Mitochondria and Lipid Defects in Hereditary Progranulin-Related Frontotemporal Dementia
Jon Ondaro, Jose Luis Zúñiga-Elizari, Mónica Zufiría, Maddi Garciandia-Arcelus, Ángela Sánchez Molleda, Miren Zulaica, Miguel Lafarga, Javier Riancho, Adolfo López de Munaín, Fermin Moreno, Francisco Javier Gil-Bea, Gorka Gerenu

TL;DR
This study finds that a genetic mutation in GRN causes mitochondrial and lipid issues in cells from frontotemporal dementia patients, which could lead to new treatments.
Contribution
The study identifies mitochondrial dysfunction and lipid metabolism disruption as novel features of GRN-related frontotemporal dementia.
Findings
GRN-mutant fibroblasts show mitochondrial swelling and reduced respiration.
Lipid metabolism is disrupted in GRN-mutant fibroblasts with lipid droplet accumulation.
rhPGRN supplementation improves lysosomal and mitochondrial abnormalities in these cells.
Abstract
What are the main findings? FTD GRN-mutant fibroblasts replicate lysosome-related features observed in FTD patients.Fibroblasts from heterozygous GRN c.709-1G>A mutation carriers show altered mitochondrial function.Fibroblasts from heterozygous GRN c.709-1G>A mutation carriers exhibit disrupted lipid metabolism. FTD GRN-mutant fibroblasts replicate lysosome-related features observed in FTD patients. Fibroblasts from heterozygous GRN c.709-1G>A