The Complete Chloroplast Genomic Characteristics and Phylogenetic Analysis of Abutilon theophrasti Medicus
Changli Chen, Xiahong Luo, Ziyi Zhu, Xingcai An, Junyuan Dong, Qingqing Ji, Tingting Liu, Lina Zou, Shaocui Li, Jikang Chen, Xia An

TL;DR
This paper analyzes the chloroplast genome of Abutilon theophrasti to understand its genetic structure and evolutionary relationships with other Malvaceae plants.
Contribution
The study provides a complete chloroplast genome sequence and phylogenetic analysis of A. theophrasti, revealing insights into its genetic evolution.
Findings
The chloroplast genome of A. theophrasti is 160,440 bp with a GC content of 36.89% and a typical tetrad structure.
Codon preference analysis shows leucine is the most frequently used amino acid, with 31 codons having RSCU > 1.
Phylogenetic analysis shows A. theophrasti is most closely related to A. indicum and forms a clade with M. cathayensis and Malva crispa.
Abstract
To clarify the phylogenetic relationship between Abutilon theophrasti M. and other Malvaceae plants, the chloroplast genome of A. theophrasti was assembled, annotated, and analyzed. The complete chloroplast genome was sequenced using the Illumina NovaSeq 6000 platform. Bioinformatics methods were employed to systematically analyze its genomic structure, repetitive sequences, nucleic acid diversity, and codon preference. Additionally, a phylogenetic tree was constructed by integrating chloroplast genomic sequences from other Malvaceae species. The results showed that the chloroplast genome of A. theophrasti was 160,440 bp in length with a GC content of 36.89%, exhibiting a typical tetrad structure. A total of 130 coding genes were annotated, including 85 mRNA genes, 37 tRNA genes, and 8 rRNA genes, with no pseudogenes detected. Codon preference analysis indicates that leucine (Leu) is…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
