The Effects of BRCA1 and BRCA2 Promoter Methylation on Clinicopathological Characteristics and Clinical Outcomes in HGSOC
Katarina Živić, Ivana Boljević, Milica Nedeljković, Milana Matović, Radmila Janković, Miljana Tanić

TL;DR
This study examines how BRCA1 and BRCA2 gene methylation affects ovarian cancer patients' clinicopathological features and survival outcomes.
Contribution
The study reports the prevalence of BRCA1/2 promoter methylation in a Serbian HGSOC cohort and its association with clinicopathological features.
Findings
Full BRCA1/2 promoter methylation was found in 4.1% and 0.45% of patients, respectively.
Methylation was associated with younger age of onset compared to BRCA1/2-mutated cases.
No strong survival advantage was observed for methylated cases in the follow-up analysis.
Abstract
Ovarian cancer is a highly lethal disease. Tumors with a deficiency in the homologous recombination repair pathway (HRD) resulting from mutations in BRCA1/2 genes have a favorable response to platinum-based chemotherapy and targeted therapy with PARP inhibitors (PARPi) mediated by synthetic lethality. Promoter methylation of BRCA1/2 genes was previously associated with HRD, but little is known about whether it translates to clinical benefit. Here, we evaluated the prevalence of BRCA1/2 promoter methylation in HGSOC patients from Serbia and examined their clinicopathological characteristics and the effect on progression-free and overall survival. Using methylation-specific PCR, we screened for hypermethylation in the promoter region of BRCA1/2 genes in a cohort of 244 patients. We found fully methylated BRCA1 and BRCA2 promoter in 4.1% and 0.45% of patients, and 23.36% and 11.21%…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
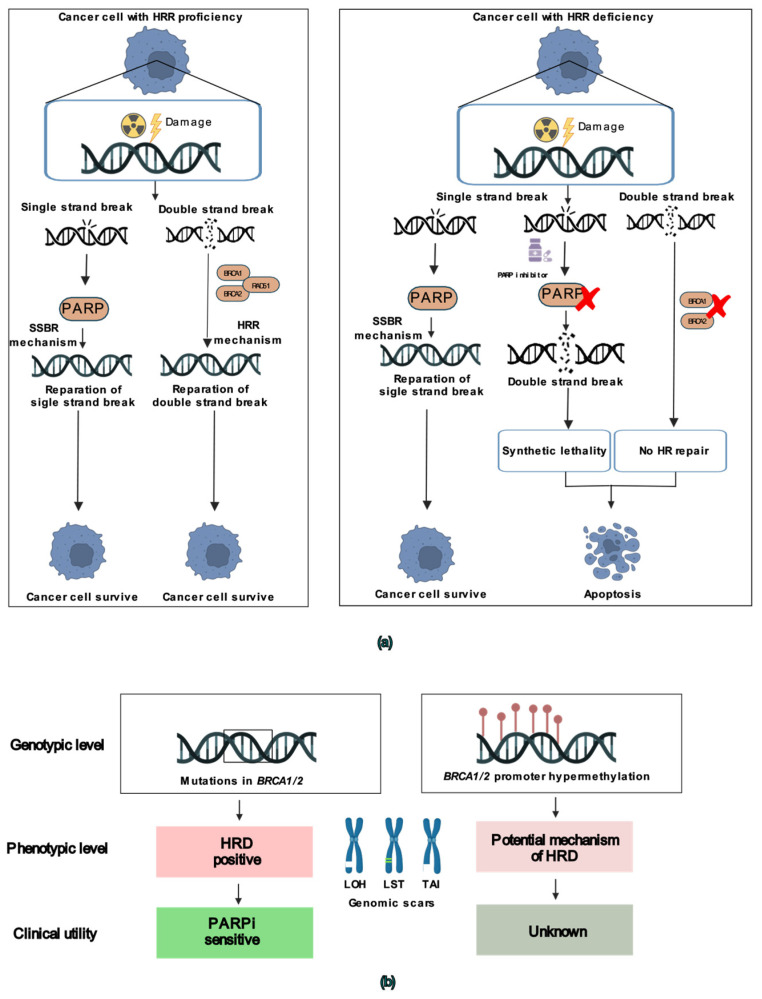 Figure 1
Figure 1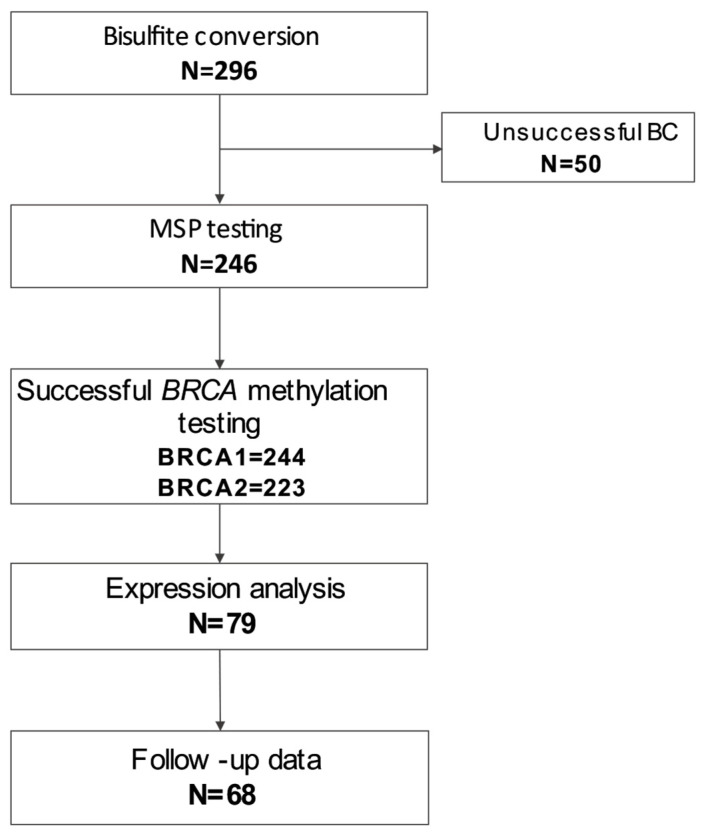 Figure 2