mutation carriers show altered mitochondrial function. Fibroblasts from heterozygous GRN c.709-1G>A mutation carriers exhibit disrupted lipid metabolism. What are the implications of the main findings? FTD GRN-mutant fibroblasts provide a useful initial system to investigate lysosomal dysfunction and associated metabolic alterations relevant to FTD.These metabolic changes suggest that pathways related to mitochondrial and lipid homeostasis may…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
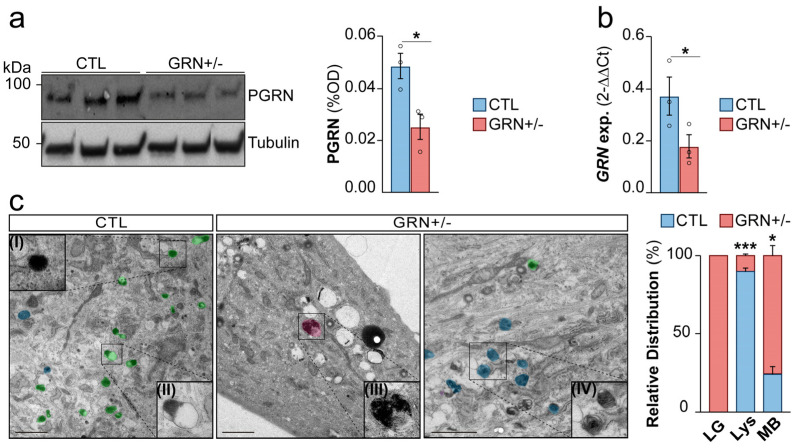 Figure 1
Figure 1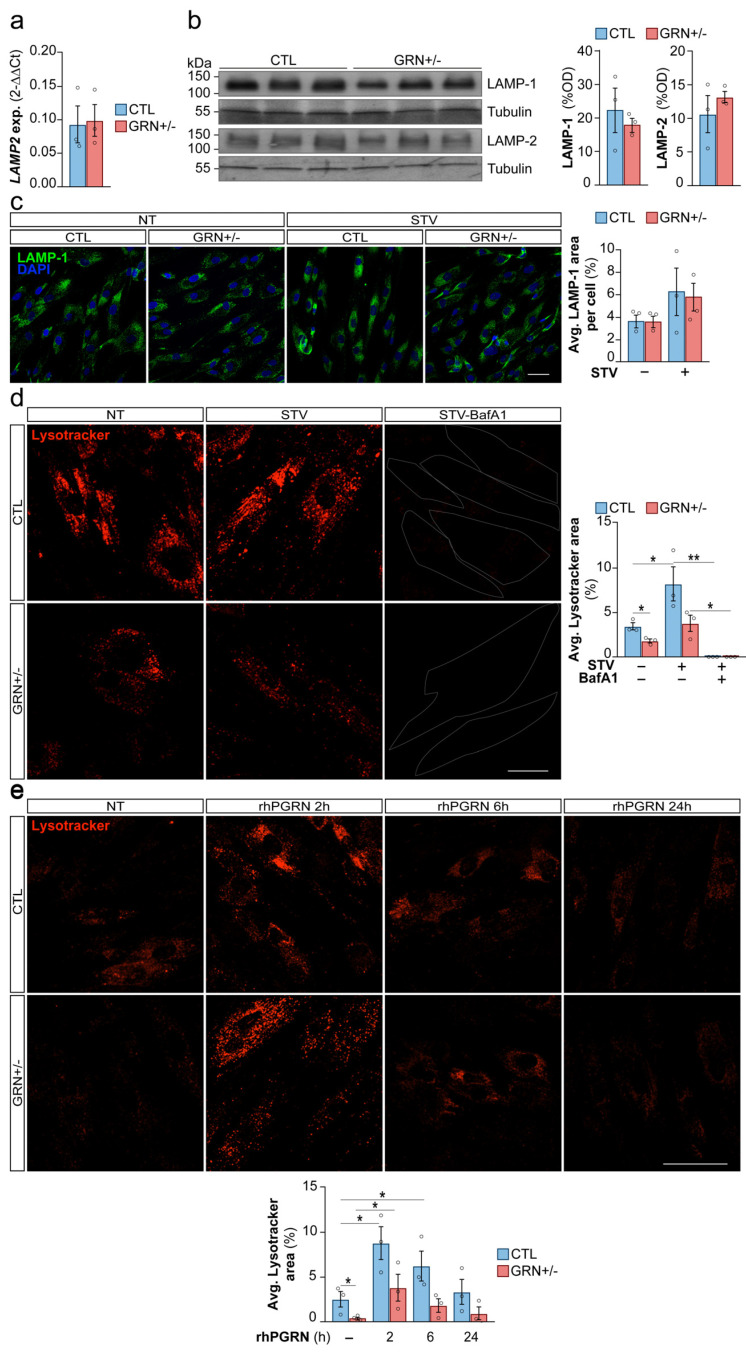 Figure 2
Figure 2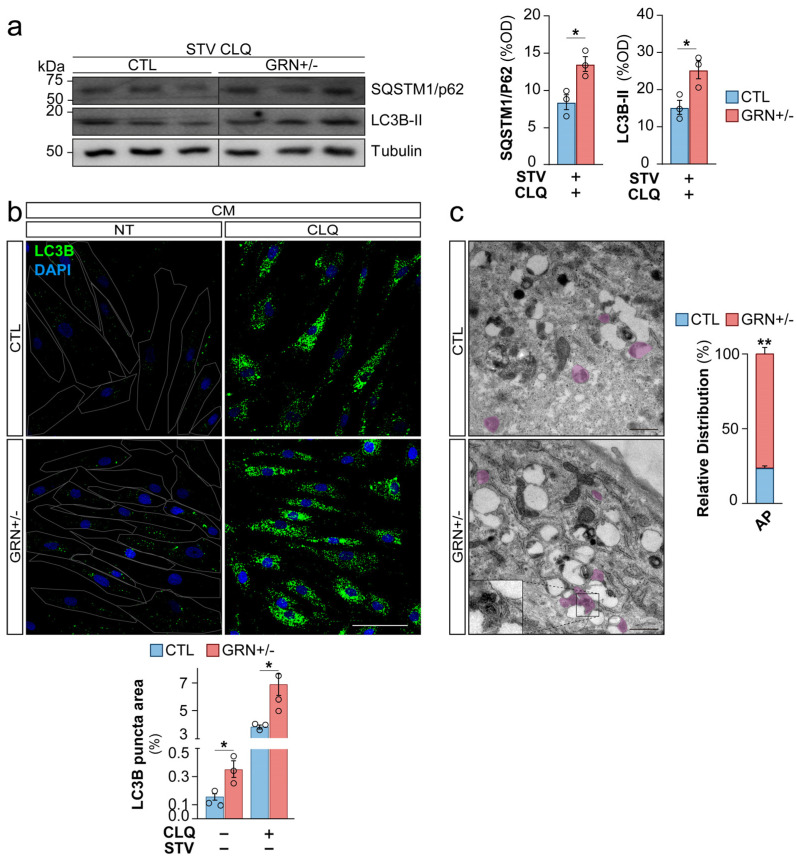 Figure 3
Figure 3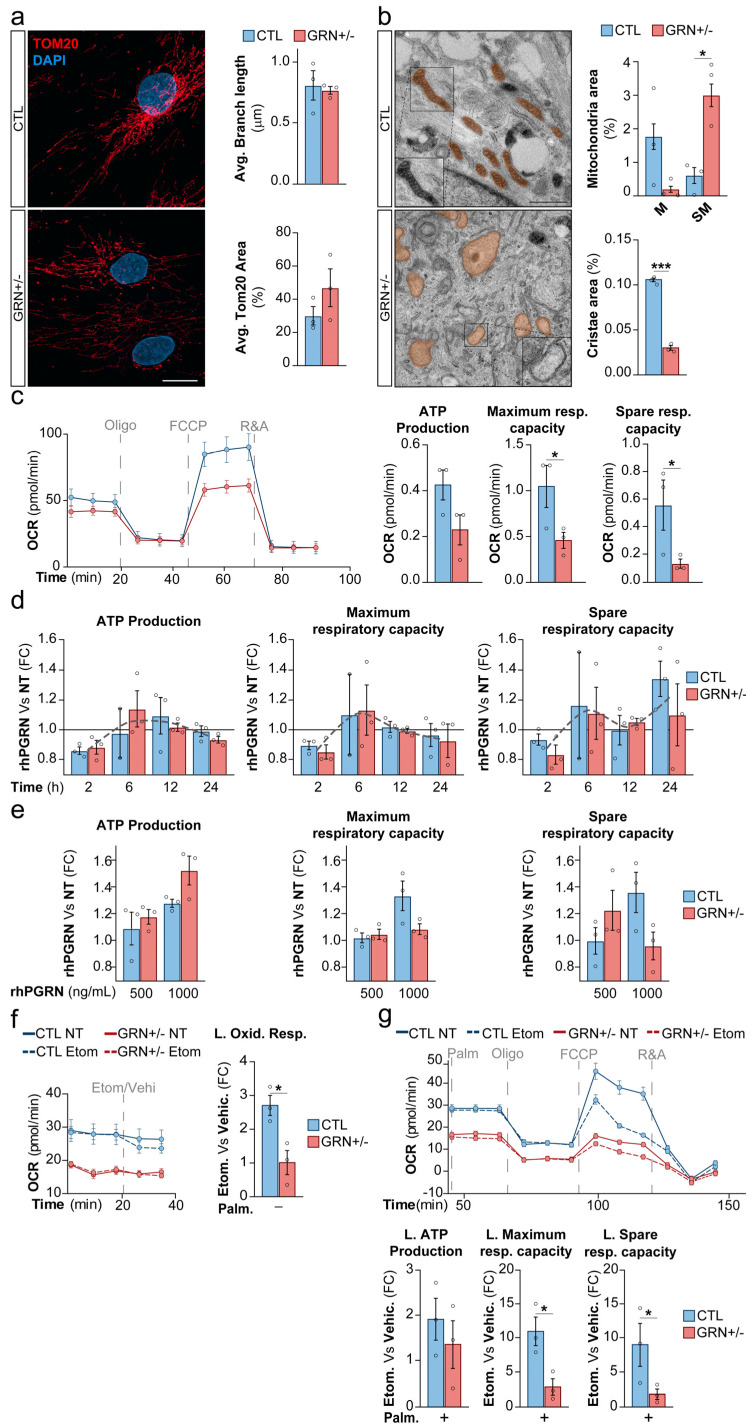 Figure 4
Figure 4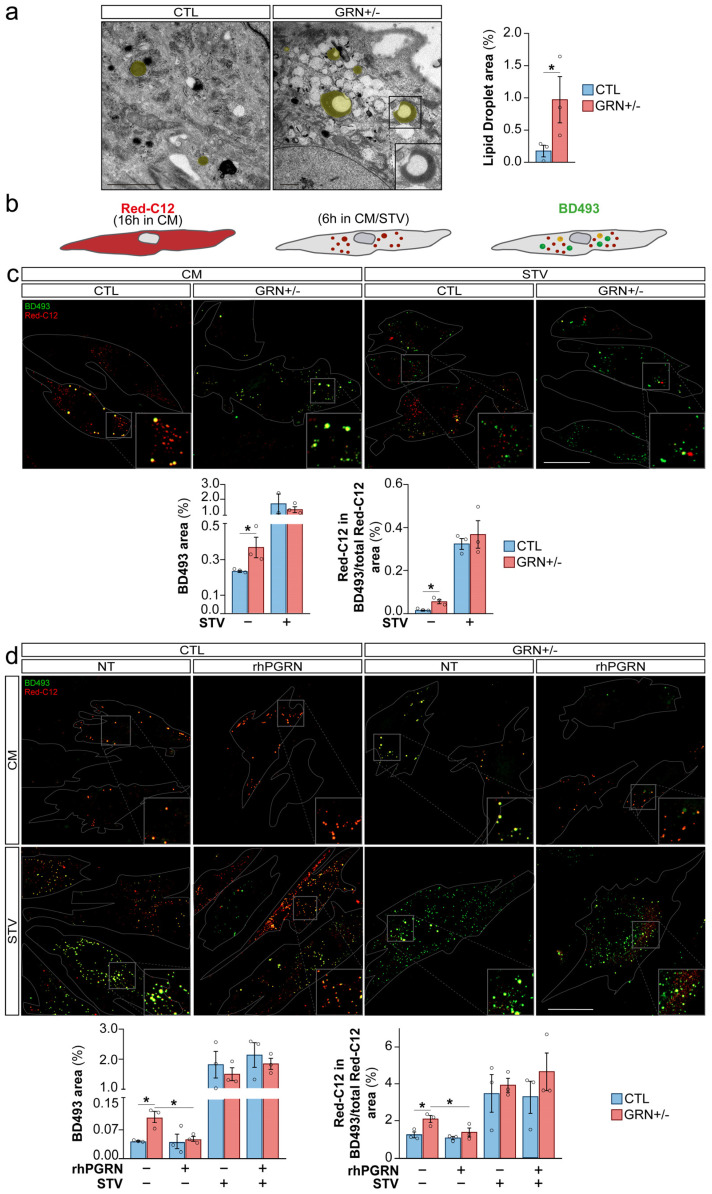 Figure 5
Figure 5- —MICIU/AEI/10.13039/501100011033
- —ERDF/EU
- —European Union
- —European Union NextGenerationEU/PRTR
- —Instituto de Salud Carlos III (ISCIII)
- —European Union through the European Regional Development Fund (ERDF)
- —CIBERNED—Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas
- —Diputación Foral de Gipuzkoa
- —EiTB Maratoia
- —Fundació La Marató
- —IKUR strategy of the Basque Government
- —Basque Government Department of Health
- —Department of Education of the Basque Government
- —Roche “Stop Fuga de Cerebros” program
- —Spanish Ministry of Science and Innovation