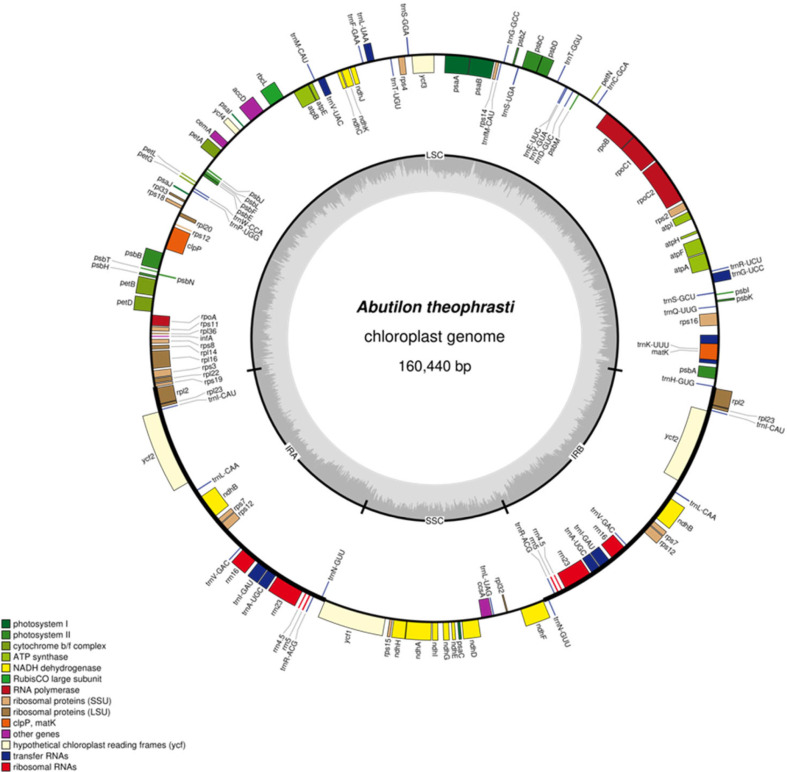 Figure 1
Figure 1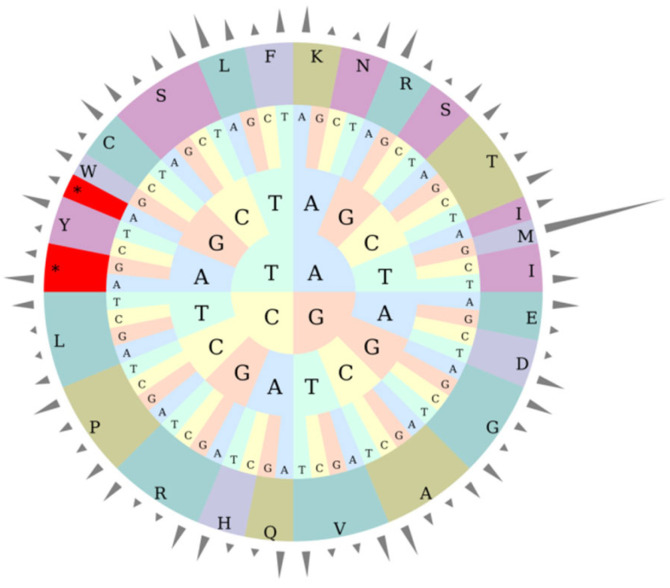 Figure 2
Figure 2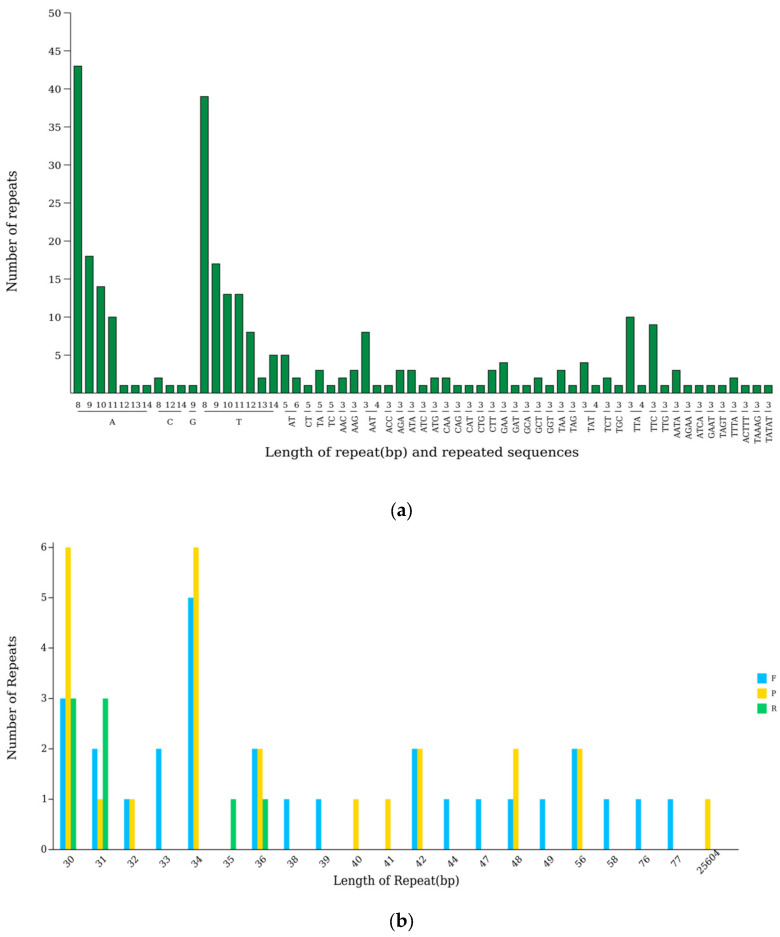 Figure 3
Figure 3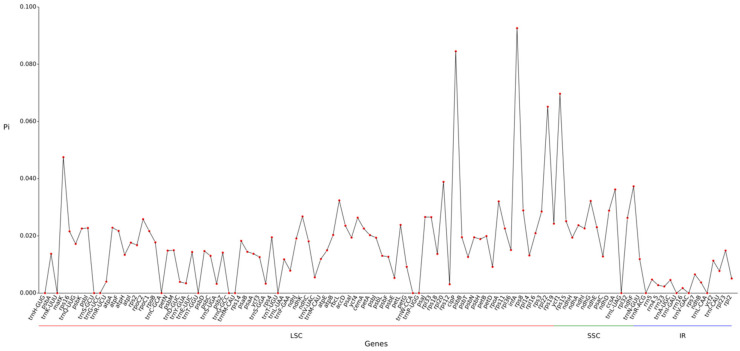 Figure 4
Figure 4 Figure 5
Figure 5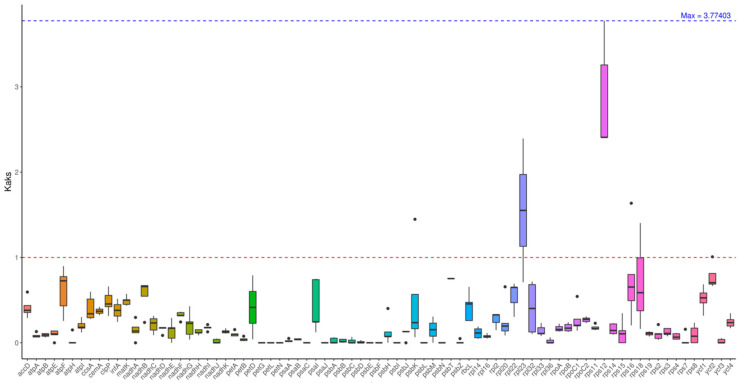 Figure 6
Figure 6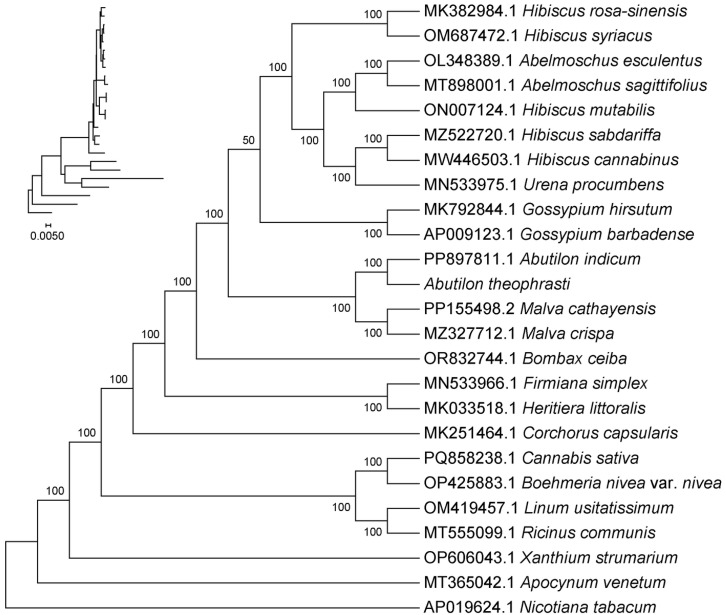 Figure 7
Figure 7- —the Agriculture Science and Techonolgoy Innovation Program
- —National Crop Germplasm Sharing and Service Plantform for Bast and Leaf Fiber Crops
- —China Agriculture Research System for Bast and Leaf Fiber Crops
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Photosynthetic Processes and Mechanisms · Plant Diversity and Evolution
1. Introduction
Abutilon theophrasti Medicus, commonly known as green hemp or white hemp, etc., is an annual herb native to China and widely distributed in temperate and tropical regions [1]. Historically, it has been valued for medicinal and ornamental purposes, as well as a source of fiber and oil crop [2], boasting substantial economic and medicinal potential. The stem bark fibers of A. theophrasti are tough, making it a novel natural textile material [3,4]. The entire plant and seeds are used in traditional Chinese medicine, characterized by a bitter taste and neutral nature, with effects of clearing heat, promoting diuresis, detoxifying, and removing cataracts. It is frequently employed to treat dysentery, abscesses, cataracts, and other conditions [5]. Modern research has identified active components such as flavonoids and alkaloids in A. theophrasti, which exhibit anti-inflammatory and antioxidant properties, though its specific pharmacological mechanism requires further investigation [6].
Plant chloroplasts are semi-autonomous genetic organelles with a double-membrane structure [7]. By converting light energy into ATP and carbohydrate energy, they directly determine crop yields. Adverse environmental factors such as drought, flood, salinity, extreme temperature, nutritional imbalance, and pathogen/virus infections can disrupt photosynthetic function, leading to significant yield reduction. Chloroplasts possess three types of membranes: inner, outer, and thylakoid membranes, each equipped with specific ion channels and transport proteins that efficiently mediate the transmembrane transport of nutrients, solutes, and metabolites [8]. They play crucial roles in processes such as photosynthesis and carbohydrate metabolism [9]. In recent years, advances in high-throughput technology have deepened research on plant chloroplast genomes. The majority of higher plants have a typical tetrameric chloroplast genome structure consisting of a large single copy region (LSC), a small single copy region (SSC), and two inverted repeat regions (A and B). IRA and IRB consist of four parts [10,11]. The chloroplast genome is maternally inherited and highly conserved, unaffected by the nuclear genome. These characteristics make chloroplast genomes ideal for studies on plant phylogeny and genetic diversity analysis [12,13], with significant applications in the research of plant system evolution, the analysis of photosynthetic molecular mechanisms, and genetic engineering [14].
