FGFR2-Rearranged Biliary Tract Cancer: Biology, Resistance Mechanisms, and Emerging Therapeutic Strategies
Xin Xin, Ruoyu Miao

TL;DR
This paper reviews FGFR2-related biliary tract cancer, focusing on biology, resistance to treatments, and new strategies to improve patient outcomes.
Contribution
The paper provides a comprehensive overview of FGFR2 alterations in biliary tract cancer and emerging strategies to overcome treatment resistance.
Findings
FGFR2 rearrangements are actionable targets in biliary tract cancer, particularly in intrahepatic cholangiocarcinoma.
Acquired resistance to FGFR inhibitors is often driven by secondary mutations and bypass signaling pathways.
Next-generation inhibitors and combination therapies are being explored to overcome resistance and improve treatment outcomes.
Abstract
Biliary tract cancer is a rare and aggressive cancer with limited treatment options and poor survival outcomes. In recent years, alterations in fibroblast growth factor receptor 2 (FGFR2) have been identified in a subset of patients, particularly those with intrahepatic cholangiocarcinoma. These genetic changes drive tumor growth but also create an opportunity for targeted treatment. Several drugs that inhibit FGFR2 have shown meaningful clinical benefit; however, most patients eventually develop resistance, leading to disease progression. This review summarizes current knowledge of FGFR2-related cancer biology, available targeted therapies, and the main mechanisms by which resistance develops. We also discuss emerging treatment strategies, including next-generation drugs, combination approaches, and the use of blood-based testing to monitor disease evolution. Improving understanding of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
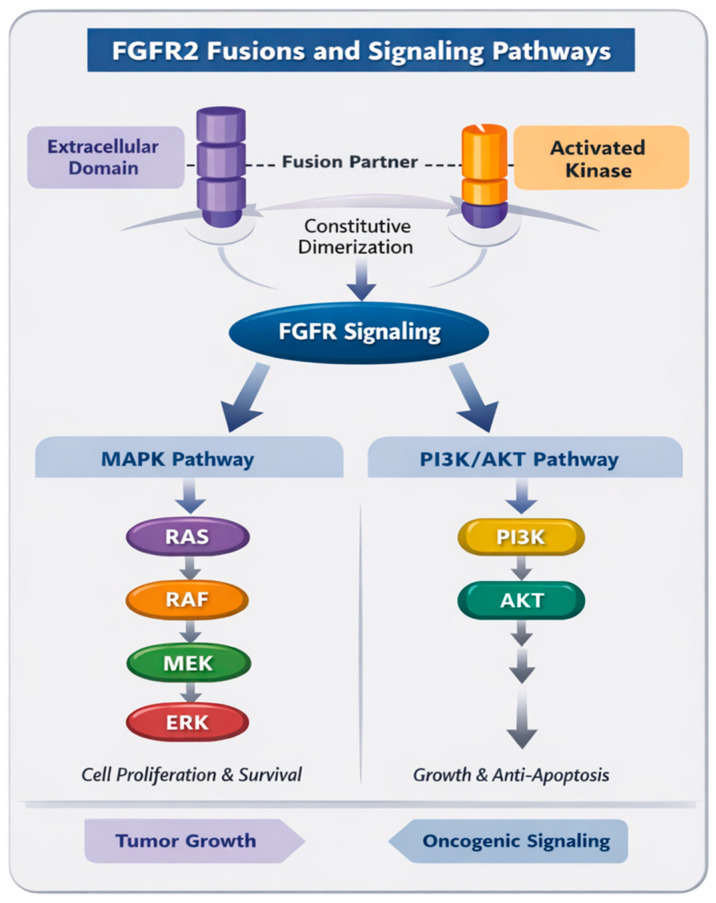 Figure 1
Figure 1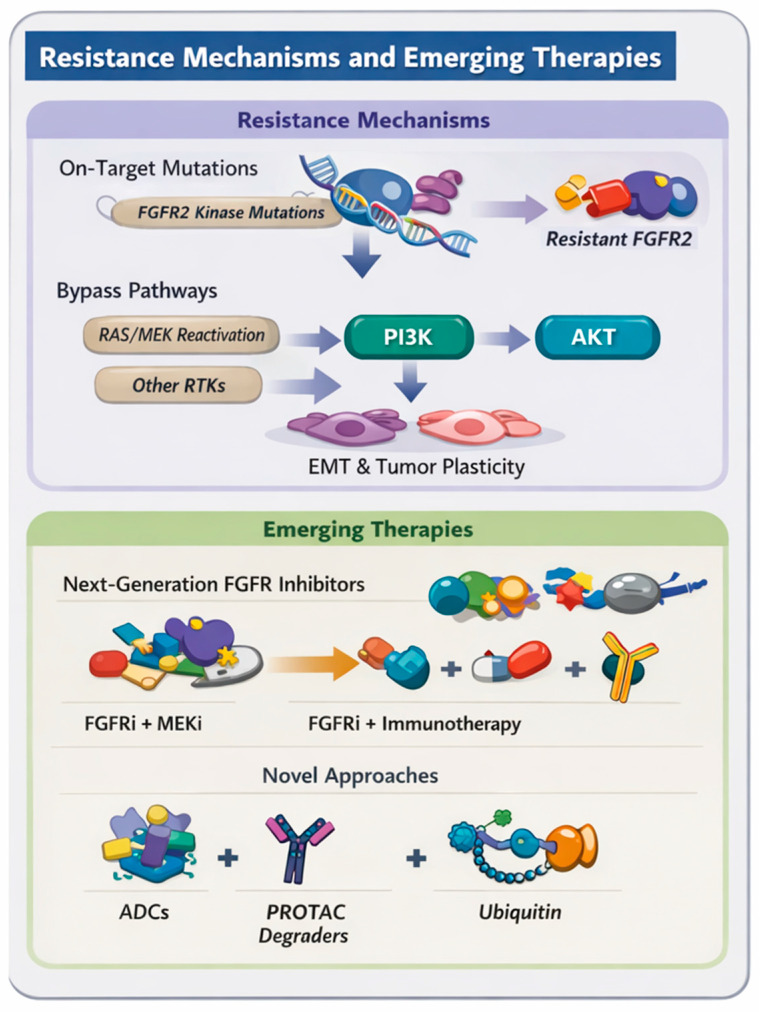 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFibroblast Growth Factor Research · Cholangiocarcinoma and Gallbladder Cancer Studies · IgG4-Related and Inflammatory Diseases
1. Introduction
Biliary tract cancers (BTCs) comprise a heterogeneous group of malignancies arising from the epithelium of the intrahepatic and extrahepatic bile ducts as well as the gallbladder. Among these, intrahepatic cholangiocarcinoma (iCCA) has emerged as a subtype with an increasing incidence worldwide and a particularly poor prognosis [1,2]. Despite the introduction of gemcitabine and cisplatin as first-line standard chemotherapy for advanced BTC, the median overall survival (OS) remains below one year [3]. Incorporating immune checkpoint inhibitors (ICIs), such as durvalumab or pembrolizumab, into this regimen has yielded only modest improvements, extending median OS to just over 12 months [4,5,6]. Molecular profiling has significantly advanced the understanding of iCCA, revealing several actionable alterations such as fibroblast growth factor receptor 2 (FGFR2) fusions, isocitrate dehydrogenase 1 (IDH1) mutations, B-Raf proto-oncogene (BRAF) V600E mutations, and human epidermal growth factor receptor 2 (HER2) amplifications [7,8]. Among these, FGFR2 rearrangements are particularly notable: they are considered lineage-defining events and predict marked sensitivity to selective FGFR inhibition [9,10,11].