- —IKERBASQUE Research Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAmyotrophic Lateral Sclerosis Research · Genetic Neurodegenerative Diseases · Mitochondrial Function and Pathology
1. Introduction
Frontotemporal dementia (FTD) is a neurodegenerative disorder and the second leading cause of dementia in individuals under 65 years of age, characterized by progressive impairments in behavior and language [1]. Despite ongoing progress, there are no effective treatments for FTD. Age is considered the most important risk factor for FTD and the majority of cases are sporadic, meaning there is no clear family history. However, approximately 40% of patients of FTD do have a family history of dementia, suggesting a significant genetic component in the development of the disease [2]. Mutations in several genes including MAPT, GRN, FUS, VCP, and C9orf72 are known to cause familial frontotemporal dementia (fFTD). Among them, mutations in the granulin (GRN) gene account for 5–20% of the total fFTD cases [3]. The heterozygous mutations lead to a haploinsufficiency of its encoding protein progranulin (PGRN), resulting in a production of a truncated PGRN protein [4] and triggering neurodegeneration. One of these FTD-causing mutations was discovered 15 years ago in a cluster of Basque families carrying a unique ancestral genetic substitution (c.709-1G>A) in the GRN gene [5]. This mutation is representative of the majority of GRN-associated FTD cases, as most GRN mutations result in loss of function and lead to disease through GRN haploinsufficiency.
PGRN is a secreted glycoprotein composed of 7.5 GRN segments (granulins A-G and paragranulin), that play significant roles in several cellular processes [6]. Although the precise function of PGRN is not yet fully understood, accumulating evidence suggests its critical role in lysosomal function [7,8,9]. Intracellularly, full-length PGRN predominantly localizes within the lysosome, where it undergoes proteolytic cleavage by lysosomal proteases, resulting in the generation of individual GRN peptides [10]. These peptides are believed to regulate lysosomal enzymes such as cathepsin D [11] and glucocerebrosidase [12,13]. Dysfunction of lysosomal enzymes leads to impaired lysosomal function and is associated with lysosomal storage disorders (LSDs). Homozygous GRN mutations, resulting in complete loss of PGRN, cause neuronal ceroid lipofuscinosis (NCL), a type of LSD. NCL shares pathological features with FTD due to GRN mutation (FTD-GRN), including the accumulation of lipofuscin granules [14] and dysfunctional lysosomes [15]. Consequently, lysosomal dysfunction is widely recognized as a key contributor to neurodegeneration in FTD-GRN [9].