Figure 2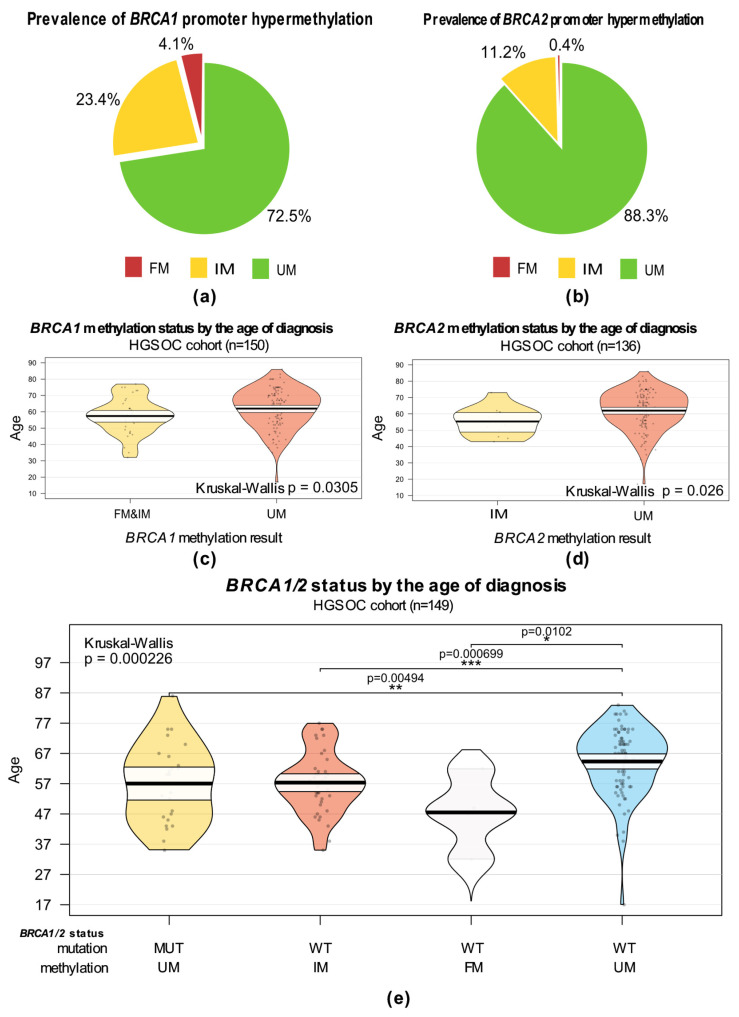 Figure 3
Figure 3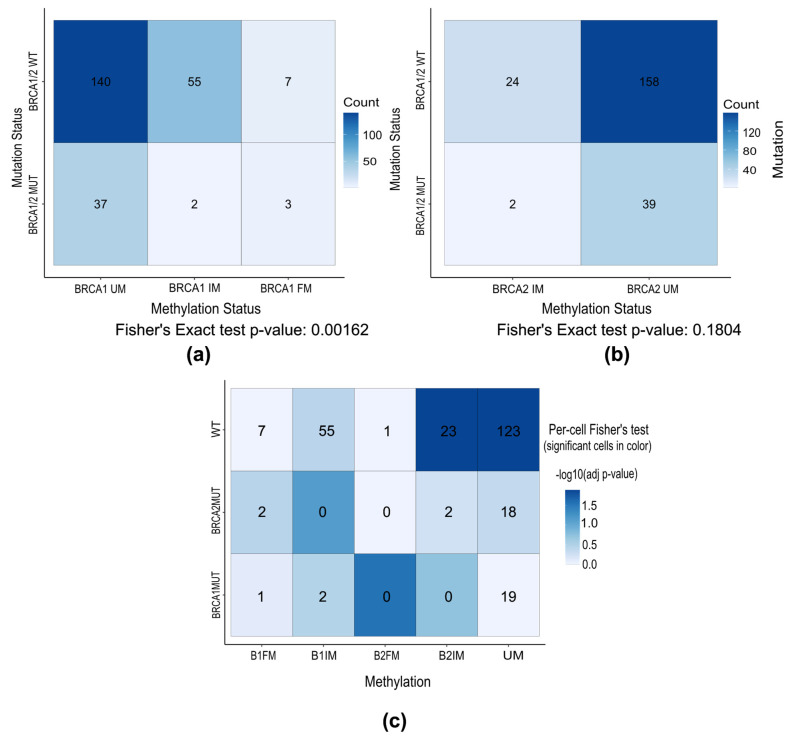 Figure 4
Figure 4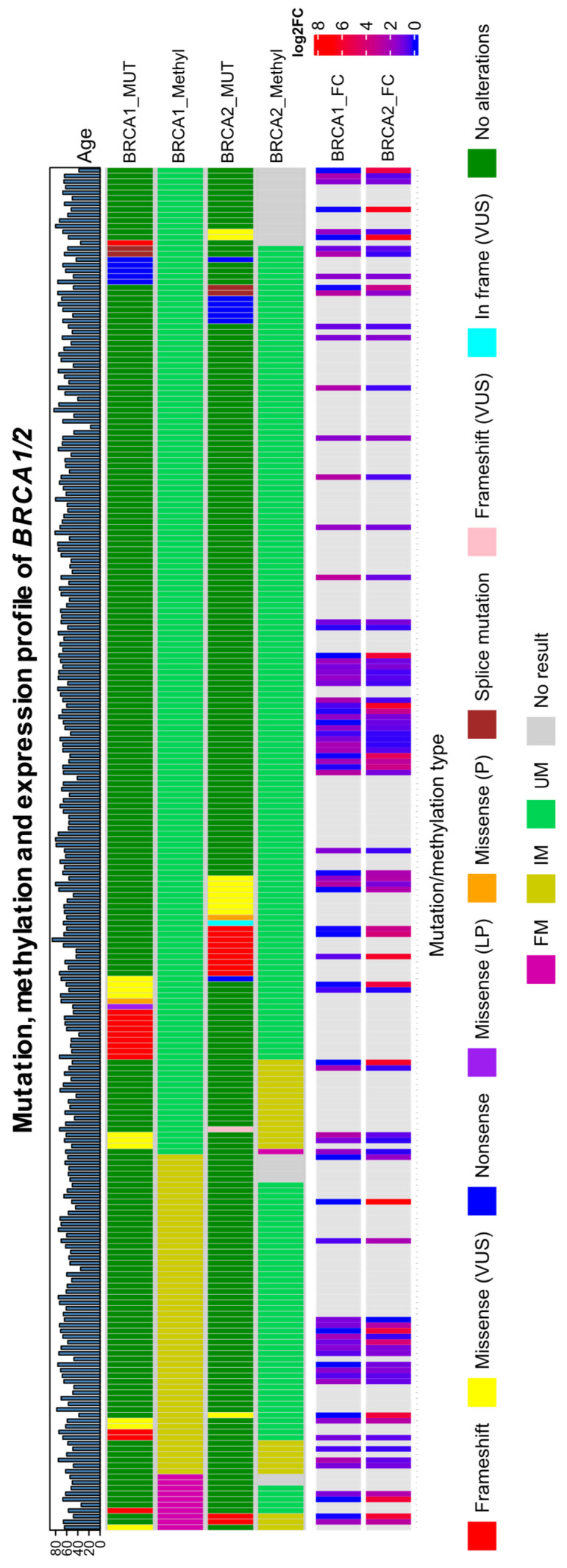 Figure 5
Figure 5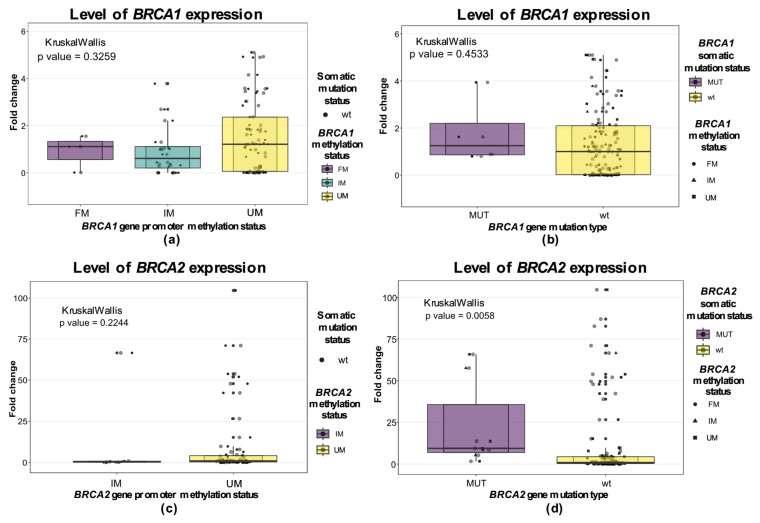 Figure 6
Figure 6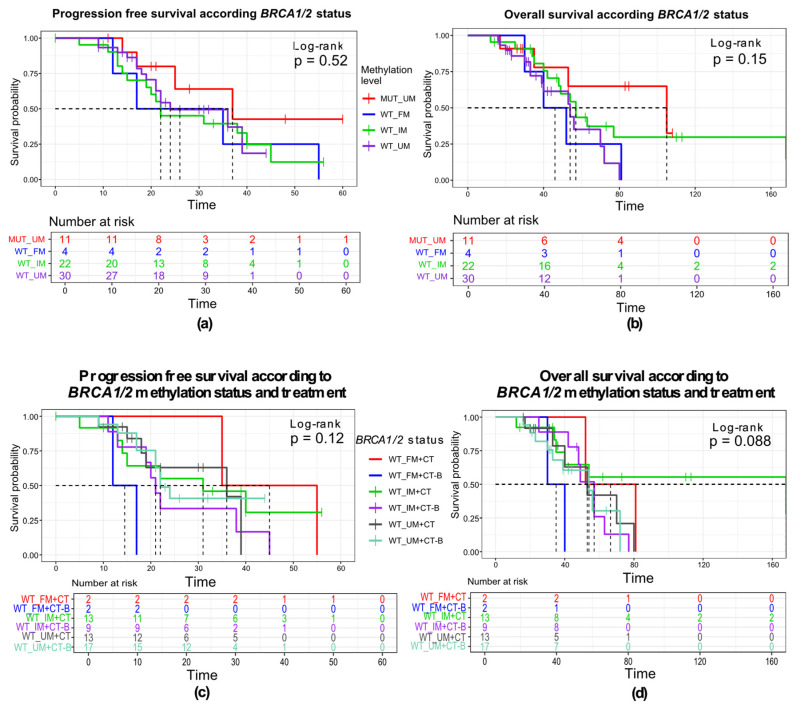 Figure 7
Figure 7- —Ministry of Education, Science, and Technological Development
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPARP inhibition in cancer therapy · Ovarian cancer diagnosis and treatment · BRCA gene mutations in cancer
1. Introduction
Ovarian cancer (OC) remains the most lethal gynecological cancer, and Europe has the highest age-standardized incidence in the world of 6.6 [1]. This disease is typically presented in advanced stages, due to limited screening techniques and the absence of specific symptoms [2]. Ovarian cancer is a heterogeneous disease with several histological subtypes that harbor different molecular characteristics [2]. Epithelial ovarian cancer (EOC) is the most common one, with the high-grade serous subtype representing the vast majority of all cases [3,4].
The influence of germline and somatic mutations in BRCA1 and BRCA2 tumor suppressor genes is well established in the origin of ovarian cancer. The lifetime risk of developing OC is 28–66% in patients harboring BRCA1 alterations, and 16–27% in those carrying BRCA2 mutations [5,6,7]. BRCA1 and BRCA2 proteins are necessary for the repair of double-strand DNA breaks (DSBs) and interstrand cross-links (ICLs) by homologous recombination [8]. Homologous recombination cannot be performed in the cells with mutated BRCA1 or BRCA2, and DSBs are repaired through alternative error-prone mechanisms, which may cause genetic instability in that cell and malignant transformation. DSBs can be repaired in mammalian cells by non-homologous end joining (NHEJ), which is potentially error-prone because nucleotide changes are accommodated at the rejoining sites, or single-strand annealing (SSA), where homology is between extended stretches of single-stranded DNA (ssDNA) [8]. The resulting homologous recombination deficiency (HRD) is manifested by a specific “genomic scar” characterized by the loss of heterozygosity (LOH), large-scale transitions (LSTs), and telomeric allelic imbalance TAI.
Patients with BRCA1/2 mutations show better response to platinum-based chemotherapy generating ICLs, as well as to targeted treatment with poly (ADP-ribose) polymerase inhibitors (PARPis) by the mechanism of synthetic lethality (Figure 1a) [9,10]. The prevalence of germline and somatic mutations in BRCA1/2 genes varies, ranging between 17 and 25%, while HRD phenotype was shown to be present in approximately half of OC patients and was recently introduced as an independent biomarker for therapy with PARPi [11,12].
Beyond BRCA1/2 mutations, mutations in other homologous recombination repair genes or silencing by epigenetic mechanisms may also cause HRD. Hypermethylation of CpG islands in regulatory regions of tumor suppressor genes is already established for many malignancies [13]. In ovarian cancer, several studies reported BRCA1 promoter hypermethylation in ~10% of cases, and more rarely, BRCA2 promoter hypermethylation [11]. As BRCA1/2 promoter methylation was shown to be associated with BRCAness-like phenotype and to be a 100% predictor of HRD [14], it was postulated that the treatment with platinum-based therapy and PARPi would be as successful in tumors with this type of epigenetic alteration and could be used as an independent biomarker. However, there is a lack of concordance among studies of clinical benefit conferred by a potentially reversable epigenetic inactivation of BRCA1/2.