Fibroblast growth factor receptors (FGFRs) are a family of receptor tyrosine kinases that regulate key cellular processes, including cell proliferation, differentiation, survival, and angiogenesis through downstream signaling cascades such as Rat sarcoma(RAS)/mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT), and signal transducer and activator of transcription (STAT) pathways [12]. Aberrant FGFR2 signaling—most commonly arising from gene fusions with partners that induce ligand-independent dimerization—drives oncogenesis by constitutively activating the downstream pathways [13,14]. These findings have led to a therapeutic paradigm shift in BTC, transforming a subset of this historically chemotherapy-refractory disease into a molecularly targetable entity.
The approval of selective FGFR inhibitors, including pemigatinib and futibatinib, has substantially expanded treatment options for patients with FGFR2 fusion-positive BTC [15,16]. However, secondary resistance typically develops within 12 months of therapy, representing a critical clinical challenge [17,18]. Understanding the molecular mechanisms of resistance and developing strategies to circumvent them are essential for extending therapeutic benefit and improving survival in this molecularly defined subset of BTC.
2. Molecular Biology and Clinical Significance of FGFR2 Alterations
2.1. Structure and Function of FGFR2
The FGFR2 gene, located on chromosome 10q26, encodes a transmembrane receptor tyrosine kinase comprising three extracellular immunoglobulin-like domains, a single transmembrane helix, and an intracellular tyrosine kinase domain [12]. FGFR2 consists of three extracellular immunoglobulin-like domains (D1–D3), an acidic box that modulates ligand binding, a single-pass transmembrane helix, and an intracellular split tyrosine kinase domain. Alternative splicing of the D3 domain generates the IIIb and IIIc isoforms, which display distinct ligand-binding specificities and tissue-restricted expression patterns, contributing to epithelial–mesenchymal signaling diversity [19,20].
Physiological activation of FGFR2 requires binding of fibroblast growth factors (FGFs) in the presence of heparan sulfate proteoglycans, which stabilize ligand-receptor complexes and promote receptor dimerization. This induces trans-autophosphorylation of intracellular tyrosine residues, allowing recruitment of adaptor proteins such as fibroblast growth factor receptor substrate 2 (FRS2) and growth factor receptor-bound protein 2 (GRB2) [19,21]. Activated FGFR2 engages multiple downstream signaling pathways including RAS/rapidly accelerated fibrosarcoma kinase (RAF)/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK), PI3K/AKT/mechanistic target of rapamycin (mTOR), phospholipase C γ (PLCγ), and Janus kinase (JAK)/STAT signaling, which collectively regulate cellular proliferation, survival, differentiation, migration, and angiogenesis [21,22]. Oncogenic activation of FGFR2 can arise through multiple mechanisms, such as gene amplification, activating mutations, or chromosomal rearrangements resulting in FGFR2 fusions, the latter representing the dominant oncogenic mechanism in BTC (Figure 1).