The lysosome is a dynamic organelle that coordinates waste recycling with nutrient sensing and metabolic regulation, ensuring cellular homeostasis and survival. To fulfill this role, the lysosome interacts with other cellular structures exchanging contents and information by establishing membrane contact sites [16].
As observed in some pathological conditions, lysosomal dysfunction induces alterations in mitochondria and lipid metabolism [17,18,19], reflecting the close relationship between these mechanisms. Under physiological conditions, their coordination is essential for cellular adaptation, reconfiguration of nutrient utilization, and maintenance of metabolic balance during periods of energy scarcity. Lysosomal dysfunction can, in some cases, mimic this state, inducing a starvation (STV)-like metabolism. This reconfiguration involves a shift away from glycolysis towards mitochondrial β-oxidation of fatty acids (FAs) derived from triacylglycerides (TAG) or cholesterol esters stored within lipid droplets (LDs) [20]. During energy stress, macroautophagy is enhanced to recycle cellular components and supply the cell with essential materials to prioritize vital biological processes [21]. Within the lysosome, membrane lipids are broken down into FAs and released. These FAs can serve as a source of energy production for the mitochondria or may be re-esterified and packaged into new LDs in the endoplasmic reticulum, leading to a paradoxical increase in LDs [22,23]. This mechanism helps to manage the excessive influx of FAs into the mitochondria and prevents lipotoxicity [24,25].
Based on these considerations, we hypothesized that GRN haploinsufficiency associated with FTD-GRN, may lead to secondary alterations in organelles, such as mitochondria and lipid trafficking, ultimately causing detrimental metabolic consequences. To this end, we used human fibroblasts derived from FTD-GRN patients carrying the c.709-1G>A mutation.
2. Materials and Methods
2.1. Human Samples
Six skin biopsies were analyzed, including samples from GRN c.709-1G>A mutation carriers (GRN+/−) and healthy control (CTL) relatives. All patients were diagnosed and treated at Donostia University Hospital according to established consensus criteria [26]. Biopsies and genetic analyses were performed with written informed consent and approval from the Ethics Committee of the Government of the Basque Country.
2.2. Cell Culture Conditions
Primary fibroblasts from CTL donors and GRN c.709-1G>A mutation carriers were cultured in cell medium (CM), DMEM (Gibco, Thermo Fisher Scientific, Paisley, UK) supplemented with FBS (Gibco), GlutaMAX (Thermo Fisher Scientific), and penicillin/streptomycin (Gibco) at 37 °C and 5% CO_2_. STV conditions were induced using supplemented EBSS for 6 h. Cells were treated with chloroquine (CLQ) (Sigma-Aldrich, Darmstadt, Germany), bafilomycin A1 (BafA1) (Selleckchem, Munich, Germany) or recombinant human progranulin (rhPGRN) (R&D Systems Europe Ltd., Abingdon, Oxfordshire, UK) at the indicated concentrations and times. The vehicle was referred to as non-treated (NT).
2.3. mRNA Expression/Processing Analysis
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and reverse-transcribed with SuperScript VILO (Thermo Fisher Scientific). qRT-PCR was performed using SYBR Green on a CFX384 Touch Real-Time PCR Detection System (Bio-Rad Europe GmbH, Basel, Switzerland), with GAPDH as the reference gene and relative expression calculated by the 2^−ΔΔCt^ method.
2.4. Protein Extraction and Western Blotting
Primary fibroblasts were lysed, and proteins were separated by SDS–PAGE and transferred to nitrocellulose membranes. Membranes were probed with specific primary antibodies and HRP-conjugated or fluorescent secondary antibodies. Signals were detected using the iBright imaging system (Thermo Fisher Scientific), quantified with Image Studio Lite (LI-COR Biosciences, Lincoln, NE, USA), and normalized to β-tubulin.
2.5. Immunofluorescence
Primary fibroblasts were cultured on Ibidi slides, fixed with paraformaldehyde, and blocked/permeabilized with Bovine serum albumin (BSA) and Triton X-100. Cells were incubated with primary antibodies against LC3B, LAMP-1, or TOM20, followed by Alexa Fluor–conjugated secondary antibodies and DAPI nuclear staining. Images were acquired using a Zeiss LSM 900 confocal microscope (Carl Zeiss, Jena, Germany), and image and colocalization analyses were performed with ImageJ (version 1.54, National Institutes of Health, Bethesda, MD, USA).