The prevalence of hereditary BRCA1/2 mutations is known to be population-specific, ranging from 5 to 10% in Western European populations to as high as 26–50% in Slavic and Asian populations [15,16,17]. However, little is known regarding the prevalence of epigenetic alterations among non-western European populations. In this study, we aimed to evaluate the prevalence of BRCA1/2 promoter methylation in a cohort of ovarian cancer patients who underwent tumor BRCA1/2 mutation testing at the Institute for Oncology and Radiology of Serbia (IORS). Additionally, expression analysis was performed to provide insight into gene silencing, as well as the impact of different genetic and/or epigenetic alterations on the level of mRNA. In a subgroup of patients, we evaluated the impact of BRCA1/2 promoter methylation on progression-free survival (PFS) and overall survival (OS) rate, as well as on different clinicopathological characteristics.
2. Materials and Methods
2.1. Patients and Samples
The study group was a retrospective cohort drawn from consecutive ovarian cancer patients, unselected for family history, who underwent reflex BRCA1/2 tumor testing at the Institute for Oncology and Radiology of Serbia (IORS) from 2019 to 2023. The eligibility criteria were present information about the BRCA1/2 mutation status and enough isolated DNA for further methylation analysis. A total of 296 samples were selected for BRCA1/2 promoter hypermethylation testing by methylation-specific PCR (MSP) from exceeding diagnostic FFPE tumor material. One group of 50 samples was excluded because of unsuccessful pretreatment of DNA prior to MSP; in total, 246 samples were tested by MSP. One subgroup of 79 methylation-tested samples, for which there was sufficient leftover FFPE blocks for RNA isolation, was used for testing the expression level of BRCA1/2 genes by qRT-PCR technology. For a total of 68 patients treated at IORS, we were able to obtain clinicopathological data from the National Health Repository to be placed into an anonymized study database. The study was conducted in accordance with the Declaration of Helsinki and approved by the local Ethics Committee (No. 01-1/2023/702). In Figure 2, the flowchart of the study design is represented.
2.2. DNA and RNA Extraction
FFPE blocks obtained from primary surgery from women with serous ovarian cancer were used to extract DNA and RNA. Tumors were fixed and prepared by local pathology departments after material was gathered from patient surgeries conducted at IORS and/or medical facilities throughout Serbia. The tumor tissue percentage (TTC%) was determined by a resident pathologist, and the identified tumor-rich tissue areas ware macro-dissected. DNA was extracted using QIAamp FFPE Tissue Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s instructions. In short, after the macro-dissection of FFPE blocks, xylol and absolute ethanol were used for deparaffinization, ATL buffer and proteinase K were used for lysis for 1 h at 56 °C and 1 h at 90 °C, and isolation and purification of DNA were performed in QIAamp MinElute columns. Elution volume was 50–100 µL of nuclease-free water. Quantification was performed using the Qubit HS/BR dsDNA kit. RNA material was extracted by RNeasy FFPE Kit (Qiagen, Hilden, Germany) according to the manufacturer’s recommendation. The deparaffinization procedure was performed with xylol and ethanol, PKD buffer and proteinase K, with incubation steps on 37 °C, 56 °C, and 80 °C, after which RNA was extracted on columns and eluted with 17 µL of nuclease-free water. The derived RNA was quantified using the BioSpec-nano Spectrophotometer (Shimadzu Biotech, Kyoto, Japan).
2.3. Bisulfite Conversion
The conversion of unmethylated cytosine was performed by bisulfite treatment [18]. Bisulfite conversion and subsequent purification was performed using one of the available kits—EZ-96 DNA Methylation-Lightning™ MagPrep, EZ-96 DNA Methylation-Direct MagPrep and EZ-96 DNA Methylation-Direct (Zymo Research, Irvine, CA, USA), according to the respective kit protocols. The success of bisulfite conversion was confirmed by the amplification of samples with primers for housekeeping gene Actinβ (FW: 5′-TGGTGATGGAGGAGGTTTAGTAAGT-3′; REV: 5′-AACCAATAAAACCTACTCCTCCCTTAA-3′) on Light Cycler 480 Instrument (Roche, Basel, Switzerland) machine under the following conditions: one cycle of denaturation at 95 °C for 10 min, followed by 40 cycles of the 15 s at 95 °C and 1 min at 60 °C, using Power SYBR Green Master Mix (Applied Biosystems Thermo Fisher Scientific, Waltham, MA, USA), or by PCR protocol using AmpliTaq Gold 360 master mix (AppliedBiosystems, Thermo Fisher Scientific, Waltham, MA, USA) and subsequent visualization on 2% agarose gel electrophoresis.
2.4. Methylation Status Analysis
A methylation-specific PCR (MSP) reaction was used to detect the methylation profile. For this purpose, we utilized two sets of primers for each of the tested genes. The unmethylated DNA was amplified using one set of primers, and the methylated DNA was amplified using another set of primers. All primers were extracted from the literature [19]. For BRCA1 gene methylation analysis, primer pairs were as follows: methylated forward 5′-TCGTGGTAACGGAAAAGCGC-3′, methylated reverse 5′-AAATCTCAACGAACTC ACGCCG-3′, unmethylated forward 5′-TGGTTTTTGTGGTAATGGAAAAGTGT-3′, unmethylated reverse 5′-CAAAAAATCTCAACAAACTCACACCA-3′ [20]; and for BRCA2 gene methylation analysis: methylated forward 5′-GCGGTAGAGGCGGAGTC-3′, methylated reverse 5′-CGAAATAAACTAACAAAAACCG-3′, unmethylated forward 5′-ATTAGGTGGTAGAGGTGGAGTT-3′, unmethylated reverse 5′-CCAAAATAAACTAA CAAAAACCA-3′ [21]. The PCR amplification was performed with bisulfite-treated DNA as a template, using commercial PCR master mix AmpliTaq Gold 360 (AppliedBiosystems, Thermo Fisher Scientific, Waltham, MA, USA), and cycling conditions were as follows: denaturation at 95 °C for 10 min, followed by 40 cycles of the 30s at 95 °C, 30 s at 60 °C, 45 s at 72 °C, and one cycle of final elongation of 7 min at 72 °C for BRCA2 analysis; for BRCA1, we used the program and protocol previously described by Ben Gacem et al. [19]. Commercial positive (fully methylated) and negative (fully unmethylated) controls were tested in the same way alongside the samples (Human Methylated and Unmethylated DNA Set, Zymo Research, Irvine, CA, USA). PCR products were visualized on 2% agarose gel by electrophoresis or with high sense and/or 1000DNA chips on Agilent 2100 Bioanalyzer instrument (Agilent, Santa Clara, CA, USA). Samples were referred to as FM—fully methylated—if there was only one band in the agarose gel in the well with the PCR product with the primers for the methylated sequence. Those with only one band in the agarose gel in the well with the PCR product with the primers for the unmethylated sequence were referred to as UM—unmethylated. Samples with both bands were classified as IM—intermediately methylated—which may represent either the allele-specific methylation in tumor samples, subclonal fully methylated tumor tissue, or the clonal fully methylated tumor cells confounded with unmethylated normal stroma and/or immune infiltrate.