2.2. Prevalence and Oncogenic Drivers in BTC
Large-scale genomic profiling efforts have established that FGFR2 rearrangements are a defining molecular event in a substantial subset of iCCA, occurring in approximately 10–16% of cases across Western and Asian cohorts, while remaining rare in extrahepatic cholangiocarcinoma (eCCA) and gallbladder cancer (GBC) [23,24]. Among recurrent genomic alterations in iCCA, IDH1 mutations are observed at a similar frequency (approximately 10–15%), whereas alterations such as BRAF V600E mutations, neurotrophic tyrosine receptor kinase (NTRK) fusions, and anaplastic lymphoma kinase (ALK)/c-ros oncogene 1 (ROS1) rearrangements are distinctly uncommon. In contrast, erb-b2 receptor tyrosine kinase 2 (ERBB2 or HER2) amplification is infrequent in iCCA but more prevalent in eCCA and GBC, underscoring the anatomic specificity of actionable drivers within biliary tract cancer (Table 1) [9,10,25]. Such differences likely reflect heterogeneity in etiologic risk factors and underlying liver disease.
Clinically, FGFR2 fusion-positive iCCA has been associated in several retrospective series with younger age at diagnosis, female predominance, and, in some cohorts, a comparatively less aggressive clinical course [26]. However, these observations may be influenced by selection effects, referral patterns, and treatment-related factors, and should therefore be interpreted cautiously. From a therapeutic standpoint, FGFR2 fusions uniquely define a molecularly enriched disease subset supported by multiple selective FGFR inhibitors demonstrating reproducible objective responses and durable disease control across independent phase II trials [5,10,27]. This depth of clinical validation, together with the clear lineage restriction of FGFR2 rearrangements to iCCA, distinguishes FGFR2 from other targets and underpins its current role as one of the most therapeutically actionable drivers in BTC.
More than 100 distinct fusion partners have been identified to date, including BICC1, PPHLN1, KIAA1598 (SHROOM3), and TACC3, most of which contribute oligomerization domains that drive constitutive receptor dimerization. These rearrangements preserve the FGFR2 kinase domain while removing regulatory regions, resulting in ligand-independent activation of downstream oncogenic signaling pathways [13,14,28]. Available evidence suggests that many FGFR2 fusions share common oncogenic properties driven by constitutive kinase activation, regardless of fusion partner; however, biological heterogeneity likely exists, and the extent to which specific fusion partners influence signaling output, tumor biology, or therapeutic sensitivity remains an active area of investigation.
2.3. Downstream Signaling and Biological Effects
Constitutive activation of FGFR2 fusion proteins leads to persistent, ligand-independent stimulation of downstream oncogenic signaling, most prominently the MAPK (RAS/RAF/MEK/ERK) and PI3K/AKT/mTOR axes, thereby promoting sustained cellular proliferation, survival, and resistance to apoptosis (Figure 1) [20,29,30]. Beyond tumor cell-intrinsic effects, aberrant FGFR2 activation may further modulate the tumor microenvironment (TME) by inducing pro-angiogenic factors, including VEGF, and engaging in paracrine crosstalk with endothelial cells and cancer-associated fibroblasts, thereby facilitating stromal remodeling, epithelial–mesenchymal transition (EMT), and invasive behavior [30,31]. Integrative genomic and transcriptomic analyses have demonstrated that FGFR2 fusion-positive iCCA constitutes a distinct molecular lineage characterized by low tumor mutation burden and an immune-cold phenotype with limited cytotoxic T-cell infiltration and low expression of immune checkpoint molecules [32,33], which may partly explain the limited clinical activity of ICIs observed in this subtype.
2.4. Clinical Significance
FGFR2 fusions represent a validated predictive biomarker for response to FGFR-targeted therapies in iCCA. Across multiple phase II studies, selective FGFR inhibitors, including pemigatinib, infigratinib, and futibatinib, have demonstrated reproducible antitumor activity in patients with FGFR2 fusion-positive iCCA, with objective response rates (ORR) of approximately 20–40% and median progression-free survival (PFS) of 6–9 months. These outcomes compare favorably with those historically reported for cytotoxic chemotherapy in advanced biliary tract cancer. However, it is important to emphasize that these data derive predominantly from single-arm trials and basket study subsets, and that any comparisons with historical chemotherapy cohorts are inherently indirect. Differences in patient selection, prior lines of therapy, molecular enrichment, and assessment methodologies limit cross-trial interpretability, and preclude definitive conclusions regarding relative efficacy [15,16,34,35]. Although responses to FGFR inhibition can be durable, most patients ultimately develop acquired resistance. Nevertheless, the consistent clinical benefit observed across multiple agents has validated FGFR2 as a therapeutic target and transformed the treatment paradigm for this molecularly defined subgroup. Accordingly, comprehensive genomic profiling should therefore be routinely performed for all patients with advanced iCCA to identify FGFR2 fusions and other actionable alterations and inform targeted therapy selection [36].