2.6. Live Cell Imaging
Lysosomal activity was assessed using LysoTracker™ Red (Thermo Fisher Scientific)–labeled fibroblasts imaged by confocal microscopy. Fatty acid pulse–chase assays were performed using BODIPY 558/568 C12 (Red-C12) (Life Technologies Europe BV, Bleiswijk, The Netherlands) to track lipid incorporation into LDs and trafficking to mitochondria, with LDs labeled by BODIPY 493/503 (BD493) (Life Technologies) and mitochondria by MitoTracker™ Green (Invitrogen, Thermo Fisher Scientific, Breda, The Netherlands). Live-cell imaging was conducted on a Zeiss LSM 900 confocal microscope (Carl Zeiss), with cells maintained at 37 °C in a controlled CO_2_ atmosphere. Image quantification of LDs, lysosomes, and FAs distribution was performed using ZEN 3.1 Blue (Carl Zeiss), ImageJ (NIH), and Cellpose-based cell segmentation [27].
2.7. Seahorse XF-96 Metabolic Flux Analysis
Oxygen consumption rates (OCRs) were measured using a Seahorse XF-96 Analyzer (Agilent Technologies GmbH, Waldbronn, Germany). Primary fibroblasts were seeded in XF-96 plates and, when indicated, exposed to STV conditions prior to analysis. Mitochondrial function was assessed using the XF Cell Mito Stress Test with sequential injections of oligomycin, FCCP, and rotenone/antimycin A to calculate basal respiration, ATP production, and maximal and spare respiratory capacities.
Fatty acid oxidation (FAO) dependency was evaluated using the Seahorse FAO assay with palmitate-BSA, followed by sequential injections of etomoxir and mitochondrial inhibitors. OCR values were normalized to cell number, and metabolic parameters were calculated using Wave Agilent Software 2.4.2 (Agilent Technologies).
2.8. Transmission Electron Microscopy
Primary fibroblasts were fixed in 3% glutaraldehyde and washed before processing for transmission electron microscopy (TEM) at the Principe Felipe Research Center. Ultrathin sections (70 nm) were imaged at the Core Facility for Polymer Characterization (UPV/EHU, San Sebastian, Spain). Quantification of lysosomes, autophagosomes, fingerprint structures, LDs, and mitochondrial area was performed in 10–15 cells per sample and expressed relative to total cell area. Mitochondrial cristae were analyzed following established methods [28].
2.9. Statistical Methods
Data are shown as mean ± SEM from at least three independent experiments. Statistical analyses were performed using GraphPad Prism 10.0.0 (GraphPad Software, San Diego, CA, USA). Two-group comparisons used the Mann–Whitney U test, while multi-factor comparisons were analyzed by two-way ANOVA with Tukey’s post hoc test. Significance is indicated as ns (p > 0.05), * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
3. Results
3.1. Skin Fibroblasts from FTD-GRN Patients Reproduce the Neuropathological Hallmarks Observed in FTD Brains
Heterozygous pathogenic GRN variants cause an approximately 50% reduction in both PGRN mRNA and protein levels [29,30]. This feature was reproduced in GRN+/− fibroblasts, showing significantly reduced PGRN protein levels (p < 0.05) (Figure 1a, uncropped images in Figure S1) and mRNA levels when compared with CTL fibroblasts (p < 0.05) (Figure 1b). Based on this finding, we investigated whether this reduction could induce FTD-like pathology in patient fibroblasts.
FTD-GRN patients also showed an accumulation of lipofuscin granules and lysosomal deposits [14]. These pathological features are typically associated with NCL, a disease caused by the complete absence of PGRN. To explore these pathological features, both CTL and GRN+/− fibroblasts were examined by TEM. GRN+/− fibroblasts showed a prominent accumulation of lysosome-related storage material, with a significantly higher proportion of multilamellar bodies compared to the CTLs (p < 0.05) (Figure 1c, detail in Figures S2–S4). Furthermore, dense lipofuscin granules, which were not present in CTLs, were observed in GRN+/− fibroblasts (Figure 1c, detail in Figures S2–S4). Additionally, the number of electron-dense lysosomes was reduced in the GRN+/− fibroblasts in comparison to CTLs (p < 0.05) (Figure 1c, detail in Figures S2–S4). We then sought to quantitatively validate these putative lysosomal defects and to investigate whether they were a direct consequence of PGRN deficiency.
3.2. Skin Fibroblasts from FTD-GRN Patients Exhibit Impaired Lysosomal Function
As the accumulation of multilamellar bodies indicates lysosomal dysfunction, and this is consistently observed in animal and cellular models of PGRN deficiency [31,32,33], we investigated lysosomal function in GRN+/− fibroblasts. First, we assessed lysosomal load by measuring the protein levels of two lysosomal structural proteins, LAMP-1 and LAMP-2, and the mRNA levels of LAMP2, and found no differences between GRN+/− and CTL fibroblasts (Figure 2a,b, uncropped images in Figure S5). Furthermore, immunofluorescence analysis revealed no differences in lysosomal localization between GRN+/− and CTL fibroblasts under both CM and STV conditions. The similar LAMP-1 levels in GRN haploinsufficient and CTL fibroblasts under STV condition suggest that these mutant cells retain the ability to synthesize lysosomal structures comparably to non-mutant cells (Figure 2c).
