miRNA-Mediated Regulation of γ-Globin to β-Globin Switching: Therapeutic Potential in β-Hemoglobinopathies
Daniah Alotaibi, Falak Aldagdog, Sajidah Alramadhan, Basmah Almuhaidib, Nada Asiri, Leena Almodhi, Manar Alshabaan, Razan Alborhan, Chittibabu Vatte, Shamim Shaikh Mohiuddin, Amein K. Alali, Alawi Habara

TL;DR
This paper reviews how miRNAs regulate the switch from fetal to adult hemoglobin, offering potential therapies for β-hemoglobinopathies like sickle cell disease.
Contribution
The paper highlights novel miRNA-mediated regulation and nanoparticle delivery strategies for reactivating fetal hemoglobin.
Findings
miRNAs like miR-144 and miR-26b modulate hemoglobin switching by targeting transcription factors.
Nanoparticle-based delivery systems show promise for therapeutic miRNA application in β-hemoglobinopathies.
Abstract
Erythropoiesis is a tightly regulated developmental process that requires the switch from fetal hemoglobin (HbF) to adult hemoglobin (HbA). In β-hemoglobinpathies such as SCD and β-thalassemia, disease severity is influenced by the fetal-to-adult hemoglobin switch because persistence or induction of HbF will ameliorate the clinical manifestations. miRNAs play an essential role in regulating this switch by modulating the expression levels of key transcription factors, such as BCL11A, KLF1, and MYB, which repress γ-globin expression. Multiple miRNAs have been identified as potential modulators of the hemoglobin switch, including miR-144, miR-486, miR-26b, and miR-15a. The molecular interactions between miRNA and γ-to β-globin switch have the potential for new therapeutic interventions that aim to reactivate HbF expression to ameliorate β-hemoglobinopathies such as SCD and β-thalassemia.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
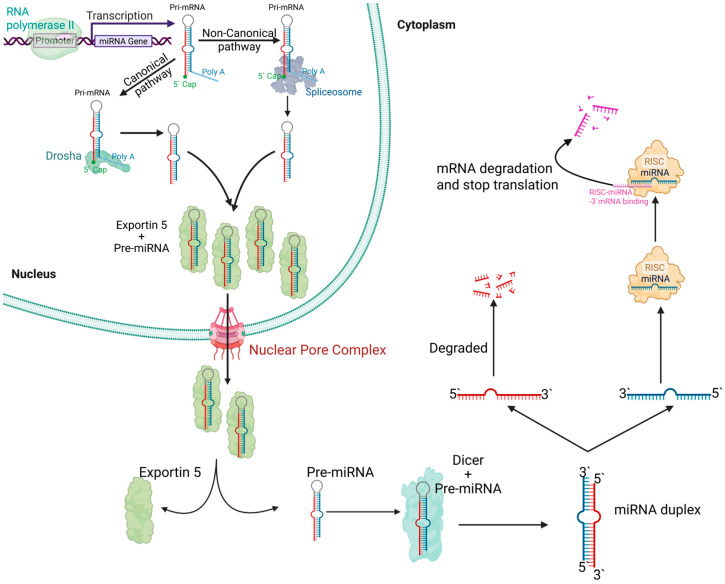 Figure 1
Figure 1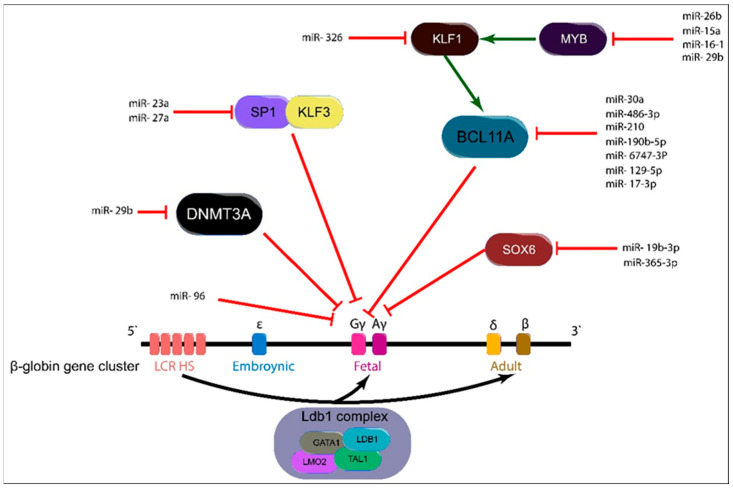 Figure 2
Figure 2- —Imam Abdulrahman Bin Faisal University, Dammam
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHemoglobinopathies and Related Disorders · MicroRNA in disease regulation · Kruppel-like factors research
1. Introduction
β-hemoglobinopathies are genetic disorders affecting the structure or production of hemoglobin (Hb), the oxygen-carrying protein in red blood cells. These disorders arise due to mutations in the HBB or HBA genes, leading to abnormal Hb variants or reduced Hb synthesis. Sickle cell disease (SCD) is an inherited hematologic disorder caused by a single-nucleotide mutation in the HBB gene, which encodes the β-globin subunit of hemoglobin (HBB: c.20A > T) [1,2,3,4,5]. This mutation leads to the substitution of glutamic acid with valine at position six of the β-globin chain (Glu6Val), resulting in the production of sickle hemoglobin (HbS) instead of normal adult hemoglobin (HbA). The disease typically follows an autosomal recessive pattern, where individuals with two mutated copies of HBB (β^s^/β^s^ genotype) develop sickle cell anemia (SCA), whereas those with one mutated and one normal allele (β^S^/β^A^ genotype) are carriers, commonly referred to as having sickle cell trait, and usually do not exhibit disease symptoms [1,2,3,4,6]. Nevertheless, some reports described sickle cell disease caused by a cis “double” mutation on the same HBB allele, for example, HbS-Oman, Hb-Jamaica Plain, and Hb-São Paulo [7,8,9], which are rare and typically have an autosomal dominant inheritance pattern.