3. Clinical Development of FGFR Inhibitors in Biliary Tract Cancer
3.1. Overview of FGFR-Targeted Agents
Therapeutic development of FGFR inhibitors has largely centered on small-molecule tyrosine kinase inhibitors (TKIs) that target the ATP-binding pocket of the FGFR kinase domain. These agents include reversible, ATP-competitive inhibitors, such as pemigatinib, infigratinib, derazantinib, and erdafitinib, as well as irreversible covalent inhibitors exemplified by futibatinib, which forms a covalent bond with a conserved cysteine residue within the FGFR kinase domain. Although these compounds differ in their biochemical potency and selectivity profiles across FGFR1-4, they share a common mechanism of suppressing aberrant FGFR-driven signaling through inhibition of downstream pathways. Across multiple phase I-II clinical trials, these agents have demonstrated reproducible and clinically meaningful antitumor activity in pretreated patients with FGFR2 fusion-positive cholangiocarcinoma (CCA). The consistent clinical activity of these agents, together with the relative lack of activity in tumors lacking FGFR2 rearrangements, has established FGFR2 fusions as validated oncogenic drivers and therapeutic targets [15,16,27,34,37,38,39,40,41]. Table 2 summarizes the approved and investigational FGFR inhibitors for BTC.
3.2. Key Clinical Trials
FIGHT-202 was a pivotal phase II trial evaluating the selective FGFR1-3 inhibitor pemigatinib in patients with previously treated, locally advanced or metastatic CCA [15,38]. Among 108 patients with FGFR2 fusions or rearrangements, the ORR was 37.0% (95% confidence interval [CI]: 27.9–46.9), with a median duration of response (DOR) of 9.1 months (95% CI: 6.0–14.5). No objective responses were observed among patients with other FGF/FGFR alterations or without FGF/FGFR alterations. Median PFS was 7.0 months (95% CI: 6.1–10.5) while median OS was 17.5 months (95% CI: 14.4–22.9) among patients with FGFR2 fusions or rearrangements. The most common treatment emergent adverse events (TEAEs) were hyperphosphatemia, alopecia, diarrhea, stomatitis, dysgeusia, and fatigue [38]. The randomized phase III FIGHT-302 trial (NCT03656536) was designed to compare pemigatinib with gemcitabine plus cisplatin in the first-line treatment of advanced CCA with FGFR2 rearrangements [42]. According to publicly available ClinicalTrials.gov records, the study is listed as discontinued, with the most recent update posted on 24 August 2025, citing enrollment challenges in the context of an evolving first-line treatment landscape.
A phase II single-arm study of the selective FGFR1-3 inhibitor infigratinib in patients with previously treated CCA harboring FGFR2 fusions or rearrangements demonstrated an ORR of 23.1% (95% CI: 15.6–32.2), a median PFS of 7.3 months (95% CI: 5.6–7.6), and a median OS of 12.2 months (95% CI: 10.7–14.9) [34]. Despite the promising clinical activity, the subsequent development program was later discontinued, and the U.S. Food and Drug Administration (FDA) approval for infigratinib in this indication was voluntarily withdrawn following a strategic and commercial review by the sponsor.
Similarly, the multikinase FGFR1-3 inhibitor derazantinib (ARQ 087) demonstrated moderate clinical activity in patients with FGFR2 fusion- or rearrangement-positive iCCA in the phase II FIDES-01 study, with an ORR of 21.4% (95% CI: 13.9–30.5) and a median DOR of 6.4 months (95% CI: 3.9–9.2). Median PFS and OS were 7.8 months (95% CI: 5.5–8.2) and 15.5 months (95% CI: 11.8–21.9), respectively, with a manageable toxicity profile [27,39]. Although subsequent evaluation in metastatic urothelial cancer (FIDES-02) did not demonstrate sufficient clinical benefit [43], and development was ultimately discontinued following strategic reprioritization, early results in iCCA supported FGFR2 as a biologically relevant therapeutic target.
FOENIX-CCA2 was a single-arm, global phase II trial evaluating futibatinib (TAS-120), an irreversible covalent FGFR1-4 inhibitor, in patients with previously treated iCCA harboring FGFR2 fusions or rearrangements [16,40]. Among 103 evaluable patients, the ORR was 42% (95% CI: 32–52), with a median DOR of 9.7 months (95% CI: 7.6–17.0). Median PFS was 9.0 months (95% CI: 6.9–13.1), and median OS was 21.7 months (95% CI: 14.5-not reached [NR]). The most common side effects were hyperphosphatemia, alopecia, and dry mouth [40]. Notably, futibatinib also demonstrated activity in a subset of patients previously treated with reversible FGFR inhibitors, supporting its ability to overcome certain acquired resistance mutations [44,45]. FOENIX-CCA3 (NCT04093362) was a randomized phase III study evaluating futibatinib in the first line setting for patients with FGFR2-rearranged iCCA. Based on ClinicalTrials.gov (accessed on 28 January 2026) registry information, the trial is currently listed as discontinued, with the most recent update posted on 7 February 2025, noting recruitment challenges after changes in the standard-of-care landscape. Subsequent clinical development has focused on dose and exposure optimization, including the ongoing FOENIX-CCA4 study (NCT05727176).