2.5. Real-Time PCR
To detect the expression level of BRCA1/2 genes, we performed a quantitative real-time PCR reaction. Reverse transcription was performed using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Vilnius, Lithuania). Expression measuring was performed by quantitative real-time PCR on Light cycler 480 Instrument (Roche, Basel, Switzerland) using Power SYBR Green Master Mix (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA). For this reaction, we used primers that were previously successfully used and were retrieved from PrimerBank (https://pga.mgh.harvard.edu/primerbank/ (accessed on 16 June 2023)). Primer pairs were as follows: for BRCA1 forward 5′-ACCTTGGAACTGTGAGAACTCT-3′, reverse 5′-TCTTGATCTCCCACACTGCAATA-3′; for BRCA2 forward 5′-TGCCTGAAAACCAGATGACTATC-3′, reverse 5′-AGGCCAGCAAACTTCCGTTTA-3′; housekeeping GAPDH gene forward 5′-GACAGTCAGCCGCATCTTCT-3′, reverse 5′-GCGCCCAATACGACCAAATC-3′. PCR conditions included initial denaturation at 95 °C for 2 min, followed by 45 cycles of 15 s at 95 °C and 1 min at 60 °C. Missing Ct values were overwritten with the maximum number of cycles in the reaction. Further, ΔCt was calculated relative to GAPDH, as the subtraction of Ct values of the gene of interest and Ct of GAPDH. ΔΔCt was calculated as the subtraction of ΔCt and the median ΔCt value for the gene. Finally, fold change was calculated as a negative exponential of the ΔΔCt value, according to the formula 2^−ΔΔCt^, and used in statistical analysis.
2.6. Statistics
Descriptive statistics were used to ascertain the patients’ clinical and demographic characteristics. Overall survival (OS) was computed from the date of the initial diagnosis to the date of death or the loss of follow-up. The period between the initial diagnosis date and the date of recurrence or the most recent follow-up was used to compute progression-free survival (PFS). Progression-free interval (PFI) was calculated from the date of the last chemotherapy cycle to the date of progression/recurrence or loss of follow-up. Using Fisher’s exact test, we examined the association of promoter methylation and mutation of the BRCA1 and BRCA2 genes, as well as the association with clinicopathological data. The Shapiro–Wilk test was employed for determining the normality of distribution, and the differences in age distribution across groups were tested by the Kruskal–Wallis test, with the post hoc Dunn test. We used the same Kruskal–Wallis test to assess whether expression levels differ within the methylation and/or mutation groups. The Kaplan–Meier technique was used to plot survival functions and the log-rank test was used to test for differences in survival between groups. Cox proportional hazard regression models were used to perform multivariable analysis of OS, adjusting for mutation status, age, patient group, and therapy approach using the “survival” version 3.8-3 R package. Post hoc power analysis was performed using the “powerSurvEpi” version 0.1.5. R package. Statistical analysis was performed using RStudio (version: 4.3.1). Two-tailed p-values less than 0.05 were considered statistically significant.
3. Results
3.1. Patient Characteristics
The cohort tested for hypermethylation consisted of both newly diagnosed (ND) and relapsed (R) cases of HGSOC patients due to evolving clinical criteria used to refer patients for BRCA1/2 tumor mutation testing as companion diagnostics for PARPi therapy. BRCA1/2 methylation testing was always performed on FFPE blocks obtained during primary cytoreductive surgery or biopsy prior to first-line treatment, thus providing the baseline methylation status of these tumors.
The cohort of 296 samples selected for BRCA1/2 promoter hypermethylation testing consisted of 186 relapsed and 109 newly diagnosed cases of ovarian cancer. After bisulfite conversion, a total of 246 successfully converted samples were tested by MSP. The tested group consisted of 190 BRCA1/2 benign (wild-type—WT), 21 BRCA1 pathogenic/likely pathogenic, 21 BRCA2 pathogenic/likely pathogenic, and 14 BRCA1/2 VUS cases of ovarian cancer patients that were treated as WT. Two patients were excluded, as the MSP testing was unsuccessful for both BRCA1 and BRCA2. The median age at testing was 62 (range: 17–86), and 61 at diagnosis (available for 150 patients—newly diagnosed and relapsed with the available information about disease history). Most cases (~90%) were presented in the advanced stage (FIGO category III and IV). Regarding the treatment approach, 58% of patients, whose clinicopathological data we were able to obtain, were treated with chemotherapy in combination with targeted maintenance therapy with either bevacizumab or olaparib. Response to platinum-based treatment was described as sensitive if more than 12 months passed from the last chemotherapy cycle to progression; it was described as partial if PFI was between 6 and 12 months; and patients with PFI < 6 months were resistant. Patients and clinical characteristics are presented in Table 1.
3.2. Prevalence of Promoter Hypermethylation in BRCA1/2 Genes
A total of 296 DNA samples were bisulfite converted. Bisulfite conversion treatment was not successful in 50 samples; hence, the hypermethylation of BRCA1/2 promoter regions was tested in a total of 246 patients, with successful BRCA1 hypermethylation testing in 244 (99.19%) samples and BRCA2 hypermethylation testing in 223 (90.65%) samples.
We evaluated the percentage of patients who had a fully methylated (FM), intermediately methylated (IM), or unmethylated (UM) promoter region of the BRCA1 and BRCA2 genes based on the presence of a band in the agarose gel electropherogram after methylation-specific PCR (Supplementary Figure S1). While a fully methylated or unmethylated result implies the clonal homozygous methylation status of both alleles, the intermediate level of methylation reflects the presence of cells with both unmethylated and methylated CpGs in the promoter but does not provide information on the level of methylation, its origin, zygosity, or clonal architecture. Fully methylated promoter was found in a small number of samples; for BRCA1 it was 4.1% of samples (10/244), and for BRCA2 it was 0.45%, only in one sample (1/223). Intermediately methylated BRCA1 and BRCA2 promoters were found in 23.36% (57/244) and 11.21% (25/223) of samples, respectively. The rest of the samples were found to have fully unmethylated promoter: 72.5% (177/244) for BRCA1 and 88.34% (197/223) for BRCA2 (Figure 3a,b).