However, we observed a significant reduction in the staining of Lysotracker Red DND-99, a dye used to label acidic organelles in live cells, in the GRN+/− fibroblasts compared to CTL fibroblasts (p < 0.05). After six hours of STV, the number of active lysosomes increased as expected in CTL fibroblasts when compared to the CM fibroblasts (p < 0.05), but not in GRN+/− fibroblasts. However, in both CTL (p < 0.01) and GRN+/− fibroblasts (p < 0.05), treatment with BafA1, an inhibitor of the vacuolar H^+^-ATPase, resulted in inhibition of lysosomal function when compared to NT fibroblasts (Figure 2d, uncropped images in Figure S6). This analysis in lysosomal function, revealed that, despite the equal number of lysosomal structures, the functional units are reduced in GRN+/− fibroblasts due to impaired lysosomal acidification, which may explain the observed accumulation of storage material in TEM images. To evaluate whether lysosomal dysfunction is directly linked to insufficient PGRN, we used 500 ng/mL of rhPGRN as a proof-of-concept for protein replacement in our cell model. We assessed Lysotracker Red DND-99 staining at 2, 6, and 24 h to monitor lysosomal acidification. The peak of Lysotracker staining manifested at 2 h post treatment was consistently observed in both CTL and GRN+/− fibroblasts when compared to NT fibroblasts (p < 0.05) (Figure 2e, uncropped images in Figure S7). Notably, the 2 h rhPGRN treatment in GRN+/− fibroblasts restored lysosomal function comparable to those observed in untreated CTLs (Figure 2e). This observation supports a strong association between PGRN levels and lysosomal acidification in fibroblasts from FTD-GRN patients.
3.3. Skin Fibroblasts from FTD-GRN Patients Display Abnormalities of Autophagy
Lysosomes play a pivotal role in the resolution of the autophagy to facilitate the enzymatic degradation of the cellular cargo. In fact, alterations in autophagic flux have been described in several studies of PGRN deficiency [32,33]. When fibroblasts are cultured under CM, they do not show a significant level of basal autophagic activity. This observation is supported by the low levels of lipidated microtubule-associated protein 1A/1B light chain 3 (LC3B-II), a well-established marker of autophagy-related structures, under CM (Figure S8a). Therefore, these findings suggest that autophagy is not essential under CM conditions in fibroblasts. Consistently, we observed an increase in the level of LC3B-II in fibroblasts after 5 h of lysosomal inhibitor CLQ, and no significant difference between samples derived from CTL individuals and FTD-GRN patients (Figure S8b). Therefore, under CM conditions, it becomes challenging to detect a potential alteration in autophagy due to GRN haploinsufficiency.
On the contrary, a prominent activation of autophagosome biogenesis can be detected under STV conditions. Moreover, fibroblasts were cultured in STV medium for 6 h prior to the assay and treated with CLQ to measure the autophagy flux in these cells by measuring LC3B-II levels and sequestosome 1 (P62/SQSTM1), an ubiquitin-binding scaffold protein that links LC3B to autophagic degradation. We observed a higher accumulation of LC3B-II and P62/SQSTM1 in GRN+/− fibroblasts when compared to CTL fibroblasts (p < 0.05) (Figure 3a, uncropped images in Figure S9).
To corroborate this finding, we performed a quantitative analysis of autophagosomes through LC3B immunofluorescence, a technique that is more sensitive than Western blot analysis, which revealed a significant increase in LC3B-II positive puncta within the GRN+/− fibroblasts compared to CTL fibroblasts (p < 0.05), under CM condition (Figure 3b). These findings suggest an increase in autophagosome formation in fibroblasts derived from FTD-GRN patients, which could be interpreted as a compensatory feedback mechanism for lysosomal dysfunction, similar to what has been proposed in LSDs [34]. Furthermore, analysis of TEM images revealed a striking accumulation of autophagosome structures, approximately threefold higher, in GRN+/− fibroblasts compared to CTL fibroblasts cultured under CM conditions (p < 0.05) (Figure 3c, detail in Figures S10 and S11). Taken together, these data allow us to conclude that autophagosomes accumulate in GRN+/− fibroblasts. In the context of the lysosomal dysfunction observed in these cells, one possible explanation is the activation of a compensatory response aimed at increasing autophagosome formation. However, as autophagic flux was not directly measured, this interpretation remains tentative, and alternative mechanisms (such as impaired autophagosome degradation) cannot be excluded. Nonetheless, GRN-mutant fibroblasts recapitulate key lysosome-related features of FTD and therefore provide a useful model for investigating lysosomal dysfunction and downstream lysosome-associated alterations relevant to the disease.