The selective pan-FGFR inhibitor erdafitinib was evaluated in the phase II RAGNAR basket trial in previously treated patients with FGFR1-4-altered advanced non-urothelial solid tumors [46]. In an exploratory subgroup of 35 patients with CCA, preliminary results showed an ORR of 60.0% (95% CI: 42.1–76.1) with a median DOR of 5.6 months (95% CI: 2.8–8.3). Responses were observed in patients harboring FGFR fusions and activating mutations. Disease control was achieved in nearly all treated patients. Median PFS and OS were 8.4 months (95% CI: 5.5–9.7) and 18.7 months (95% CI: 8.9–not evaluable [NE]), respectively. Safety profile was similar to that of other FGFR inhibitors, with the most common TEAEs being hyperphosphatemia, diarrhea, and stomatitis [41]. These findings suggest promising activity of erdafitinib in selected FGFR-altered CCA.
Collectively, these data establish FGFR inhibition as a clinically meaningful therapeutic strategy in molecularly selected BTCs and support incorporation of FGFR testing into routine diagnostic workflows.
4. Mechanisms of Resistance to FGFR Inhibition
4.1. Overview
Despite initial responses, virtually all patients treated with FGFR TKIs eventually experienced disease progression, reflecting the development of acquired resistance. The median duration of response across clinical trials is approximately 6–9 months, with some variability by agent [27,34,38,40,41]. Molecular analyses of post-progression tumor biopsies and circulating tumor DNA (ctDNA) have delineated two principal categories of resistance mechanisms: on-target mutations within the FGFR2 kinase domain and off-target or bypass pathway alterations that restore downstream signaling independently of FGFR2 (Table 3 and Figure 2) [17,18,47,48].
4.2. On-Target Resistance Mechanisms
The most frequent mechanism of acquired resistance to FGFR inhibition in FGFR2 fusion-positive CCA involves the emergence of secondary mutations within the FGFR2 kinase domain that impair inhibitor binding affinity. These alterations typically arise under selective pressure from ATP-competitive FGFR inhibitors and cluster within structurally and functionally critical regions of the kinase domain, such as the ATP-binding cleft, the gatekeeper residue, and the molecular brake motif that normally maintains kinase autoinhibition [17,49].
Recurrent gatekeeper substitutions at valine 565 (V565F/L/I) increase steric hindrance within the ATP-binding pocket and reduce the binding affinity of reversible FGFR inhibitors [17,49,50]. In parallel, molecular-brake mutations involving N550D/K/H, E566A, or K659M destabilize the inactive conformation of FGFR2, enhance constitutive receptor phosphorylation, and sustain downstream MAPK and PI3K signaling despite continued drug exposure [49,50,51]. Importantly, distinct resistance mutations confer heterogeneous effects on inhibitor sensitivity, highlighting the structural diversity of on-target resistance mechanisms [17].
Acquired resistance is further complicated by its frequently polyclonal nature. Post-progression analyses have demonstrated the coexistence of multiple FGFR2 kinase-domain mutations within individual patients, sometimes varying across metastatic sites or evolving dynamically over time [48,49,52]. This spatial and temporal heterogeneity reflects convergent evolutionary pressure and underscores the biological complexity of resistance development in FGFR2-driven disease.
4.3. Off-Target and Bypass Signaling Mechanisms
Not all resistance to FGFR inhibition is mediated by structural alterations of FGFR2. A subset of tumors develops off-target resistance mechanisms that bypass FGFR dependency through activation of alternative growth and survival pathways capable of re-engaging downstream signaling [53,54]. In this context, tumor cells maintain proliferative signaling despite sustained pharmacologic FGFR blockade, representing a mechanistically distinct form of resistance from kinase-domain mutation-driven escape.
The most commonly implicated bypass mechanism involves reactivation of the MAPK pathway, driven by acquired alterations in RAS or BRAF or by upregulation of alternative receptor tyrosine kinases such as MET, epidermal growth factor receptor (EGFR), or ERBB2 [55,56,57]. These changes restore ERK phosphorylation independently of FGFR2 activity and support continued tumor growth.
In addition to genomic alterations, tumors may adopt non-genetic adaptive programs. Transcriptional and epigenetic reprogramming, including epithelial–mesenchymal transition (EMT), has been observed in resistant models and clinical samples [58,59]. EMT is characterized by reduced epithelial marker expression and increased levels of mesenchymal regulators such as TWIST1, SNAIL, and ZEB1, promoting phenotypic plasticity and diminished dependence on FGFR2 signaling [60].
4.4. Role of Circulating Tumor DNA in Resistance Detection
Serial monitoring of circulating tumor DNA (ctDNA) has substantially enhanced the ability to characterize resistance evolution in patients receiving FGFR-targeted therapy. Multiple studies specifically in FGFR2-rearranged iCCA have demonstrated that ctDNA profiling can detect emergent FGFR2 kinase-domain mutations prior to radiographic progression, often providing clinically meaningful lead time and revealing marked intrapatient heterogeneity of resistance mechanisms [61].
In addition, important insights have come from basket trial populations that included, but were not limited to, CCA. In the FIGHT-207 basket study of pemigatinib in advanced solid tumors harboring FGFR1-3 alterations, which enrolled a subset of patients with CCA, longitudinal ctDNA analysis demonstrated that secondary FGFR2 mutations emerged almost exclusively in patients who initially derived clinical benefit, supporting the role of selective therapeutic pressure in shaping resistance [1].