3.3. Epigenetic Alterations and Clinicopathological Characteristics
The clinicopathological features based on BRCA1/2 methylation status in the tested cohort are represented in Table 2, and the ones based on overall BRCA1/2 aberration status are presented in Supplementary Table S1.
The mean age of diagnosis was 62 for both BRCA1 and BRCA2 methylation-tested groups. By comparing the age distribution at diagnosis according to the methylation test result, shown in Figure 3c,d, we found that cases with methylated promoters of BRCA1 or BRCA2 genes are more common in younger patients (p = 0.026 and p = 0.0305, respectively). However, the difference lost its significance (p = 0.0618) when FM and IM cases are considered separately for BRCA1 (Supplementary Figure S2a). However, the number of FM cases in BRCA1 is very small for adequate comparison and for BRCA2 we found hypermethylation in only one case aged 62 at testing. This case was included in the IM group for age statistics. We observed similarities in age distribution between BRCA1/2-mutated and BRCA1/2 intermediately methylated cases, with BRCA1/2 fully methylated patients being even younger (p = 0.000226) (Figure 3e). Distribution of age at testing for these groups is presented in Supplementary Figures S2 and S3.
Of 68 patients whose family history we were able to extract, 19 patients had a family history of either ovarian or breast cancer, and it was observed that the BRCA1 gene is preferably unmethylated in this group of patients (two-sided Fisher’s test, p = 0.0277). However, no significant difference was found for BRCA2 methylation status in relation to family history. There was no significant association of BRCA1/2 methylation status and FIGO stage (Fisher’s exact test p-values of 0.573 and 0.147 for BRCA1 and BRCA2 methylation cohorts, respectively). We found a significant enrichment of tumors with both BRCA1 and BRCA2 baseline methylation among patients who had a relapse (p = 0.002 and p < 0.001, respectively). This group consisted exclusively of patients who had an initial good response to platinum chemotherapy, unlike the newly diagnosed group, which potentially consisted of both platinum-sensitive and platinum-resistant tumors.
3.4. Co-Occurrence Analysis of Genetic and Epigenetic BRCA1 and BRCA2 Inactivation
Using Fisher’s exact test, we tested for association between promoter methylation and mutation status in BRCA1/2 genes to confirm if hypermethylation and mutations are mutually exclusive events. We found that BRCA1 promoter methylation (FM and IM observed together as BRCA1_M) and BRCA1/2 mutations are mutually exclusive (one-sided, alternative = “less”; p = 0.008; OR = 0.31). Association remains significant even when BRCA1 methylation status is represented through all three categories as FM, IM, and UM (Fisher’s exact two-sided test; p = 0.0016), which is represented in Figure 4a. BRCA1/2 mutation status is also represented in three categories: BRCA1_MUT, BRCA2_MUT, and WT (two-sided Fisher’s exact test, p = 0.0055). A fully methylated BRCA1 promoter was found in two samples in co-occurrence with BRCA1 VUS and pathogenic BRCA1 alteration. And in two samples harboring pathogenic and likely pathogenic BRCA2 variants, a fully methylated BRCA1 promoter was detected. In three of these cases, BRCA2 methylation status was described as intermediately methylated, and in BRCA1-mutated it was described as unmethylated.
No significant association was found between BRCA2 promoter methylation and BRCA1/2 mutation status (two-sided Fisher’s exact test, p = 0.1804 and p = 0.3711 when BRCA1/2 mutations were represented in three categories) (Figure 4b), as well as between BRCA1 and BRCA2 methylation status (two-sided Fisher’s exact test, p = 0.1399). Most cases were wild-type and unmethylated for BRCA1/2. OncoPrint representation of all BRCA1/2 aberrations found in patients in the HGSOC cohort from this study, as well as fold change rate of expression level, are shown in Figure 5.
3.5. Methylation Effects on Gene Expression
To verify if methylation and/or mutations have an impact on expression level, we performed quantitative RT-PCR of BRCA1 and BRCA2 mRNA. Expression level analysis was performed for 81 samples. Amplification of the housekeeping gene GAPDH was successful for 79 samples, for which we further performed statistical analysis.
We analyzed the expression levels of the tested samples in relation to their methylation and mutation types. While we failed to discover a statistically significant difference between expression levels in relation to promoter methylation status for either of these two genes, there was a notably larger variance in BRCA1 and BRCA2 expression in the unmethylated groups (Figure 6). Observing BRCA1/2 expression levels in relation to BRCA1/2 mutation type, the results show a significantly higher expression level for BRCA2 gene expression in the BRCA2-mutated group in comparison to WT samples (Kruskal–Wallis p = 0.0058; Figure 6d). However, the mutations in the BRCA1 gene did not affect the expression of the same gene. Level of expression according to overall BRCA1/2 aberration is presented in Supplementary Figure S4.
3.6. Exploratory Analysis of BRCA1/2 Promoter Methylation on Survival
From the whole BRCA1/2 methylation cohort, for 68 patients, we had available clinicopathological follow-up data for exploratory survival analysis. We did not observe any selection bias for age, stage, or molecular features between the full cohort and the follow-up subset (Supplementary Table S3). The strong survival benefit of PARPi therapy observed for BRCA1/2-mutated HGSOC found in several clinical trials (SOLO1: HR = 0.55; PAOLA-1: HR = 0.60) motivated us to look for comparable effect sizes [22,23]. Our study was powered to detect only hazard ratios of under 0.5 due to the prevalence of the analyzed covariates (roughly 40% for combined FM and IM cases), at 80% power and alpha of 0.05. However, the study is underpowered (~40% power) due to large class imbalance to discern a comparable effect in FM vs. IM and FM cases and should be considered as exploratory. The range of follow-up was from 12 to 170 months, with a median follow-up of 42 months (3.5 years). We examined possible differences in OS and PFS rates according to BRCA1/2 baseline methylation status in the BRCA1/2 WT subgroup to test if any of these alterations have an impact on patient survival. We found no evidence of impact either for BRCA1 methylation or for BRCA2 methylation on progression-free survival (log-rank p = 0.87 and p = 0.55 for the BRCA1 and BRCA2 genes, respectively), or on overall survival (log-rank p = 0.34 and p = 0.94, respectively; Supplementary Figure S5).