The Malvaceae family is rich in species, exhibiting considerable morphological and ecological diversity. As a medium-to-large family in the plant kingdom, it holds significant ecological and economic importance [15,16]. A. theophrasti belongs to the Malvaceae subfamily, which is the most species-rich and representative core subfamily of Malvaceae [17], and is one of its economically and medicinally valuable species. However, the phylogenetic position of A. theophrasti within the Malvaceae family and its evolutionary relationship with related species remain unclear. Chloroplast genomes can provide abundant genetic information for plant evolution studies. In this study, A. theophrasti was used as the experimental material, and high-throughput sequencing technology and bioinformatics methods were employed to sequence, assemble, and annotate its chloroplast genome, followed by in-depth analysis of its structural characteristics and functional genes. Additionally, phylogenetic analysis was conducted to clarify the evolutionary position of A. theophrasti in the Malvaceae. The results provide a theoretical foundation for the phylogenetic study of Malvaceae, as well as the protection and utilization of germplasm resources.
2. Results
2.1. Basic Characteristics of A. theophrasti M. chloroplast Genome
The chloroplast genome of A. theophrasti exhibits a typical tetrad structure, with a length of 160,440 bp. It comprises two identical inverted repeat regions (IRa and IRb, each 25,604 bp), a large single-copy (LSC) region of 89,084 bp, and a small single-copy (SSC) region of 20,148 bp (Figure 1, Table 1). Base composition analysis showed that the contents of A, C, G, and T in the chloroplast genome of A. theophrasti were 31.19%, 18.66%, 18.23%, and 31.92%, respectively. The total GC content in the genome is 36.89%, with IRa and IRb regions (both 42.96%) showing higher GC content than the LSC (34.64%) and SSC (31.38%) (Table 1).
2.2. Functional Annotation of A. theophrasti M. Chloroplast Genes
A total of 130 genes were annotated in the chloroplast genome of A. theophrasti, including 85 mRNA genes, 37 tRNA genes, and 8 rRNA genes with no pseudogenes identified. These genes primarily function in photosynthesis, self-replication, and auxiliary metabolism, such as protein processing and membrane structure maintenance. The functions of some genes remain uncharacterized (Table 2). Regarding gene copy numbers: 73 mRNAs and 23 tRNAs are present as single copies; 6 mRNAs, 7 tRNAs, and 4 rRNAs are duplicated. Intron analysis showed that 11 mRNAs and 8 tRNAs contain one intron, while 4 mRNAs contain two introns (Table 2).
2.3. Codon Usage Bias Analysis
Excluding stop codons, the chloroplast genome of A. theophrasti contains 20,772 codons encoding amino acids. Leucine (Leu) is the most frequently used amino acid (2408 codons), followed by isoleucine (Ile, 1978 codons) and serine (Ser, 1697 codons).
Further analysis of relative synonymous codon usage degree (RSCU) shows 31 codons with RSCU > 1, among which 29 codons end with A or U. There are 33 codons with RSCU < 1, and 30 codons ending in G or C. Notably, tryptophan (Trp) is encoded by a single codon (UGG) with a RSCU value of 1. Among all codons, the methionine (Met) codon AUG has the highest RSCU value (6.9867), followed by the leucine (Leu) UUA (1.9884) and alanine (Ala) GCU (1.7776) codons. The methionine (Met) codon GUG has the lowest RSCU value (0.0133) (Table 3).
Combined with the codon circular diagram, it visually displays the distribution of codons corresponding to each amino acid, providing insights into codon usage preferences in the chloroplast genome of A. theophrasti (Figure 2).
2.4. Repeated Sequence Analysis
The A. theophrasti chloroplast genome contains 61 scattered repeat sequences, including 28 forward (F), 25 palindromic (P), 8 reverse (R), and 0 complementary (C) sequences. Among them, the lengths of most scattered repeat sequences are distributed in the range of 30 to 77 bp, with 30 bp repeats being the most common (12 repeats), followed by 34 bp repeats (11 copies). In addition, one dispersed repeat of 25,604 bp was identified (Figure 3a).
Simple repeat sequences (SSRs) are short tandem repeats of 1 to 6 nucleotides. A total of 288 SSRs were detected in the A. theophrasti chloroplast genome, distributed as follows: 205 in LSC regions, 43 in SSC regions, and 40 in the IR regions. From the perspective of the genetic components in different regions. There are 37 SSRs in exons, 41 in introns, and 127 in Intergenic regions in the LSC regions; the number of SSRs in exons, introns, and gene spacers was 23, 1, and 19 in the SSC regions; the number of SSRs in exons, introns, and gene spacers is 17, 8, and 15 in the IR regions. Among these SSRs, the single-nucleotide repeat types are the most abundant. The number of A repeats ranges from 8 to 14, and the quantity is between 1 and 43. The number of T repetitions also ranges from 8 to 14, and the quantity is between 2 and 39. There are a certain number of dinucleotide repeats, such as AT/TA, and numerous types of trinucleotide repeats, such as TTA/TTC, that exist. There are also small amounts of tetranucleotide repeats, pentanucleotide repeats, and hexanucleotide repeats present.
