^fl/fl, Alb^ mice, and Arhgdia ^fl/fl^ mice served as controls.
4.4. HFD Diets Feeding, AAV8 Injection, and Pharmacological Treatment
The MAFLD model was established by feeding 8-week-old C57/BL6N male mice at a high-fat diet (HFD; 60 kcal% fat, 20 kcal% protein, 20 kcal% carbohydrate; D12492; Research Diets, New Brunswick, NJ, USA) for 12 weeks in the evaluation of the efficiency of compound TR08 (10 mg/kg/d, og) in the model and the TR08 treated group. CCl_4_ was diluted in olive oil at a ratio of 1:9 to prepare a 10% (v/v) solution. Mice received injections at a dose of 5 µL/g body weight (equivalent to 0.5 µL/g of pure CCl_4_) twice per week for 4 weeks. A GAN diet (40 kcal% Fat, 20 kcal% Fructose and 2% Cholesterol; D09100310; Research Diets, New Brunswick, NJ, USA) was administered for 12 weeks in the Arhgdia^fl/fl,Alb^ mice group, and Arhgdia^fl/fl^ mice group. Mice received a tail vein injection of AAV8 (Vigene Biosciences, Jinan, China) at a dose of 1 × 10^12^ vg/mL. The treatment for all experimental groups consisted of either the AAV8 construct or a control plasmid. Mice that were fed with a normal control (NC) diet (Q031, Shanghai Xietong Biologic Science & Technology, Shanghai, China) served as controls. Blood was collected from the retro-orbital plexus under isoflurane anesthesia. The collected whole blood was centrifuged for subsequent biochemical analysis.
4.5. Cell Culture and Treatment
The HepG2 cell line was cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 100 μg/mL streptomycin, and 100 U/mL penicillin at 37 °C in a humidified atmosphere containing 5% CO_2_. To establish a cellular model of lipid accumulation, a modeling agent was prepared by complexing palmitic acid (PA) and oleic acid (OA) with 5% bovine serum albumin (BSA) to a final concentration of 5 mM for each fatty acid. This mixture was then applied to the culture system at a final concentration of 1 μM. Cells were subsequently treated with TR08 at a final concentration of 20 μM for 24 h. For in vitro coculture assays, HepG2 cells (obtained from Percell, Wuhan, China) were used. To investigate the function of Arhgdia, HepG2 cells at 60–70% confluence were transfected with either si-Arhgdia or a scramble control using Lipofectamine 3000 (L3000015, Thermo Scientific, Waltham, MA, USA) in serum-free medium according to the manufacturer’s protocol. After 6 h of transfection, the medium was replaced with complete medium to minimize cytotoxicity. Protein expression was evaluated 48 h post-transfection by Western blotting.
4.6. Western Blot Analysis
HepG2 cells were seeded in a 6-well plate and liver lysates were subjected to sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis, transferred to nitrocellulose or polyvinylidene difluoride (PVDF) membranes, and then blotted with antibodies. The immunoblots were visualized by chemiluminescence using an enhanced chemiluminescence Western blotting system (Bio-Rad Laboratories, Shanghai, China). The antibodies used in this study are indicated in Table 1. Quantification was performed by measuring the band intensities using Image J software version 2.14.0.
4.7. Glucose and Insulin Tolerance Tests
The oral glucose tolerance test (OGTT) was conducted one week before euthanasia. After a 16 h fast, the animals received a glucose solution (2 g/kg body weight) by oral gavage. For the insulin tolerance test (ITT), the animals were fasted for 6 h and then administered insulin (0.75 U/kg body weight) via intraperitoneal injection. Venous tail blood samples were collected at 0, 30, 60, 90, and 120 min post-administration, and blood glucose levels were measured using a glucose meter (OneTouch Verio, Johnson & Johnson, New Brunswick, NJ, USA).
4.8. Biochemical Analysis
The serum levels of aspartate AST, ALT, total TG, and total TC and Glucoses (GLU) were determined with an automated biochemical analyzer (Hitachi 7600, Tokyo, Japan), while TG (A110-1-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China) levels, TC (A111-1-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China) and hepatic FFA (A042-2-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China) levels were measured using commercial assay kits in accordance with the manufacturer’s instructions.
4.9. Immunohistochemistry Analysis
RhoGDI protein expression was assessed in liver tissues from eight patients and normal controls using Immunohistochemistry. Paraffin-embedded sections were subjected to antigen retrieval and incubated overnight at 4 °C with a primary antibody against RhoGDI (1:200, #EPR3773, Abcam, Waltham, MA, USA). Following incubation with a horseradish peroxidase-conjugated secondary antibody at room temperature for 1 h, immunoreactivity was visualized with 3,3′-diaminobenzidine substrate and counterstained with hematoxylin. Stained sections were examined and analyzed under a light microscope.