At the molecular level, HbS undergoes polymerization under hypoxic conditions, which causes red blood cells (RBCs) to become rigid and deformed into the characteristic sickle shape [1,2,3,4,10]. These misshapen RBCs exhibit reduced deformability and increased adhesiveness, which promotes vascular occlusion and ischemic injury in tissues and organs [1,2,3,4,10]. The mechanical fragility of sickled cells leads to chronic hemolysis, reducing RBC lifespan and contributing to anemia and compensatory erythropoiesis in the bone marrow. The repetitive cycles of vaso-occlusion and hemolysis drive a state of systemic inflammation, endothelial activation, and oxidative stress, which exacerbate the disease burden and contribute to progressive organ damage [11,12]. Additionally, recurrent microvascular occlusion followed by reperfusion results in ischemia–reperfusion (I/R) injury, which drives endothelial activation, inflammation, and multi-organ pathology [11,12,13,14,15]. Reoxygenation converts xanthine oxidoreductase to xanthine oxidase, generating reactive oxygen species and promoting leukocyte recruitment and vascular dysfunction. I/R injury extends beyond the occluded microvascular bed: the lungs are the principal site of damage, with additional involvement of the liver and heart [13,14]. In line with these considerations, nationwide inpatient data from 2020 indicate that SCD patients presenting with acute coronary syndrome are younger [6]. While in-hospital mortality and time to percutaneous coronary intervention (PCI) mirror those of non-SCD patients, SCD is associated with a markedly increased risk of coronary artery dissection during PCI, warranting careful procedural planning [6].
Similarly, β-thalassemia is an autosomal recessive hemoglobinopathy caused by diverse mutations in the HBB gene, resulting in reduced (β^+^) or absent (β^0^) synthesis of β-globin chains [16,17]. Rare autosomal-dominant forms also occur, typically due to unstable β-globin variants; for example, hemoglobin Dieppe (HBB:c.383A > G; β127Gln → Arg), which produces a dominant β-thalassemia phenotype through globin instability [18]. This deficiency leads to an imbalance between α- and β-globin chains, with excess α-globin chains precipitating within erythroid precursors and mature red blood cells. Such precipitation causes ineffective erythropoiesis, apoptosis of erythroid precursors, severe hemolytic anemia, and iron overload secondary to increased intestinal iron absorption and repeated blood transfusions in severe cases [16,17]. Individuals with severe forms of β-thalassemia are often transfusion-dependent, requiring lifelong supportive care, and experience substantial morbidity from chronic anemia, ineffective erythropoiesis, extramedullary hematopoiesis, and organ damage due to iron accumulation [16,17].
miRNAs are small, non-coding RNA molecules, about 20 to 23 nucleotides long [19,20], that regulate gene expression post-transcriptionally by binding to target mRNAs to inhibit translation or trigger degradation [19,20]. They are initially transcribed as long primary transcripts, pri-miRNAs, that can be produced by either RNA polymerase II, which generates capped and polyadenylated transcripts, or RNA polymerase III, which uses distinct promoter and termination elements for transcription [19,20]. There are two main pathways for miRNA processing: the canonical pathway and the non-canonical pathway (Figure 1). In the canonical pathway, pri-miRNAs fold into characteristic hairpin structures and are processed in the nucleus by the Drosha-DGCR8 microprocessor complex to produce precursor miRNAs (pre-miRNAs) [19,20]. These pre-miRNAs are exported to the cytoplasm, where the RNase Dicer cleaves them into a miRNA duplex, one strand of which is then loaded into the RNA-induced silencing complex (RISC) to mediate gene silencing [19,20]. In contrast, non-canonical pathways bypass one or more of these standard processing steps by employing alternative mechanisms, such as splicing-dependent formation of pre-miRNA hairpins or direct Ago2-mediated cleavage, to generate mature miRNAs that are also incorporated into RISC. Despite these differences in biogenesis, both pathways converge on producing functional miRNAs that regulate gene expression via translational repression or mRNA degradation [19,20].