Among the 288 SSRs, the top three most frequent types are A (8 repeats, 14.93%, 43 copies), T (8 repeats, 13.54%, 39 copies), and T (9 repeats, 5.90%, 18 copies) (Figure 3b).
2.5. Nucleic Acid Diversity and Boundary Analysis
Nucleotide diversity (Pi) analysis of 113 gene regions in the chloroplast genome of A. theophrasti yielded an average Pi value of 0.0170 (Figure 4). Regional Pi values varied as follows: SSC region (0.0275) > LSC region (0.0177) > IR regions (0.0048), indicating higher conservation in the IR regions. Thirty-seven highly variable regions (Pi ≥ 0.02) were identified: 27 in the LSC region 27 l [e.g., infA (0.0926), clpP (0.0845), rpl22 (0.0651)] and 10 in the SSC region [e.g., ycf1 (0.0697), ccsA (0.0362), ndhF (0.0373)]. The most variable locus is infA (0.0926) in the LSC region.
Expansion and contraction of the IR boundaries are the key factors driving chloroplast genome size variation. The boundary analysis results indicated that the chloroplast genomes of six Malvaceae plants and one Tiliaceae plant revealed four conserved boundaries: JLB (LSC/IRb), JSB (IRb/SSC), JSA (SSC/IRa), and JLA (IRa/LSC) (Figure 5). Genes near these boundaries include rps19, rpl2, rpl22, ycf1, ndhF, trnN, and trnH. In Malvaceae plants, the JLB boundary is located within the rps19 coding region in all species except G. barbadense and H. cannabinus. The chloroplast genome of H. cannabinus differs by 327–343 bp from the other five species, while the five species differ among themselves by 6–16 bp. The JSB boundary is located within the ycf1 gene region in all species except G. barbadense and M. cathayensis. A small segment (45–961 bp) of the ycf1 is in IRb, with the majority (4787–5652 bp) in SSC. For G. barbadense and M. cathayensis, the JSB boundary lies between trnN and ndhF, 396 bp and 619 bp from trnN in IRb, respectively. The JSA boundary is located between ndhF and trnN except for G. barbadense, H. syriacus, and M. cathayensis. The ndhF gene coding regions in SSC are 62 bp, 62 bp, and 106 bp for these three exceptions, respectively. For H. syriacus and M. cathayensis, the JSA boundary is within ycf1: 10 bp in SSC and 608 bp in IRa (H. syriacus); 5385 bp in SSC and 312 bp in IRa (M. cathayensis). The JLA boundary is located 0–13 bp to the left of the trnH coding region in LSC across all species.
For Corchorus capsularis (Tiliaceae), the JLB boundary is located within rps19, differing by 6–16 bp from most species except H. cannabinus. The boundary of JSB is between trnN and ycf1, with greater positional differences from other species. The JSA boundary is located between ndhF and trnN, differing by 11 to 55 bp from A. theophrasti, A. indicum, and H. cannabinus. The JLA boundary is 0 bp left of trnH in LSC. In summary, chloroplast genomes of Abutilon species are highly conserved and stable, with consistent IR boundary patterns (e.g., rps19 crossing the LSC/IRb and ycf1 crossing the IRb/SSC). These patterns serve as a unique phylogenetic signal for distinguishing genera.
2.6. Nucleic Acid Diversity Pi Analysis
Ka/Ks analysis of chloroplast genes between A. theophrasti M. and six related species showed an overall average ratio of 0.24 (Figure 6). Most genes (e.g., atpA, psaA, etc.) had Ka/Ks < 1, indicating purifying selection and functional conservation. Ten genes, atpH, petG, petL, petN, psaC, psaJ, psbF, psbI, psbL, and psbN, had Ka/Ks = 0, reflecting extreme conservation. Among the highly variable genes, rps12 (vs. AP009123) had the highest Ka/Ks ratio (3.77); rpl23 (vs. MK251464) also had Ka/Ks > 1, indicating positive selection. Interfamily comparisons showed a slightly higher Ka/Ks ratio than intrafamily comparisons (Malvaceae family only), consistent with phylogenetic differentiation.