Together, these CCA-specific cohorts and basket trial analyses establish ctDNA as a powerful, minimally invasive tool to capture the temporal and polyclonal nature of resistance to FGFR inhibition, although the relative sensitivity of plasma-based assays and the generalizability of findings across tumor types must be carefully considered.
5. Strategies to Overcome Resistance and Future Directions
5.1. Sequential and Next-Generation FGFR Inhibitors
Sequential use of FGFR inhibitors, often informed by emerging resistance mechanisms, has become an important exploratory therapeutic strategy in FGFR2-rearranged iCCA. Patients progressing on reversible ATP-competitive inhibitors who develop on-target kinase domain mutations may derive benefit from switching to irreversible inhibitors. Futibatinib, which forms a covalent bond with a conserved cysteine residue in the FGFR2 kinase domain, retains activity against multiple secondary FGFR2 mutations, although incomplete mutation coverage remains a challenge [16,62].
Beyond currently approved agents, ongoing structure-guided drug development and medicinal chemistry efforts aim to refine FGFR inhibition and address the molecular heterogeneity of resistance in FGFR2-driven cancers by leveraging multiple complementary strategies. One active direction is the design of next-generation reversible inhibitors with structure-optimized ATP-binding pockets and hinge interactions to better accommodate steric hindrance introduced by gatekeeper and molecular-brake mutations such as V565 and N550 variants [63,64,65]. In parallel, investigational covalent FGFR inhibitors are being optimized to improve engagement of conserved residues within the kinase domain and maintain potency against a broader array of resistant alleles while balancing selectivity and toxicity [66,67,68]. Recent structure-based scaffold redesign has yielded selective FGFR2/3 inhibitors with improved biochemical profiles against wild-type and mutant kinases, underscoring how rational scaffold modification can enhance both selectivity and resistance coverage [69]. Several next-generation inhibitors and allele-specific compounds are currently in early clinical development [70].
Tinengotinib (TT-00420) is a spectrum-selective, multikinase inhibitor with potent activity against FGFR1-3 along with Aurora A/B, VEGFR, JAK1/2, and CSF1R that has demonstrated clinical activity in patients with FGFR-altered CCA, including those previously treated with FGFR inhibitors [71]. In a phase II study (NCT04919642), tinengotinib showed objective responses across FGFR alteration subgroups, albeit with limited sample sizes. ORR was 6.3% (95% CI: 0.2–30.2) in patients with FGFR2 fusions and primary FGFR inhibitor resistance, 30.0% (95% CI: 6.7–65.3) in patients with FGFR2 fusions and acquired resistance, 23.1% (95% CI: 5.0–53.8) in other FGFR alterations, but 0% in FGFR wild type. Exploratory analyses suggested improved outcomes in FGFR inhibitor-naïve patients compared with those previously exposed, underscoring the continued impact of prior FGFR therapy on subsequent treatment efficacy [72].
More selective FGFR2 inhibitors have been developed to enhance on-target potency while minimizing off-target toxicity. Lirafugratinib (RLY-4008), a highly FGFR2-selective, covalent inhibitor, exhibits robust preclinical activity against common resistance mutations and avoids FGFR1- and FGFR4-mediated toxicities [67]. Early clinical data from the phase I/II ReFocus study (NCT04526106) demonstrated substantial activity in FGFR inhibitor-naïve FGFR2 fusion- or rearrangement-positive CCA [73], with more modest responses in patients previously treated with FGFR inhibitors, consistent with a resistance-driven disease biology [74]. Tasurgratinib (E7090), an irreversible, ATP competitive FGFR1-3 inhibitor, has also shown promising early activity in FGFR2-rearranged CCA in a phase I study (NCT02275910), although data remain preliminary [75]. SURF201 (NCT06160752) is investigating TYRA-200, an oral covalent FGFR1/2/3 inhibitor, in patients with advanced iCCA and other solid tumors with primary activating alterations in FGFR2 and on-target acquired FGFR2 resistance mutations [68]. Collectively, these next-generation agents illustrate evolving strategies to extend clinical benefit through improved kinase selectivity and broader coverage of resistance mutations.
5.2. Combination Strategies Targeting Bypass Pathways
Given the frequent activation of alternative signaling routes following FGFR inhibition, rational combination therapies have emerged primarily from preclinical and early translational studies as an attractive strategy to delay or overcome acquired resistance. Combination approaches have focused on simultaneously targeting FGFR2 and key downstream or parallel signaling nodes that sustain tumor growth despite receptor blockade.
In preclinical models of FGFR2 fusion-positive CCA, combinations of FGFR inhibitors with agents targeting the MAPK pathway or selected receptor tyrosine kinases have demonstrated synergistic antitumor activity, including enhanced growth suppression and increased apoptotic responses in vitro and in vivo [53,57]. These studies provide proof-of-concept that dual-pathway inhibition may delay the expansion of resistant subclones. However, clinical validation of these approaches remains limited, and their safety, tolerability, and efficacy require prospective evaluation in early-phase clinical trials.
In parallel, combination strategies incorporating inhibitors of the PI3K/AKT/mTOR axis have been investigated in preclinical FGFR-driven CCA models. Co-targeting FGFR and PI3K–mTOR signaling has been shown to restore sensitivity to FGFR inhibition and enhance cytotoxic responses, including induction of autophagic cell death in resistant models [54,76]. While these findings highlight the therapeutic potential of multi-pathway suppression, PI3K–mTOR-based combination approaches remain investigational and should currently be pursued within carefully designed clinical trials.