In β-hemoglobinopathies, precise control of globin gene expression is essential for balancing HbF and HbA production. Recent research has demonstrated that epigenetic mechanisms and specific miRNAs are crucial in regulating the developmental switch between γ and β-globin. Certain miRNAs target key transcriptional repressors, such as BCL11A and MYB, as well as epigenetic modifiers like DNA methyltransferases, thereby relieving the suppression of γ-globin expression and enhancing HbF production [21,22,23,24,25]. Moreover, distinct miRNA expression profiles in erythroid cells not only provide insight into the molecular pathways governing hemoglobin switching but also serve as sensitive biomarkers for disease severity in conditions like sickle cell disease. These findings have significant clinical implications, as miRNA-based therapeutic strategies that elevate HbF levels can potentially ameliorate clinical symptoms and improve outcomes in patients with β-hemoglobinopathies [26,27]. Additionally, miRNAs are involved in a broader circRNA–miRNA–mRNA network [25,28]; however, we do not elaborate on miRNA-circRNA interactions here and instead center this review on how miRNAs modulate the γ-to β-globin transition alongside established transcription factors.
2. miRNA-Mediated Upregulation of HbF Expression
In recent years, considerable attention has focused on the role of miRNAs in upregulating HbF expression by modulating key transcription factors that normally repress γ-globin gene expression. Transcription factors such as BCL11A, SOX6, MYB, KLF1, and the SP1-KLF3 axis are critical regulators of the HbF-to-HbA switch; under normal conditions, they suppress HbF levels during adult erythropoiesis [29,30,31]. However, specific miRNAs have been identified that target these repressors, thereby relieving their inhibitory effects and promoting HbF production [31]. We review the current evidence on how miRNAs that target BCL11A, SOX6, MYB, KLF1, SP1-, and KLF3 contribute to the reactivation of γ-globin expression, highlighting their potential as therapeutic agents for β-hemoglobinopathies such as sickle cell disease and β-thalassemia (Figure 2).
2.1. miRNA-BCL11A Axis
The BCL11A gene, located on chromosome 2p16.1 and comprising four exons and three introns, encodes a zinc-finger protein that functions as a transcriptional repressor [29,30]. BCL11A is the central transcription factor in the formation of the BCL11A-MBD2-NuRD complex, which represses γ-globin formation [2,32,33,34,35]. The extra-long isoform, BCL11A-XL, is the predominant variant in adult erythroblasts [33,36]. The BCL11A-XL isoform C-terminal zinc fingers (ZnF4-6) bind the HBG promoters, leading to silencing of γ-globin production [33,36]. Reduction in the BCL11A-XL isoform or disruption of ZnF4-6 derepresses γ-globin and increases HbF [33,36]. The shorter BCL11A isoforms, predominant in fetal and primitive erythroid cells, lack the XL-specific ZnF4-6 domain and therefore lack the repressive capacity to silence the HBG promoters [36].
Several miRNAs converge on BCL11A to modulate HbF levels. In β-thalassemia intermedia, miR-30a-5p binds the 3′ untranslated region of BCL11A mRNA, reducing its stability and translation and thereby increasing HbF; elevated miR-30a in erythroid precursors correlates strongly with higher HbF, underscoring the therapeutic potential of the miR-30a-BCL11A interaction [23].
Convergent evidence also supports a miR-210–BCL11A pathway. In CD34^+^-derived erythroid precursors, transfection with miR-210 induces a dose-dependent reduction in the BCL11A-XL isoform at mRNA and protein levels, with a corresponding rise in γ-globin expression [22,37]. Importantly, flow cytometry analysis shows that key erythroid markers, such as CD71 and CD235a, remain unchanged after miR-210 treatment, indicating that the modulation is specific to the globin gene switch rather than affecting overall erythroid differentiation [37]. These findings underscore the therapeutic potential of targeting miR-210 to elevate HbF levels and improve clinical outcomes in β-hemoglobinopathies like β-thalassemia.
Next, miR-486-3p regulates HbF production by directly modulating BCL11A-XL in human erythroid cells. In primary human CD34^+^-derived erythroid cell culture, miR-486-3p is minimally expressed in early progenitors but becomes significantly upregulated during erythroid differentiation [38]. Overexpression of miR-486-3p results in a ~40% reduction in BCL11A protein levels and a concomitant increase in γ-globin mRNA and protein [38]. Conversely, inhibition of miR-486-3p leads to higher BCL11A levels and reduced γ-globin expression. Furthermore, erythroid cells from β-thalassemia patients show significantly higher miR-486-3p expression, which correlates with increased HbF synthesis. These findings indicate that miR-486-3p is a potent post-transcriptional mechanism controlling HbF levels and a promising therapeutic target for β-hemoglobinopathies [38]. Additionally, miR-486-3p has a modest prediction value to differentiate patients with STEMI from those with stable ischemic heart disease [39]. miR-486-3p dual relevance to globin regulation and coronary events positions it as a plausible diagnostic and prognostic tool across β-hemoglobinopathies who suffer from acute coronary syndrome.