2.7. Phylogenetic Analysis
To clarify evolutionary relationships among Malvaleae plants, chloroplast genomic data from 24 species were downloaded from the NCBI. On this basis, N. tabacum of the Solanaceae family in the Solanales order was used as an exophyte to construct a phylogenetic tree. The results showed that A. theophrasti is most closely related to its congener A. indicum, forming a sister group followed by M. cathayensis, M. crispa, and other Malva plants, followed by G. hirsutum and G. barbadense. H. syriacus and H. rosa-sinensis, both belonging to the Malvaceae family, are more distantly related to A. theophrasti. N. tabacum, as an outgroup, is the most distant from A. theophrasti (Figure 7).
3. Discussion
Chloroplasts are semi-autonomous organelles in the plant genetic system, with a more conserved gene number, composition and arrangement than mitochondrial and nuclear genomes [18]. The chloroplast genome of A. theophrasti assembled in this study (160,440 bp, GC content 36.89%) presents a typical tetrameric structure, which is consistent with most terrestrial plants. Comparison with previously published chloroplast genomes of A. theophrasti [19,20] shows high consistency in length (160,331 and 160,446 bp) and GC content (36.9%), confirming species-specific conservation. However, the number of annotated genes in our study (130) differs from those of Lv et al. [19] (76) and Yu et al. [20] (113), likely due to variations in annotation criteria, assembly completeness, or potential intraspecific differences. Our analysis, which includes a more comprehensive examination of repetitive sequences, nucleotide diversity, IR boundary dynamics, and genome-wide selective pressure (Ka/Ks), provides an expanded resource for the genomic characterization of A. theophrasti. The following discussion integrates these prior findings to contextualize our results within a broader framework.
Potentially reflecting varietal differences. Our annotation of 130 genes suggests a more complete or differentially interpreted gene set, particularly regarding the identification of distinct transcriptional units and intron-containing genes. These differences underscore the impact of annotation pipelines on comparative genomics and highlight the need for standardized methods. Nevertheless, the core structure—a quadripartite organization with highly conserved LSC, SSC, and IR regions—remains unchanged across all studies, aligning with the characteristic stability of malvaceous chloroplast genomes noted in broader comparative studies [21].
Of the 130 annotated genes, 45 are involved in photosynthesis, highlighting chloroplasts’ central role in this process. Codon usage bias analysis showed leucine, isoleucine, and serine to be the most frequent amino acids. Relative synonymous codon usage (RSCU) is defined by comparing the actual occurrence frequency of a specific codon with its theoretical expected frequency. It is an effective tool for evaluating codon preference. An RSCU greater than 1 indicates a clear preference for the use of that codon [22]. In the chloroplast genome of A. theophrasti, there are 31 codons with RSCU greater than 1, among which 93.55% end with A or U. Similar phenomena are widespread in the chloroplast genomes of angiosperms [23,24]. SSRs are widely used for constructing genetic linkage maps, population genetic analysis, and so on. This study found that there were 61 dispersed repeat sequences and 288 SSRs in the chloroplast genome of A. theophrasti, which is consistent with winter rapeseed [25], with conserved repeat types such as forward repeats and single-nucleotide repeats. Overall, it shows a relatively high degree of conservatism. This difference reflects the differentiation in evolutionary strategies between the nuclear genome and the chloroplast genome: the nuclear genome enhances chromatin stability through satellite DNA amplification to adapt to environmental stress, while the chloroplast genome retains conserved repeat sequences to maintain the stability of photosynthetic function. It can provide potential candidate molecular markers for the study of the genetic diversity of the A. crops. Nucleic acid diversity Pi is an important indicator for measuring the degree of genetic variation within a population.
A high Pi value indicates rich genetic diversity within the population and can provide potential molecular markers for population genetics [26]. The average nucleic acid diversity (Pi) of all 113 gene regions detected in this study was 0.0170. The average nucleic acid diversity of different regions from largest to smallest was SSC (0.0275), LSC (0.0177), and IR (0.0048), indicating that IR was more conserved compared to the other two regions. Among them, the top four sites with the highest nucleic acid diversity are infA, clpP, and rpl22 in the LSC region, and ycf1 in the SSC region in sequence. These highly variable sites can be used as molecular markers for species identification in the Malvaceae family. During the evolution of plant genomes, the expansion or contraction of the IR region is the main driving force for the structural variation of chloroplast genomes, which can provide molecular evidence for species identification and phylogenetic studies [27].