Patients were grouped according to their mutation and methylation status. Survival analysis was performed for a group of 68 patients, 18 of whom were long-term survivors with survival of more than 60 months. That group consisted of four BRCA1/2-mutated, nine BRCA1/2-methylated cases, one with both aberrations, and four BRCA1/2 mutation- and methylation-free cases (Figure 7a,b). Four out of five mutated cases were treated with PARPi, which may be the possible reason for prolonged survival. We did not observe a significant difference in PFS between the groups (log-rank, p = 0.52), nor in the overall survival (log-rank, p = 0.15). Stratifying the WT group according to treatment and BRCA1/2 methylation status, we can observe an unfavorable effect of maintenance treatment with bevacizumab in both of the groups. Median PFS was 37 months for the BRCA1/2-mutated group and 26 months for the fully methylated. On the other hand, BRCA1/2 wild-type tumors with IM promoter median PFS was 22 months, and for patients without any BRCA1/2 alterations, the median PFS was the shortest (24 months).
Expectedly, in Cox regression univariable analysis, patients with relapsed OC show 2.45 times higher risk of progression compared to newly diagnosed patients, but without reaching statistical significance (p = 0.064, HR, 2.45; 95% CI 0.95–6.33). We found no statistically significant impact on progression-free or overall survival in patients for clinicopathological characteristics (FIGO stage, grade, age), nor for the methylation status of BRCA1 and BRCA2 genes. Observing treatment approach in the univariable model, we found an unfavorable effect of combination treatment with maintenance bevacizumab and chemotherapy on the overall survival of patients (HR, 2.47; 95% CI 1.16–5.22; p = 0.018), compared to chemotherapy only (Figure S6; Supplementary Table S2). The five-year overall survival rate in this cohort was 43.3% (95% CI, 31.1–60.1%). The highest 5-year survival rates were in the BRCA1 and BRCA2-mutated patient groups (50% and 66.7%; 95% CI, 12.5–100%, and 37.9–100%, respectively). The comparison to the BRCA1/2 WT group did not reach statistical significance (p = 0.2 and 0.07, respectively). None of the other BRCA1/2 aberrations showed a significant difference in survival rate compared to BRCA1/2 WT. BRCA1- and BRCA2-methylated patients showed similar 5-year survival rates of 44.6% and 40% (95% CI, 24.9–79.9% and 13.7–100%, respectively). BRCA WT cases showed the lowest 5-year survival rate of 35.1% (95% CI, 17–72.4%).
Performing multivariable Cox regression analysis, we adjusted analysis for BRCA1/2 methylation and BRCA1/2 mutation status, patient group, type of treatment, and age at diagnosis. Results showed significance only for overall survival in the chemotherapy–bevacizumab combination of therapy (p = 0.023; HR, 2.54; 95% CI, 1.14–5.68) (Table 3).
4. Discussion
Epigenetic inactivation of BRCA1/2 genes is postulated to be a marker of response to platinum-based and PARPi systemic therapy by conferring HRD. In this study, we aimed to evaluate the prevalence of BRCA1/2 promoter methylation in the Serbian population and its association with clinicopathological features, as well as to explore the effects on patient survival.
There is a wide range of reported frequencies of BRCA1/2 methylation in ovarian tumors in the literature, which may be attributed to population diversity, type of ovarian cancer studied, techniques used for the detection of methylation, and the number of CpG islands covered, as well as the type of material used. For quantitative methods such as quantitative MSP or methylation array, the choice of cut-off methylation level is of particular importance, where some studies have used percentages as low as 5% to call methylation [24,25,26]. Qualitative methods such as MSP or methylation-specific High Melting Resolution (MS-HRM), while lacking a precise methylation level, offer a simple and cost-effective way to determine the presence or absence of methylation and are attractive as complementary tests to BRCA1/2 mutation testing. The prevalence of homozygous promoter hypermethylation (FM) found in our study cohort (4.1% and 0.45% for BRCA1 and BRCA2, respectively) was comparable to the prevalence of BRCA1 methylation of 6.4% reported in the US population by Swisher et al. using MSP [24], yet lower than the reported range of 6.4–73.7% for BRCA1 promoter hypermethylation in OC patients in the meta-analysis conducted by Kalachand et al., where different methods and cut-off values were used to call hypermethylation [24]. Conversely, BRCA2 methylation is a rather rare event, with different studies reporting diverse prevalence, ranging from none or only one case of BRCA2 hypermethylation to 21% [21,25,27,28,29,30,31]. Overall BRCA1/2 dysfunction in this cohort was found in 49.6%, counting BRCA1/2 hypermethylation (FM and IM cases) and BRCA1/2 genomic alterations (pathogenic and likely pathogenic variants). Similar results were observed by Kalachand et al., with 40% overall aberrations of BRCA1/2 in HGSOC [24].
Recent studies demonstrated that the HRD phenotype may also be induced by BRCA1 promoter methylation, suggesting its use as a predictive biomarker from PARP inhibitor therapy [14,24,31,32,33]. Information about PARPi treatment in BRCA1-methylated ovarian cancer is limited, with the ARIEL-2 study reporting a positive effect of methylation on the response to PARPi rucaparib [34]. They demonstrated higher loss of heterozygosity (LOH), which is connected with a higher genomic instability score (GIS) associated with HRD, in one proportion of BRCA1-hypermethylated cases [35]. However, studies researching BRCA2 promoter methylation in response to PARPi are lacking.
An important finding in our cohort was that the patients’ age of diagnosis of ovarian cancer was strongly affected by BRCA1/2 status. We found similar representation of age in BRCA1/2-mutated cases without methylation and BRCA1/2 wild-type cases which are intermediary methylated (IM). Notably, the group without mutations but with fully methylated BRCA1/2 had the earliest age of onset, which is in concordance with the finding from a meta-analysis performed by Kalachand et al. [24]. Additionally, none of the methylated cases had previous family history of breast or ovarian cancer, which was observed as well in a study conducted by Baldwin et al. [36]. Hypermethylation is thought to occur almost exclusively in a sporadic manner, suggesting that the methylation of the BRCA1/2 genes might be employed as a pre-test when the existence of a hereditary nature is suspected [37,38]. Our results confirm reports that BRCA1 methylation and BRCA1/2 mutation are mutually exclusive events and that methylation is not likely a “second hit” in Knudson’s two-hit hypothesis [31,34,39,40,41].