Another relevant miRNA, miR-190b-5p, significantly upregulates HbF production. In pediatric β-thalassemia, miR-190b-5p is markedly upregulated in peripheral blood and shows a significant inverse correlation with BCL11A mRNA levels while positively correlating with increased HbF. Luciferase reporter assays confirmed that miR-190b-5p directly binds to the 3′-UTR of BCL11A, leading to its downregulation and consequent enhancement of HbF production. Additionally, ROC analysis indicates that hsa-miR-190b-5p has a diagnostic value comparable to HbA2, and combining both markers further improves diagnostic accuracy. These findings underscore the clinical relevance of miR-190b-5p as a potential biomarker and therapeutic target for modulating HbF levels in pediatric β-thalassemia [40].
Also relevant is miR-6747-3p; it significantly upregulates HbF production in β-thalassemia. miR-6747-3p was found to be markedly upregulated in patients with β-thalassemia major relative to healthy controls, with its expression positively correlating with increased HbF levels. Functional assays in HUDEP-2 and K562 demonstrated that overexpression of miR-6747-3p not only impairs cell proliferation and promotes apoptosis but also accelerates erythroid differentiation, leading to enhanced γ-globin expression. Mechanistically, miR-6747-3p directly binds to the 546–552 nucleotide region of the BCL11A mRNA 3′-UTR, thereby downregulating this key transcriptional repressor of fetal hemoglobin. These findings underscore the potential of targeting the miR-6747-3p-BCL11A axis as a novel therapeutic strategy to induce HbF production in β-thalassemia [41].
Another miRNA, miR-129-5p, is upregulated in β-thalassemia and associated with higher HbF [42]. Overexpression of miR-129-5p in erythroid cell models, K562 and HUDEP-2, demonstrates that miR-129-5p directly targets the BCL11A 3′-UTR, leading to BCL11A downregulation and, in turn, inducing HbF production. Additionally, miR-129-5p promotes erythroid maturation [42]. miR-129-5p level was also found to be inversely associated with serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) [42], supporting its use as a noninvasive marker of hepatocellular injury. Taken together, these data support evaluating miR-129-5p both as a therapeutic target for HbF modulation and as a biomarker in β-hemoglobinopathies, including for monitoring liver damage.
miR-17-3p is upregulated in peripheral blood from β-thalassemia patients and positively correlates with HbF; it also shows good discrimination between patients and controls, AUC ≈ 0.88, and between high vs. low HbF cases, AUC ≈ 0.93 [43]. It directly targets BCL11A 3′UTR, lowering BCL11A mRNA and protein, thereby increasing γ-globin in K562 cells [43]. Overall, miR-17-3p is a promising candidate for HbF-inducing therapy and a potential biomarker for monitoring β-thalassemia.
2.2. miRNA-MYB-KLF1-BCL11A Axis
The MYB-KLF1-BCL11A axis is an upstream pathway that enforces the switch from HbF to HbA [44,45]. MYB indirectly regulates γ-globin by activating KLF1 [3], which then silences γ-globin by upregulating the repressor BCL11A [3,44,46] (Figure 2).
Hydroxyurea involves this axis via miR-26b; hydroxyurea elevates miR-26b, which binds to MYB mRNA at its 3′-UTR and promotes RISC-mediated decay, lowering MYB protein level, which in turn lowers KLF1 and BCL11A; γ-globin is consequently derepressed, and HbF increases [47]. Consistently, enforced miR-26b induces γ-globin in K562 erythroid cells [47,48].
Another miRNA, miR-15a/16-1, acts similarly to regulate HbF expression. Overexpression of miR-15a and miR-16-1 in primary CD34^+^ cells and K562 cells using lentiviral vectors robustly increased γ-globin and embryonic ε-globin levels. Luciferase reporter assays confirmed that these miRNAs directly target the MYB 3′-UTR, and Western blot analyses demonstrated a reduction in MYB protein [49]. Complementary shRNA-mediated knockdown of MYB similarly elevated HbF, linking miR-15a/16-1 suppression of MYB and the consequent decrease in BCL11A activation to enhanced γ-globin synthesis [49].
Another miRNA, miR-29b, inhibits the de novo synthesis of the DNMT enzyme, specifically DNMT3A, which leads to the upregulation of HbF expression in primary human CD34^+^ derived erythroid cell culture [50]. Further investigation of miR-29b revealed that it also inhibits MYB in KU812 leukemia cells, primary human CD34^+^ derived erythroid cells, and in the Townes SCD mouse model [51].