4.10. H&E Staining
For histological evaluation, liver tissue samples were fixed in 4% paraformaldehyde at room temperature for 24 h, dehydrated through a graded ethanol series, cleared in xylene, and embedded in paraffin. Consecutive sections of 5 μm thickness were cut using a microtome (Leica RM2235, Leica, Wetzlar, Germany). After deparaffinization and rehydration, the sections were sequentially stained with hematoxylin (D006-1-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China) for 5 min and eosin for 1 min. Following dehydration through an ethanol gradient and clearing in xylene, the sections were mounted with neutral resin. Stained sections were examined for histomorphology and images were captured under a light microscope (DM2500, Leica, Wetzlar, Germany).
4.11. Masson’s Trichrome Staining
Masson’s trichrome staining was performed to assess collagen deposition in liver tissues. Deparaffinized sections were mordanted in Bouin’s solution overnight, followed by sequential staining with Weigert’s iron hematoxylin (10 min), Biebrich scarlet-acid fuchsin (10 min), phosphomolybdic–phosphotungstic acid (10 min), and aniline blue (5 min). After dehydration and mounting, collagen fibers (blue), muscle fibers (red), and nuclei (blue-black) were visualized under a light microscope (DM2500, Leica, Wetzlar, Germany). The collagen area percentage was quantified using ImageJ software (version 1.53) from three randomly selected fields per section.
4.12. Oil Red O Staining
Liver cryosections (8–10 μm thick) were air-dried and fixed in pre-chilled 4% paraformaldehyde for 15 min, followed by a brief rinse in 60% isopropanol. Sections were then stained with a filtered Oil Red O working solution for 30 min at room temperature. After thorough rinsing in 60% isopropanol and distilled water to remove background dye, nuclei were counterstained with Mayer’s hematoxylin for 1 min. Finally, sections were washed under running tap water, and examined under a light microscope.
4.13. Mitochondrial Staining
HepG2 cells cultured on coverslips were incubated with MitoTracker™ Deep Red (M22672, Thermo Fisher Scientific, Waltham, MA, USA) at a working concentration of 100 nM in serum-free medium for 30 min at 37 °C in the dark. After staining, cells were washed twice with warm PBS, fixed with 4% paraformaldehyde for 15 min, and mounted with ProLong™ Gold Antifade Mountant containing DAPI. Fluorescence images were captured using a confocal microscope (Leica TCS SP8). Mitochondrial morphology and distribution were analyzed with ImageJ software.
4.14. Quantitative Real-Time PCR
Total RNA was extracted from tissue or cell samples using FreeZol Reagent (R711, Vazyme, Nanjing, China) following the manufacturer’s instructions. RNA was reverse-transcribed into cDNA using HiScript II Q RT SuperMix for qPCR (+gDNA wiper) (R223, Vazyme). Quantitative Real-Time PCR (qPCR) was performed with ChamQ SYBR qPCR Master Mix (Low ROX Premixed) (Q331, Vazyme) on a QuantStudio 5 Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). The primer sequences were as follows: forward: CGGCCAAGAGGAAGGAGTAT; reverse: GCATCCTTGTCATTGGCTTT. Relative gene expression was normalized to GAPDH and calculated using the 2^−ΔΔCT^ method.
4.15. Lipidomic Analysis
Liver tissues (20 mg) were extracted with chloroform–methanol–water (2:1:0.8, v/v/v), dried under nitrogen, and reconstituted in isopropanol–acetonitrile (1:1). Separation was performed on an Agilent 1290 Infinity II UHPLC system using a ZORBAX RRHD Eclipse Plus C18 column (Agilent, Santa Clara, CA, USA) (1.8 μm, 2.1 × 100 mm, 45 °C) with a gradient of mobile phases A (acetonitrile-water with 10 mM ammonium formate) and B (isopropanol-acetonitrile with 10 mM ammonium formate) at 0.3 mL/min over 20 min. Detection was carried out on Agilent 6545 Q-TOF (Agilent, Santa Clara, CA, USA) in positive/negative switching mode (resolution ≥ 40,000, m/z 100–1700). Lipids were identified with LipidSearch 4.2 and quantified by normalization to internal standards (SPLASH^®^ LIPIDOMIX) and tissue weight. This experiment was completed/tested by Bioprofile (Shanghai, China).
4.16. Transcriptomic Analyses
The Transcriptomic analyses were performed by LC-Bio Technology Co., Ltd. (Shanghai China). Using Trizol reagent, total RNA was isolated from the liver tissues of Arhgdia^fl/fl,Alb^ mice and Arhgdia ^fl/fl^ mice with NASH diets, as shown in Figure 1. The RNA quality was checked with Bioanalyzer 2200 (Agilent Technologies, Santa Clara, CA, USA). cDNA libraries were prepared using NEBNext^®^ Ultra™ Directional RNA Library Prep Kit, NEB Next^®^ Poly (A) mRNA Magnetic Isolation Module, and NEB Next^®^ Multiplex Oligos according to the instructions of the manufacturer (New England Biolabs, Ipswich, MA, USA). Genes with fold-change > 2.0 or <0.5 and a false discovery rate < 0.05 were considered to be significantly differentially expressed. Volcano, heat, and bubble maps were generated using the “ggplot2” packages (version 3.3.0) in R (version 3.6.3; The R Project for Statistical Computing, Vienna, Austria). Gene set enrichment analysis (GSEA) was performed using the “enrichplot” package (version 1.8.1).