IR boundary analysis revealed that the differences in IR boundaries were mainly related to the positions of rps19, ycf1, ndhF, trnH, and rpl2. The chloroplast genomes of plants within the A. are generally conserved and highly stable, with highly consistent IR boundary types, especially the patterns of rps19 crossing the LSC/IRb boundary and ycf1 crossing the IRb/SSC boundary, which are consistent with the conclusions drawn from the study of Lycium species by Zhang et al. [28]. The specificity among different genera is strong. The degree of IR boundary expansion (especially the crossing of the rps19 gene) of the Abutilon is a unique phylogenetic signal, which can be used to distinguish different genera. The expansion and contraction of IR boundaries are recognized as a major evolutionary force shaping chloroplast genome diversity in land plants [23]. Our boundary analysis identified rps19 and ycf1 as key genes straddling the LSC/IRb and IRb/SSC junctions, respectively, in A. theophrasti. This pattern provides a unique phylogenetic signal for the genus Abutilon. Notably, Zhong et al. [21] emphasized that such IR boundary dynamics can lead to gene duplication or truncation, potentially altering gene dosage and function. Therefore, the specific IR configuration we observed in A. theophrasti may not merely be a taxonomic marker but could reflect underlying evolutionary mechanisms, such as selection for optimized gene expression or a genomic signature of past hybridization events. Investigating the correlation between these structural variations and phenotypic traits or ecological adaptation presents a promising future direction.
Phylogenetic analysis indicates that A. theophrasti M. and A. indicum of the same genus form a phylogenetic branch together, that is, they have a direct "sister group" relationship. Secondly, there is another branch of the Malva plants, M. cathayensis and Malva crispa. Most closely related to A. theophrasti M. is the Gossypium. It indicates that among the plants of the Malvaceae family, the Malvaceae genus plants are the most evolutionarily close group to the A. plants, such as A. theophrasti M., followed by the Gossypium plants. H. syriacus and H. rosa-sinensis, both belonging to the Malvaceae family, have a relatively distant genetic relationship with A. theophrasti. Our phylogenetic reconstruction placed A. theophrasti in a clade with its congener A. indicum, with members of Gossypium as closely related lineages. This contrasts with the findings of Yu et al. [20], who positioned A. theophrasti at a basal node within the Malveae tribe. Such topological discrepancies are not uncommon in phylogenetic studies and may arise from several factors: (1) the selection of different outgroups and taxon sampling, which can affect root placement; (2) the choice of phylogenetic inference methods and genomic data partitions (e.g., whole genome vs. coding sequences only); and (3) the potential for incomplete lineage sorting or hybridization history within Malveae, which can confuse phylogenetic signals. Despite this difference, both studies confirm the monophyly of Malvaceae and support clear distinctions among major lineages like Malva, Gossypium, and Hibiscus. Resolving the precise phylogenetic position of Abutilon may require a phylogenomic approach incorporating data from both chloroplast and nuclear genomes across a denser sampling of the genus.
4. Materials and Methods
4.1. Sample and Sequencing
The experimental material was A. theophrasti cultivated at the Zhejiang Institute of Landscape Plants and Flowers (Zhejiang Xiaoshan Cotton and Bast Fiber Crops Research Institute, Hangzhou, China) (30°07′ N, 120°23′ E). The young leaves of healthy plants were taken, wrapped in tinfoil, and quickly frozen in liquid nitrogen. After being taken out, they were stored in a −80 °C refrigerator. Total DNA of A. theophrasti M. was extracted using the universal plant DNA extraction kit (D312) and sequenced using the Illumina NovaSeq 6000 platform.
4.2. Chloroplast Genome Assembly and Functional Annotation
Clean data were obtained by filtering the original data using fastp v0.23.4 software [29]. Chloroplast genomes were assembled using GetOrganelle v1.7.7.1 software. The CDS of chloroplasts were annotated respectively by Prodigal v2.6.3 [30], hmmer v3.1b2 [31], and Aragorn v1.2.38 [32]. Predict rRNA and predict tRNA. The chloroplast genome map was drawn using OGDRAW v1.3.1 [33] software.
4.3. Analysis of Dispersed and Simple Sequence Repeats
The repetitive sequences were identified by using the vmatch v2.3.09 software [34], and the relevant parameters were set as follows: The Hamming distance is 3, the minimum length is 30 bp, and the identification forms are four types: forward, palindromic, reverse, and complement. Simple sequence repeats (SSRs) were analyzed through MISA v1.0 software [35]. The parameter configuration is as follows: the single-base repetition should be not less than 8 times, the double-base repetition not less than 5 times, and the 3-base, 4-base, 5-base, and 6-base repetitions should occur at least 3 times.