5.3. Role of Antibody-Based and Non-TKI Strategies
Beyond small-molecule kinase inhibitors, alternative therapeutic strategies that remain largely preclinical are being explored to target FGFR2 signaling while bypassing resistance driven by kinase-domain mutations. Beyond classical inhibition, proteolysis-targeting chimera (PROTAC)-based degraders directed against the FGFR family are emerging as a promising investigational non-occupancy-based therapeutic modality [64]. Early FGFR-directed degraders have demonstrated potent depletion of FGFR2 and antiproliferative activity in FGFR-dependent models [77,78], establishing the feasibility of targeted FGFR degradation.
Although FGFR2-selective degraders and constructs engineered to address gatekeeper mutations remain in early preclinical development, the PROTAC approach offers a mechanistically distinct strategy to eliminate both wild-type and mutant FGFR2 proteins rather than merely inhibit their kinase activity [64,79]. As such, PROTAC-based FGFR targeting represents a compelling translational concept, though substantial optimization and clinical evaluation will be required before applicability in biliary tract cancer can be established.
Additional innovations include antibody-based approaches that target the extracellular domain of FGFR2, enabling ligand blockade and receptor inhibition independent of ATP-binding site conformation and potentially retaining activity despite certain kinase-domain mutations. Antibody-based FGFR2-targeted strategies have shown proof-of-concept activity in preclinical studies, but clinical development has thus far been limited, and no antibody-based FGFR2 therapies are currently approved for biliary tract cancer.
The earliest clinical agent, aprutumab ixadotin (BAY1187982), an FGFR2-directed antibody-drug conjugate (ADC), demonstrated selective cytotoxicity in FGFR2-expressing preclinical models and early signs of antitumor activity; however, dose-limiting ocular and hematologic toxicities, together with limited clinical efficacy, led to early termination of its first-in-human study in advanced solid tumors [80]. Nonetheless, its development established proof of concept for FGFR2-targeted biologics as an alternative to kinase inhibition. Parallel efforts are now focused on engineering next-generation ADCs with improved antibody backbones and refined linker-payload designs to achieve greater tumor selectivity while minimizing systemic toxicity [81,82]. Biparatopic antibodies that bind multiple non-overlapping extracellular FGFR2 epitopes demonstrate enhanced receptor blockade, internalization, and antitumor activity in preclinical FGFR2 fusion-driven CCA models, including activity against kinase-mutant variants, supporting their potential as innovative therapeutics [83]. Additional modalities, including FGFR-targeted bispecific antibodies, FGF-ligand-trap fusion proteins, and FGFR-directed CAR-T constructs, are being explored across FGFR-altered solid tumors, but data specific to FGFR2-fusion or FGFR2-mutant CCA are still limited [84,85,86]. These emerging biologic strategies offer mechanistically orthogonal approaches to ATP-competitive inhibition and may ultimately provide therapeutic options for patients with TKI-refractory FGFR2-driven disease once further preclinical and clinical evaluation is completed (Figure 2).
5.4. Integration of ctDNA-Guided Precision Oncology
The integration of ctDNA profiling into clinical workflows represents a promising advance for patients with FGFR2-rearranged iCCA. In CCA-focused studies, serial plasma analysis has enabled non-invasive detection of emergent on-target resistance mutations, delineation of polyclonal evolution, and dynamic monitoring of molecular response during FGFR inhibitor therapy [1,61].
Insights from larger basket trials that included CCA, such as FIGHT-207, further demonstrate the feasibility of longitudinal ctDNA monitoring across FGFR-altered solid tumors and support its role in identifying resistance mechanisms that may inform therapeutic sequencing or clinical trial selection. However, because such studies enroll heterogeneous tumor types, extrapolation to CCA should be performed cautiously and interpreted in the context of disease-specific datasets [87].
As ctDNA technologies mature, prospective studies focused specifically on FGFR2-rearranged CCA will be critical to define standardized testing intervals, analytic thresholds, and actionable resistance frameworks [88].
5.5. Clinical Implications and Treatment Sequencing Considerations
In current clinical practice, FGFR inhibitors are typically employed in patients with advanced FGFR2-rearranged iHCC following progression on first-line chemotherapy, consistent with regulatory approvals and guideline recommendations. Selection among available FGFR inhibitors is influenced by prior FGFR inhibitor exposure, toxicity profiles, and the presence of known resistance mutations.
Repeating molecular assessment at disease progression is increasingly important. Tissue biopsy remains the gold standard for comprehensive genomic characterization; however, serial ctDNA analysis offers a minimally invasive alternative that can capture emerging resistance mutations and polyclonal evolution in real time. In practice, ctDNA profiling is particularly useful when tissue acquisition is not feasible or when rapid therapeutic decision-making is required, while tissue biopsy may be preferred when histologic transformation or non-genomic resistance is suspected.
Next-generation FGFR inhibitors and rational combination strategies are most appropriately considered in the context of clinical trials, especially for patients with acquired resistance to first-generation FGFR inhibitors. As prospective data mature, integrating resistance-guided sequencing strategies may further refine personalized treatment approaches for FGFR2-rearranged disease.