Some miRNAs work through KLF1 directly; an example of this is miR-326, which targets the KLF1 3′-UTR, lowering KLF1 protein; because KLF1 activates BCL11A, this downshifts BCL11A and can derepress γ-globin and increase HbF [24] (Figure 2). In K562 cells, miR-326 overexpression increased HbF. In primary CD34^+^ progenitors, miR-326 reduced KLF1 protein and BCL11A mRNA but did not significantly change γ- or β-globin transcripts; however, it reduced erythroid maturation (lower glycophorin-A), indicating context-dependent effects. In β-thalassemia major reticulocytes, higher miR-326 was positively correlated with HbF [24], consistent with a role in stress erythropoiesis and supporting its evaluation as a therapeutic target and biomarker.
2.3. miRNA-SOX6-BCL11A Axis
The SOX6 gene is located on chromosome 11p15.2 [52]. SOX6 is an HMG-box transcription factor that collaborates with BCL11A to enforce γ-globin silencing during definitive erythropoiesis [52,53]. In adult human erythroid progenitors, SOX6 and BCL11A are coexpressed, physically interact, co-occupy the β-globin cluster, and act together to repress γ-globin transcription [54]. Consistent with a role in the mature erythroid program, SOX6 overexpression accelerates human erythroid differentiation, highlighting its importance in late erythropoiesis [54]. Conversely, lowering SOX6 can derepress γ-globin. Lentiviral RNAi knockdown increased γ-globin in K562 cells and in erythroblasts derived from normal donors and from patients with β-thalassemia major without impairing maturation, and CRISPR/Cas9 disruption of the SOX6 binding domain in K562 cells raised γ-globin mRNA levels [54,55,56].
In a mouse fetal liver model, complete loss of BCL11A led to significant upregulation of miR-365-3p, which directly targets the 3′ UTR of SOX6, reducing its expression and derepressing embryonic and fetal globin genes [57]. In human HUDEP-2 erythroid cells, overexpression of miR-365-3p similarly decreased SOX6 levels at both the mRNA and protein levels, resulting in a marked increase in fetal globin expression [57]. Additionally, luciferase reporter assays in HeLa cells confirmed that miR-365-3p directly binds to a conserved seed region within the SOX6 3′ UTR. These results delineate an evolutionarily conserved regulatory pathway involving BCL11A, miR-365-3p, and SOX6 that modulates hemoglobin switching and may offer new therapeutic avenues for β-hemoglobinopathies [57].
Another miRNA involved in the SOX6 pathway is miR-19b-3p. It modulates HbF levels in β-thalassemia. miR-19b-3p was significantly up-regulated in β-thalassemia carriers with high HbF, and this upregulation correlates with a marked downregulation of SOX6 expression. Functional assays, including RNA immunoprecipitation and dual-luciferase reporter experiments, confirm that miR-19b-3p directly binds to the 3′-UTR of SOX6, leading to its post-transcriptional repression. This interaction relieves the inhibitory effect of SOX6 on fetal globin gene expression, thereby contributing to HbF induction [58].
2.4. miRNA-SP1/KLF3 Axis
Specificity protein 1 (Sp1) is a transcription factor. It binds GC/GT boxes across the β-globin locus control region (LCR) and the ε/γ/β promoters and represses β-like globin gene expression [59]. In undifferentiated cells, Sp1 occupancy helps recruit HDAC1, keeping histones less acetylated and chromatin closed; upon differentiation, Sp1 becomes phosphorylated and dissociates from HS2–HS4 and the γ-globin promoter, allowing FKLF2 to recruit the histone acetyltransferase and increase histone acetylation, thereby enabling β-like globin gene activation [59].
miR-23a and miR-27a promote the expression of globin genes within the β-globin locus by directly repressing the transcriptional repressors SP1 and KLF3 [60]. As these miRNAs increase, SP1/KLF3 occupancy across the locus decreases, relieving repression and increasing ε- and γ-globin in K562 cells, whereas in primary CD34^+^ erythroid cultures the dominant effect is β-globin induction [60]. Because KLF3 also binds and represses the promoter of the miR-23a/27a/24-2 cluster, miR-23a further lowers KLF3, creating a positive feedback loop that amplifies both miRNA levels and globin gene expression [60]. These differing responses reflect the developmental stage-specific programming of the β-globin locus; K562 cells exhibit an embryonic/fetal-like (ε/γ-biased) phenotype, whereas CD34^+^ cultures mature toward the adult program (β-predominant).