4.17. Mitochondrial Respiration Analysis
HepG2 cells were plated in an XF96 plate (W21715; Seahorse Biosciences, North Billerica, MA, USA) at a density of approximately 1.0 × 10^4^ cells/well. The cells were equilibrated in DMEM containing 10 mmol/L glucose and 1 mmol/L sodium pyruvate. Hepatic mitochondrial respiration was measured by Seahorse Bioscience XFe 96 Extracellular Flux Analyzer. The baseline oxygen consumption rate (OCR) was recorded, which was followed by continuous injections of the following pharmacologic inhibitors through ports in the XF Assay cartridges: oligomycin (1 mM), an inhibitor of adenosine triphosphate (ATP) synthase that enabled measurements of ATP-coupled oxygen consumption through oxidative phosphorylation (OXPHOS); carbonyl cyanide 4-trifluoromethoxy phenylhydrazone (FCCP) (1.5 μM), an uncoupling agent that produced maximum electron transport and therefore enabled measurement of maximum OXPHOS respiration capacity; and a mixture of antimycin A (1 mM) and rotenone (1 mM), which are mitochondrial complex I and III inhibitors, respectively. The injections were performed during continuous oxygen measurements using Seahorse XFe96 Extracellular Flux Analyzer (Seahorse Biosciences, USA) according to the manufacturer’s instructions.
4.18. Statistical Analysis
Statistical analyses were performed using GraphPad Prism software (version 10.1.2). Continuous parameters were compared using one-way analysis of variance (ANOVA), except for the non-normally distributed data, which were compared using a Kruskal–Wallis H test. Categorical variables were compared using the chi-square test. For the animal and in vitro studies, the data were presented as the mean ± standard error of the mean. An unpaired, two-tailed Student t test was used for two-group comparisons, and one-way ANOVA was used for comparisons among multiple groups, which was followed by a least significant difference (LSD) post hoc test for the differences between any two groups. The asterisks in the figures indicate statistical significance as follows: * p < 0.05; ** p < 0.01.
5. Conclusions
This study demonstrate that hepatocyte-specific Arhgdia deletion confers protection against the progression of MASLD by reducing hepatic lipid accumulation and attenuating fibrotic development. Additionally, these results indicated that RhoGDI inhibition enhances mitochondrial β-oxidation in hepatocytes. These results establish RhoGDI as a critical regulator of MASLD pathogenesis and highlight its potential as a therapeutic target for metabolic liver diseases.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lim A.S. Cruz S.N. Uddin A.A. Malysheva O. Caudill M.A. Alonso C. Montilla A. Karsdal M.A. Leeming D.J. Mayorca-Guiliani A.E. Pilot trials of oral betaine in participants with non-alcoholic fatty liver disease and elevated alanine aminotransferase Hepatology 2025 Erratum in Hepatology 2026, 83, E 9210.1097/HEP.000000000000147741467716 · doi ↗ · pubmed ↗
- 2Neuschwander-Tetri B.A. Non-alcoholic fatty liver disease BMC Med.2017154510.1186/s 12916-017-0806-828241825 PMC 5330146 · doi ↗ · pubmed ↗
- 3Younossi Z.M. Non-alcoholic fatty liver disease—A global public health perspective J. Hepatol.20197053154410.1016/j.jhep.2018.10.03330414863 · doi ↗ · pubmed ↗
- 4Wang C. Shang Y. Kanaan G. Chai L. Li H. Qi X. Silymarin for adults with metabolic dysfunction-associated steatotic liver disease Cochrane Database Syst. Rev.20256 Cd 0155244055256910.1002/14651858.CD 015524.pub 2PMC 12186484 · doi ↗ · pubmed ↗
- 5Raza S. Rajak S. Upadhyay A. Tewari A. Sinha R.A. Current treatment paradigms and emerging therapies for NAFLD/NASH Front. Biosci. (Landmark Ed.)20212620610.2741/489233049668 PMC 7116261 · doi ↗ · pubmed ↗
- 6Saponaro F. Sestito S. Runfola M. Rapposelli S. Chiellini G. Selective Thyroid Hormone Receptor-Beta (TRβ) Agonists: New Perspectives for the Treatment of Metabolic and Neurodegenerative Disorders Front. Med.2020733110.3389/fmed.2020.00331 PMC 736380732733906 · doi ↗ · pubmed ↗
- 7Sheka A.C. Adeyi O. Thompson J. Hameed B. Crawford P.A. Ikramuddin S. Nonalcoholic Steatohepatitis: A Review JAMA 20203231175118310.1001/jama.2020.229832207804 · doi ↗ · pubmed ↗
- 8Jensen M.C.H. Holm C. Jørgensen K.J. Schroll J.B. Treatment for women with postpartum iron deficiency anaemia Cochrane Database Syst. Rev.202412 Cd 0108613967055010.1002/14651858.CD 010861.pub 3PMC 11639052 · doi ↗ · pubmed ↗