4.4. Chloroplast Genomic Nucleic Acid Diversity and Boundary Analysis
Chloroplast genomes of six species of Malvaceae and Tiliaceae plants were downloaded from NCBI, including A. indicum (PP897811.1), G. barbadense (AP009123.1), Corchorus capsularis (MK251464.1), H. cannabinus (MW446503.1), H. syriacus (OM687472.1), and M. cathayensis (PP155498.2); Global alignment and analysis of homologous gene sequences among different species were conducted using MAFFT v7.427 (--auto mode) software, and nucleic acid diversity (nucleic acid diversity, Pi value) was calculated using the dnasp5 tool [36]. Using sets of Shen cloud platform tools CPJSdraw (http://cloud.genepioneer.com:9929/#/tool/alltool/detail/296, accessed on 15 October 2025), each species chloroplast genome boundary information was subjected to visualization processing. Genomic alignment was performed using the default parameters of Mauve (v2.3.1) software [34].
4.5. System Evolution Analysis
Chloroplast genomic data of 24 plant species were downloaded from the NCBI database. It includes chloroplast genomic data of 16 species from the Malvaceae family, 1 species from the Tiliaceae family, 1 species from the Cannabaceae family, 1 species from the Urticaceae family, 1 species from the Linaceae family, 1 species from the Euphorbiaceae family, 1 species from the Asteraceae family, 1 species from the Apocynaceae family, and 1 species from the Solanaceae family. N. tabacum of the Solanaceae family was selected as the exophyte group. This is used to conduct an analysis of phylogenetic relationships. Multiple sequence alignment and evolutionary tree construction were performed, respectively, using MAFFT v7.427 (--auto mode) and RAxML v8.2.10 [37].
5. Conclusions
This study clarified the basic characteristics of the chloroplast genome of A. theophrasti: it is A typical tetrameric structure, with a total genome length of 160,440 bp, GC content of 36.89%, 130 annotated genes, and its genome codons mostly end with A/U. There are a total of 61 scattered repeat sequences and 288 SSRs. IR boundary analysis revealed that the chloroplast genomes of plants within the genus A. are generally conserved, highly stable, and have strong specificity among different genera, which are their unique phylogenetic signals and can be used to distinguish different genera. Phylogenetic analysis shows that A. indicum is most closely related to A. theophrasti. It has a relatively close evolutionary distance from the Malva, providing molecular evidence for the classification and evolutionary research of the Malvaceae family. This study clarified the conserved characteristics and evolutionary laws of the chloroplast genome of A. theophrasti, providing theoretical support for the phylogenetic research of A. theophrasti and other Malvaceae plants and the development and utilization of germplasm resources.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Meng S. Seed Germination and Differential Expression of Related Genes in Different A theophrasti Populations Master’s Thesis Chinese Academy of Agricultural Sciences Beijing, China 202110.27630/d.cnki.gznky.2021.000619 · doi ↗
- 2Kirby R.H. Vegetable Fibres. Botany, Cultivation and Utilization London and Interscience Publisher New York, NY, USA 1963 Volume 463
- 3Jia X. Gong J. Zhang X. Yang W. Hou X. Gao A. Chen Q. Li N. Ni S. Research overview of cultivation and management techniques of Abutilon theophrasti M Anim. Husb. Feed. Sci.201132515210.16003/j.cnki.issn 1672-5190.2011.02.026 · doi ↗
- 4Huang W. Yu H. Abutilon theophrasti M. fiber degumming test and performance study J. Zhongyuan Univ. Technol.201223586110.3969/j.issn.1671-6906.2012.03.013 · doi ↗
- 5Liu H. Ni S. Kang J. Luo R. Wu Y. Cui Y. Li Z. Overview of pharmaceutical research on plants of the Abutilon Northwest J. Pharm.2010256869
- 6Zhao W. Zhao C. Li C. Zhang J. Jiang H. Ren X. Study on Antiodidant Activities of Velvetleaf (Abutilon theophrasti M.) Extracts Heilongjiang Med. J.2018311187118910.14035/j.cnki.hljyy.2018.06.001 · doi ↗
- 7Zhou Y. Liu Y. Fang Y. Zhou J. Chen J. Chloroplast genome analysis and divergence time estimation of 11 Dendrobium officinale J. Zhejiang Univ. (Agric. Life Sci.)20255129130210.3785/j.issn.1008-9209.2024.08.261 · doi ↗
- 8Pottosin I. Shabala S. Transport Across Chloroplast Membranes: Optimizing Photosynthesis for Adverse Environmental Conditions Mol. Plant 2016935637010.1016/j.molp.2015.10.00626597501 · doi ↗ · pubmed ↗