2.5. miR-96-γ-Globin Axis
The miR-96-γ-globin axis operates through direct post-transcriptional repression, modulating HbF production. In adult erythroid reticulocytes, where HbF levels are low, a substantial portion of γ-globin mRNA is sequestered within AGO2-containing RNA-induced silencing complexes, a phenomenon that is markedly less pronounced in umbilical cord blood reticulocytes, which exhibit high HbF levels. Ex vivo erythropoiesis models using cord blood-derived and bone marrow-derived erythroblasts demonstrate that overexpression of miR-96 results in approximately a 50% reduction in c-globin protein expression, while knockdown of miR-96 leads to a 20% increase [61]. Moreover, luciferase reporter assays in HEK293T cells reveal that miR-96 directly binds to a highly complementary, seedless target site within the open reading frame of γ-globin mRNA. Collectively, these findings indicate that miR-96 plays a critical role in fine-tuning HbF levels by inhibiting γ-globin translation during human erythropoiesis [61]. Table 1 summarizes miRNAs and their targets.
3. Use of miRNA as Therapy for β-Hemoglobinopathies
RNA interference-based approaches using lentiviral vectors encoding short hairpin miRNA (shmiR) represent promising therapeutic strategies for β-hemoglobinopathies by achieving erythroid-specific knockdown of critical transcriptional repressors such as BCL11A. A double shmiR lentiviral vector that simultaneously targets BCL11A and ZNF410, two key repressors of γ-globin [69]. This dual approach achieves greater HbF induction than either single vector alone, reaching ~49% HbF compared with ~40% using BCL11A-only shmiR and lower levels with ZNF410-only shmiR [69]. In sickle cell patients, derived hematopoietic stem cells, HbF increased from ~32% with BCL11A shmiR to ~45% with the double shmiR, accompanied by reduced erythrocyte sickling and improved erythroid maturation [69].
Additionally, a novel bifunctional lentiviral vector incorporating shmiRs targeting both BCL11A and ZNF410 [69], combined with a modified anti-sickling β-globin (β^AS3^-globin) gene, demonstrated significant induction of HbF, robust anti-sickling effects, and reduced erythrocyte sickling in CD34^+^ hematopoietic stem and progenitor cells from SCD patients, as well as in the Berkeley sickle cell mouse model [70,71].
Currently, the only shmiR-based therapy to reach the clinic is BCH-BB694, an erythroid-restricted vector that silences BCL11A. In the ongoing NCT03282656 trial, treated patients have demonstrated reliable engraftment, durable BCL11A knockdown, and HbF induction ranging from ~20–41%, accompanied by clinical improvement and, importantly, no evidence of clonal dominance to date [21,72]. Collectively, these findings support the use of RNA interference-based silencing of BCL11A as a clinically effective strategy for inducing fetal hemoglobin production in sickle cell disease and related β-hemoglobinopathies.
Beyond vector-based RNA-interference strategies, systemically delivered miRNA therapeutics can induce HbF without genomic integration. In a Townes sickle cell mouse model, daily subcutaneous cholesterol-conjugated miR-29b for four weeks increased HbF level, expanded HbF-positive red cells, improved spleen pathology, and did not alter blood counts [73]. Mechanistically, the treatment reduced DNMT3A and demethylated the γ-globin promoter, consistent with HbF reactivation [73].
The presence of the erythroid-specific enhancer and transcript puts BCL11A at an advantage over other HbF transcription factors. Functional studies identify the long isoform, BCL11A-XL, as a key mediator of γ-globin silencing/HbF repression during erythroid maturation [30,74]. MYB is also regulated by erythroid-active distal enhancers in the HBS1L–MYB intergenic region that influence HbF levels [75], but MYB lacks the erythroid-specific isoform, which makes BCL11A a much more suitable candidate for miRNA targeting than MYB.
4. Use of Nanoparticles as a Delivery Strategy for miRNA-Based Therapies
Nanoparticles are engineered, nanoscale carriers designed to encapsulate or conjugate therapeutic cargo, such as small molecules or nucleic acids, and deliver it to specific cells or tissues [76,77,78]. Nanoparticles protect the cargo from degradation and enable targeted uptake and controlled release. Therefore, nanoparticle delivery can improve bioavailability and therapeutic efficiency while helping to limit systemic exposure and off-target effects [76,77,78].
To induce HbF safely, therapeutic nanoparticles must reach erythroid precursors in the bone-marrow niche rather than mature red cells, which are anucleate and cannot change gene expression. A practical design first biases particles toward the marrow by engaging chemokine and adhesion cues on the sinusoidal endothelium and then exploits transferrin receptor-1 (CD71) on developing erythroblasts for endocytic entry, with glycophorin A (CD235a) enhancing selective binding and retention within erythroid islands [76,77,78]. Therapeutic constructs intended to elevate HbF, such as antisense oligonucleotides directed at the BCL11A enhancer, miRNA modulators, or genome-editing systems, should be engineered for erythroid selectivity and formulated with endosomal-escape functionality to ensure productive cytosolic/nuclear entry at the site of action.