While the average BRCA1 and BRCA2 expression levels were lower in the FM and IM groups compared to UM samples, the difference was not statistically significant. For the BRCA2 gene, this result is in concordance with Pradjatmo et al.’s findings [42]. However, other studies demonstrated that BRCA1 promoter hypermethylation reduces the expression level, contrary to results from our study [24,43,44]. There are several possible explanations for this result that may stem from different levels of methylation in the IM group, technical limitations, or lack of functional effect of probed CpGs. It is important to note that MSP as a technique covers only a limited number of preselected CpG sites, not the entire regulatory region. The presence of a positive event of methylation does not confirm the same for all CpGs in the promoter region. Therefore, the lack of association of expression and methylation can come from the possibility that unscreened CpGs are not methylated, leading to gene expression. Alternatively, the large variation in BRCA1 and BRCA2 expression in unmethylated samples could be explained by other epigenetic mechanisms of gene silencing, including closed chromatin modifications, miRNA-mediated translational attenuation or mRNA degradation, or allele-specific expression driven by SNPs, causing a subset of samples in the unmethylated group to show lower levels of gene expression.
Survival analysis in relation to BRCA1/2 promoter methylation showed no favorable effect on OS or PFS in methylated versus unmethylated subgroups. Prognostic relevance for PFS and OS was not identified in the study performed by Ruscito et al. either [45]. Sahnane reported the opposite results, with a favorable outcome in BRCA1/2-methylated patients, as did Swisher et al. in the ARIEL-2 study [25,34]. However, in the further analysis performed by the ARIEL-2 consortium, Kondrashova et al. discovered that the favorable OS effect in the BRCA1-methylated group comes from homozygous BRCA1-methylated cases. Notably, in the ARIEL-2 study, Kondrashova et al. reported changes from homozygous to heterozygous BRCA1 methylation status under the pressure of treatment. Since BRCA1 methylation status can alter over the course of the disease, it may have potential as a biomarker of therapy resistance and a crucial indicator of treatment response in cases of recurrence [46,47]. Regarding the unfavorable effect of combinational treatment with bevacizumab, Fiegl et al. observed the same results [31]. This effect may be due to the criteria for selecting the patients for this type of treatment, as only the patients with higher-stage (FIGO IIIc and IV) or residual disease can be included.
Limitations of this study include the choice of technique for methylation detection, the size of testing cohort, and lack of information about methylation pattern in the complete BRCA1/2 promoter region after first-line and maintenance treatment in relapsed cases of ovarian cancer patients. Considering the sample size, class imbalance, and the number of events, this study was able to identify only large effect sizes. While we did not observe any large effects on PFS and OS, we cannot rule out moderate or modest survival benefit in BRCA1/2-methylated patients. Additionally, our follow-up cohort for survival analysis lacks a group of BRCA1/2 wt and methylated patients who received PARPi treatment to explore the effect of targeted treatment in this group of patients. MSP was the technique of choice for this study because it is a reproducible and cost-effective technique, that could easily be implemented in clinical practice. However, it is a qualitative method, and we could not obtain the exact percentage of methylation. Furthermore, not all CpG islands could be covered, which may be misleading for some of intermediary and unmethylated samples for both genes. Also, it is important to emphasize that MSP is not a suitable technique for clonal distinction, nor is it a true representation of methylation level through the entire promoter region, as in this technique a few preselected relevant CpGs are screened. Prior to the introduction of HRD as the eligibility criteria for PARPi administration, only relapsed cases with pathogenic variants in either BRCA1 or BRCA2 that already responded well to platinum were treated, which excluded this group of patients with hypermethylation. Therefore, a possible predictive role of BRCA1/2 promoter methylation for PARP inhibitor efficacy could not be evaluated in this cohort due to insufficient treatment and response data.
5. Conclusions and Future Outlook
In summary, this study demonstrated that BRCA1 and BRCA2 promoter methylation prevalence in Serbian patients is different from that reported in the existing literature. Given the early age of onset in patients with homozygous methylation of either BRCA1 or BRCA2, mutually exclusive with mutation events, and combined with an absence of positive family history, methylation analysis may serve as a pre-test for BRCA1/2 hereditary screening. The large variance observed for BRCA1/2 expression warrants an investigation of other epigenetic mechanisms as potential mediators of reduced gene expression and HRD phenotype. Finally, prospective studies with longitudinal methylation assessment and standardized treatment response data are necessary to evaluate BRCA1/2 promoter methylation as a predictive biomarker for PARP inhibitor response.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bray F. Laversanne M. Sung H. Ferlay J. Siegel R.L. Soerjomataram I. Jemal A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries CA Cancer J. Clin.20247422926310.3322/caac.2183438572751 · doi ↗ · pubmed ↗
- 2Kobel M. Kalloger S.E. Boyd N. Mc Kinney S. Mehl E. Palmer C. Leung S. Bowen N.J. Ionescu D.N. Rajput A. Ovarian carcinoma subtypes are different diseases: Implications for biomarker studies P Lo S Med.20085 e 23210.1371/journal.pmed.005023219053170 PMC 2592352 · doi ↗ · pubmed ↗
- 3Romero I. Leskela S. Mies B.P. Velasco A.P. Palacios J. Morphological and molecular heterogeneity of epithelial ovarian cancer: Therapeutic implications EJC Suppl.20201511510.1016/j.ejcsup.2020.02.00133240438 PMC 7573476 · doi ↗ · pubmed ↗
- 4Singh N. Mc Cluggage W.G. Gilks C.B. High-grade serous carcinoma of tubo-ovarian origin: Recent developments Histopathology 20177133935610.1111/his.1324828477361 · doi ↗ · pubmed ↗
- 5Sogaard M. Kjaer S.K. Gayther S. Ovarian cancer and genetic susceptibility in relation to the BRCA 1 and BRCA 2 genes. Occurrence, clinical importance and intervention Acta Obstet. Gynecol. Scand.2006859310510.1080/0001634050032462116521688 · doi ↗ · pubmed ↗
- 6Risch H.A. Mc Laughlin J.R. Cole D.E. Rosen B. Bradley L. Kwan E. Jack E. Vesprini D.J. Kuperstein G. Abrahamson J.L. Prevalence and penetrance of germline BRCA 1 and BRCA 2 mutations in a population series of 649 women with ovarian cancer Am. J. Hum. Genet.20016870071010.1086/31878711179017 PMC 1274482 · doi ↗ · pubmed ↗
- 7Whittemore A.S. Gong G. Itnyre J. Prevalence and contribution of BRCA 1 mutations in breast cancer and ovarian cancer: Results from three U.S. population-based case-control studies of ovarian cancer Am. J. Hum. Genet.1997604965049042908 PMC 1712497 · pubmed ↗
- 8Prakash R. Zhang Y. Feng W. Jasin M. Homologous recombination and human health: The roles of BRCA 1, BRCA 2, and associated proteins Cold Spring Harb. Perspect. Biol.20157 a 01660010.1101/cshperspect.a 01660025833843 PMC 4382744 · doi ↗ · pubmed ↗