5.6. Adaptive Clinical Trial Designs
Beyond current clinical practice, adaptive platform trial designs that allocate patients to treatment arms based on molecular resistance profiles represent an efficient and increasingly attractive framework for evaluating emerging therapies in precision oncology. Adaptive platform trials that allocate patients to treatment arms based on molecular resistance profiles represent an efficient and increasingly attractive framework for evaluating emerging therapies in precision oncology. Such designs facilitate the rapid assessment of combination regimens or allele-specific inhibitors in small, molecularly defined cohorts [89]. Integrating real-time ctDNA monitoring within these frameworks could enable earlier identification of molecular progression, support adaptive treatment modification, improve response durability, and generate longitudinal data to refine molecular response endpoints, although widespread clinical implementation remains investigational [90].
6. Conclusions
The discovery of FGFR2 rearrangements as a defining oncogenic driver in intrahepatic cholangiocarcinoma has transformed the therapeutic landscape of biliary tract cancer. The development of selective FGFR inhibitors such as pemigatinib and futibatinib represents a major advance in precision oncology for this rare but aggressive malignancy, supported by consistent clinical activity across multiple phase II trials and resulting in regulatory approvals. FGFR2-directed therapy is now an established molecularly guided treatment option for patients with FGFR2-rearranged disease. Nevertheless, acquired resistance—driven by heterogeneous kinase-domain mutations and bypass signaling—remains a formidable obstacle to long-term disease control.
Ongoing translational and clinical research is focused on elucidating the molecular basis of resistance, optimizing sequencing strategies, and developing next-generation or combination regimens to extend the benefit of FGFR inhibition. Incorporating serial ctDNA profiling as an emerging clinical tool may enable dynamic, mutation-guided therapy selection and earlier identification of resistance. Ultimately, multidisciplinary collaboration among clinicians, translational scientists, and trial investigators will be essential to fully realize and extend the potential of FGFR2-directed precision therapy in biliary tract cancer.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Rodon J. Damian S. Furqan M. Garcia-Donas J. Imai H. Italiano A. Spanggaard I. Ueno M. Yokota T. Veronese M.L. Pemigatinib in previously treated solid tumors with activating FGFR 1-FGFR 3 alterations: Phase 2 FIGHT-207 basket trial Nat. Med.20243016451654 Erratum in Nat. Med. 2024, 30, 2377. https://doi.org/10.1038/s 41591-024-03072-w 10.1038/s 41591-024-02934-738710951 PMC 11186762 · doi ↗ · pubmed ↗
- 2Lamarca A. Barriuso J. Mc Namara M.G. Valle J.W. Biliary Tract Cancer: State of the Art and potential role of DNA Damage Repair Cancer Treat. Rev.20187016817710.1016/j.ctrv.2018.09.00230218788 · doi ↗ · pubmed ↗
- 3Valle J. Wasan H. Palmer D.H. Cunningham D. Anthoney A. Maraveyas A. Madhusudan S. Iveson T. Hughes S. Pereira S.P. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer N. Engl. J. Med.20103621273128110.1056/NEJ Moa 090872120375404 · doi ↗ · pubmed ↗
- 4Oh D.Y. Ruth He A. Qin S. Chen L.T. Okusaka T. Vogel A. Kim J.W. Suksombooncharoen T. Ah Lee M. Kitano M. Durvalumab plus Gemcitabine and Cisplatin in Advanced Biliary Tract Cancer NEJM Evid.20221 EVI Doa 220001510.1056/EVI Doa 220001538319896 · doi ↗ · pubmed ↗
- 5Oh D.Y. He A.R. Qin S. Chen L.T. Okusaka T. Kim J.W. Suksombooncharoen T. Lee M.A. Kitano M. Burris H.A. Durvalumab plus chemotherapy in advanced biliary tract cancer: 3-year overall survival update from the phase III TOPAZ-1 study J. Hepatol.2025831092110110.1016/j.jhep.2025.05.00340381735 · doi ↗ · pubmed ↗
- 6Kelley R.K. Ueno M. Yoo C. Finn R.S. Furuse J. Ren Z. Yau T. Klumpen H.J. Chan S.L. Ozaka M. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): A randomised, double-blind, placebo-controlled, phase 3 trial Lancet 202340118531865 Erratum in Lancet 2023, 402, 964. https://doi.org/10.1016/S 0140-6736(23)01904-9; Erratum in Lancet 2024, 403, 1140. https://doi.org/10.1016/S 0140-6736(24)00545-210.1 · doi ↗ · pubmed ↗
- 7Farshidfar F. Zheng S. Gingras M.C. Newton Y. Shih J. Robertson A.G. Hinoue T. Hoadley K.A. Gibb E.A. Roszik J. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles Cell Rep.2017182780279410.1016/j.celrep.2017.02.03328297679 PMC 5493145 · doi ↗ · pubmed ↗
- 8Jusakul A. Cutcutache I. Yong C.H. Lim J.Q. Huang M.N. Padmanabhan N. Nellore V. Kongpetch S. Ng A.W.T. Ng L.M. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma Cancer Discov.201771116113510.1158/2159-8290.CD-17-036828667006 PMC 5628134 · doi ↗ · pubmed ↗