In practice, in vivo HbF induction remains limited by delivery, as the liver and spleen clear most nanoparticles before they reach the bone marrow. Even with homing ligands, marrow enrichment is modest. CD71 supports uptake but is not erythroid-specific, creating off-target sinks, whereas glycophorin A improves localization but does not drive internalization. After receptor-mediated endocytosis, endosomal escape is a major bottleneck. Current HbF-inducing constructs, such as BCL11A enhancer silencers and genome editors, are relatively large and labile, which complicates formulation and elevates safety concerns. A recent in vivo demonstration of CD117-targeted lipid nanoparticles delivering RNA directly to bone marrow hematopoietic stem and progenitor cells suggests that marrow access and receptor-mediated entry are technically achievable. However, off-target biodistribution, antibody/chemistry dependence, and rodent model constraints temper immediate translational claims [79]. Together with the high efficacy of ex vivo HSPC gene therapy [80,81], these constraints explain why in vivo nanoparticle strategies for β-hemoglobinopathies have not yet translated to the clinic. Table 2 summarizes some of the known nanoparticles targeting HSPCs.
By contrast, RBC-based nanoparticles, drug-loaded RBC carriers, RBC–membrane–coated nanoparticles, and nano-erythrosomes are optimized for prolonged intravascular residence via self-markers, such as CD47, thereby minimizing reticuloendothelial clearance [65]. They are poorly suited to HbF induction but align with adjunct management of acute vaso-occlusive crisis: long-circulating, hemocompatible carriers can deliver anti-adhesion agents that attenuate P-/E-selectin and VCAM-1–mediated interactions, antioxidants or heme-scavenging moieties that mitigate endothelial injury, deoxyribonuclease to dismantle neutrophil extracellular traps, and stimulus-responsive nitric oxide donors to improve microvascular perfusion.
5. Discussion
The regulation of Hb switching from HbF (α_2_γ_2_) to HbA (α_2_β_2_) represents a central therapeutic target in β-hemoglobinopathies such as SCD and β-thalassemia [1,12]. Mounting evidence suggests that miRNAs play a crucial role in regulating this process post-transcriptionally, acting through the coordinated repression of transcription factors and epigenetic modulators that normally silence γ-globin expression [21,22,23,34,40,42,47,48,49,51,61,66,71]. Among these, miRNAs targeting BCL11A are particularly well characterized and include miR-30a-5p, miR-210, miR-486-3p, miR-190b-5p, miR-6747-3p, miR-129-5p, and miR-17-3p consistently derepress γ-globin, highlighting BCL11A as the most intensively studied and therapeutically validated node [22,23,37,38,39,40,41,62,63,64,65].
Other critical pathways include the MYB–KLF1–BCL11A axis, where miRNAs such as miR-26b, miR-15a/16-1, miR-29b, and miR-326 modulate upstream regulators, and the SOX6 pathway, where miR-365-3p and miR-19b-3p relieve repression of γ-globin transcription [14,24,47,48,49,50,51,57,68]. Additional miRNAs, such as miR-23a/27a and miR-96, further expand the regulatory network, underscoring the complexity and redundancy of miRNA-mediated control of globin gene expression. Table 1 summarizes the miRNAs and their targeted axes.
SP1 is another transcription repressor for the β-globin locus. Sp1 helps keep chromatin closed in immature cells, so reducing Sp1 activity favors β-like globin gene activation during erythroid maturation. miR-23a and miR-27a boost globin expression by directly suppressing the repressors SP1 and KLF3, relieving repression across the locus; the exact globin genes induced (ε/γ vs. β) depend on the developmental state of the cells, and a positive feedback loop via KLF3 further amplifies this pro-globin effect [60].
By selectively repressing γ-globin silencers, miRNAs can increase HbF to levels known to mitigate hemoglobin polymerization, reduce sickling, and improve erythrocyte survival. Clinical observations have already confirmed that patients with naturally elevated HbF, whether due to genetic modifiers or stress erythropoiesis, exhibit milder disease phenotypes. Harnessing miRNAs to reproduce this protective state represents a promising strategy to modify the disease course. Moreover, the dual roles of certain miRNAs, such as miR-486-3p (with a prognostic value in cardiovascular disease) and miR-129-5p (as a biomarker of liver injury), highlight their potential as both therapeutic targets and diagnostic/prognostic tools.
The translation of these insights into therapy has been facilitated by advances in RNA interference and gene therapy technologies. Vector-based strategies encoding shmiRs against BCL11A and ZNF410, as well as lentiviral constructs like BCH-BB694 [69], demonstrate robust HbF induction in both preclinical models and clinical trials. Parallel approaches using chemically modified, systemically delivered miRNAs (e.g., miR-29b mimics) provide an alternative strategy that avoids permanent genomic modification while still inducing HbF. These complementary platforms illustrate the versatility of miRNA-based therapeutics and raise the possibility of tailored interventions based on patient-specific disease severity, comorbidities, or treatment accessibility.
Compared with ex vivo autologous HSC gene therapy, which requires stem cell harvesting, ex vivo manipulation, reinfusion, and conditioning regimens, such as myeloablation, that can cause substantial toxicity and limit broad applicability [79,81]. The miRNA-based therapy approach could be positioned as a more scalable, pharmacologic alternative if efficient erythroid/HSPC targeting is achieved [79]. Moreover, individualized manufacturing and specialized centers contribute to prohibitive costs and access barriers for ex vivo approaches, whereas drug-like miRNA platforms may better support standardized production and dose adjustment or repeat administration [79].
Nevertheless, several challenges remain. First, the pleiotropic nature of miRNAs means that off-target effects must be carefully evaluated, particularly given their roles in erythropoiesis, immune modulation, and other tissue-specific pathways. Second, the long-term safety and durability of HbF induction remain critical questions, especially for systemically delivered miRNAs. Third, inter-patient variability in miRNA expression profiles suggests that personalized strategies may be required for optimal benefit. Addressing these challenges will require integrated approaches combining molecular profiling, preclinical validation, and carefully designed clinical studies [86].
6. Conclusions
miRNAs have emerged as central regulators of globin gene expression, orchestrating a network of transcriptional and epigenetic pathways that control the HbF-to-HbA switch. Experimental and clinical data consistently demonstrate that targeting repressors such as BCL11A, MYB, KLF1, and SOX6 through specific miRNAs can effectively reactivate γ-globin expression and increase HbF levels. These findings not only deepen our understanding of hemoglobin regulation but also provide a rational framework for the development of miRNA-based therapeutics.
As the field advances, miRNA-directed therapies, whether via viral vectors or nanoparticles, hold significant promise for transforming the management of β-hemoglobinopathies. By restoring HbF, such therapies can address the root pathophysiology of SCD and β-thalassemia rather than merely alleviating symptoms. In addition, the diagnostic and prognostic potential of miRNAs as biomarkers further enhances their clinical utility. Future research should focus on optimizing delivery strategies, ensuring long-term safety, and integrating miRNA-based approaches with existing treatments such as hydroxyurea and gene editing. Ultimately, miRNA therapeutics represent a powerful and innovative avenue toward disease-modifying and potentially curative interventions for β-hemoglobinopathies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Habara A. Steinberg M.H. Minireview: Genetic basis of heterogeneity and severity in sickle cell disease Exp. Biol. Med.201624168969610.1177/1535370216636726 PMC 495038326936084 · doi ↗ · pubmed ↗
- 2Lettre G. Bauer D.E. Fetal haemoglobin in sickle-cell disease: From genetic epidemiology to new therapeutic strategies Lancet 201638725542564 Erratum for: Lancet 2016, 388, 137610.1016/S 0140-6736(15)01341-027353686 · doi ↗ · pubmed ↗
- 3Habara A.H. Shaikho E.M. Steinberg M.H. Fetal hemoglobin in sickle cell anemia: The Arab-Indian haplotype and new therapeutic agents Am. J. Hematol.2017921233124210.1002/ajh.2487228736939 PMC 5647233 · doi ↗ · pubmed ↗
- 4Kostamo Z. Ortega M.A. Xu C. Feliciano P.R. Budak E. Lam D. Winton V. Jenkins R. Venugopal A. Zhang M. Base editing Hb S to Hb G-Makassar improves hemoglobin function supporting its use in sickle cell disease Nat. Commun.202516144110.1038/s 41467-025-56578-339920120 PMC 11806015 · doi ↗ · pubmed ↗
- 5Mahadevia H. Ponvilawan B. Madan U. Sharma P. Qasim H. Shrestha A. A review on disease modifying pharmacologic therapies for sickle cell disease Ann. Hematol.202510488189310.1007/s 00277-025-06216-139828781 PMC 11971234 · doi ↗ · pubmed ↗
- 6Alharbi A. Pena C. Mhanna M. Spencer C. Bashar M. Cherian M. Abdulrahman A. Alfatlawi H. Kwak E.S. Siddique M. The impact of sickle cell disease on acute coronary syndrome and PCI outcomes: A retrospective observational study Hearts 2024523624510.3390/hearts 5020016 · doi ↗
- 7Jorge S.E. Petruk A.A. Kimura E.M. Oliveira D.M. Caire L. Suemasu C.N. Silveira P.A. Albuquerque D.M. Costa F.F. Skaf M.S. Hb S-São Paulo: A new sickling hemoglobin with stable polymers and decreased oxygen affinity Arch. Biochem. Biophys.2012519233110.1016/j.abb.2012.01.00122244832 · doi ↗ · pubmed ↗
- 8Geva A. Clark J.J. Zhang Y. Popowicz A. Manning J.M. Neufeld E.J. Hemoglobin Jamaica Plain—A sickling hemoglobin with reduced oxygen affinity N. Engl. J. Med.20043511532153810.1056/NEJ Moa 04077115470216 · doi ↗ · pubmed ↗