3.4. GRN Haploinsufficiency Causes Mitochondrial Abnormalities in Skin Fibroblasts from FTD-GRN Patients
To investigate whether GRN haploinsufficiency adversely affects cellular metabolism, given the critical interaction between mitochondria and lysosomes in metabolic processes, we analyzed the mitochondrial status in GRN haploinsufficient fibroblasts. Quantification of the mitochondrial outer membrane marker TOM20 by immunofluorescence revealed no discernible impact on either the mitochondrial surface area or mitochondrial network integrity, measured by mitochondrial branch length (Figure 4a). However, ultrastructural assessment using TEM revealed a pronounced escalation in the prevalence of swollen mitochondria (p < 0.05) and a concomitant reduction in the abundance of mitochondrial cristae within GRN+/− fibroblasts when compared to the CTL group (p < 0.005) (Figure 4b, detail in Figures S12 and S13). In GRN haploinsufficient fibroblasts, we notice a disruption specifically in the inner mitochondrial membrane (IMM), rather than the outer mitochondrial membrane (OMM). This parallels diseases resulting from IMM mutations, such as Leigh syndrome associated with alterations in SLC25A46, where IMM disturbances occur independently of OMM changes [35]. Mitochondrial cristae are essential cellular structures for energy production, containing the enzymes required for oxidative phosphorylation.
To investigate the functional capacity of these swollen mitochondria, we performed an assessment of mitochondrial respiration using the SeaHorse technology. Our findings indicate that the basal ATP production may be diminished in FTD-GRN fibroblasts, although this analysis did not reach statistical significance due to the notable variability of CTL fibroblasts (Figure 4c). However, a significant impairment in maximal respiration capacity and spare respiratory capacity was distinctly evident in GRN+/− fibroblasts (p < 0.05), attributed to a concurrent reduction in spare respiration when compared to CTL fibroblasts (p< 0.05) (Figure 4c). To elucidate the potential modulation of mitochondrial respiratory capacity by PGRN, we treated fibroblasts with 500 ng/mL of rhPGRN for varying durations. Our findings showed that rhPGRN treatment displayed the capacity to enhance mitochondrial ATP production and elevate maximal respiratory capacity and spare respiratory capacity, particularly in GRN+/− fibroblasts. Interestingly, this effect was not evident at the 2 h time point following treatment initiation, a period during which rhPGRN did exhibit functional activation of lysosomes (Figure 2e). Instead, improvements were seen at later time points, especially between 6 and 12 h (Figure 4d). In addition to these time-dependent effects, our study also found dose-dependent responses. Specifically, using rhPGRN at a concentration of 1000 ng/mL led to a noticeable increase in ATP production as early as 6 h after treatment (Figure 4e). However, this increase did not affect the maximal respiratory capacity or spare respiratory capacity (Figure 4e). These findings provide compelling evidence of marked mitochondrial abnormalities in GRN+/− fibroblasts, characterized by impaired respiratory capacity. While rhPGRN treatment partially ameliorates this defect, the effect size is modest. Notably, although basal ATP production appears sufficient, these mitochondria may be unable to meet increased energy demands under stress conditions.
After identifying alterations in mitochondrial respiration in patient-derived fibroblasts, we sought to determine whether the mitochondrial defects observed in GRN+/− fibroblasts were specific to a particular energy source. When both GRN+/− and CTL fibroblasts were subjected to a 6 h STV condition prior to SeaHorse analysis, a reduction in mitochondrial respiratory capacity was observed in both CTL and patient-derived cells (Figure S14a). Since nutrient deprivation is known to reduce mitochondrial respiration, and lysosomal dysfunction and autophagosme accumulation are also related to a STV metabolic phenotype [36], we sought to study the mitochondrial β-oxidation in PGRN-deficient fibroblasts.
To assess FAO, we inhibited mitochondrial fatty acid import using etomoxir, an irreversible inhibitor of carnitine O-palmitoyltransferase 1 (CPT1), and measured the oxygen consumption rate (OCR) using the Seahorse assay. OCR values obtained in the presence of etomoxir reflect non–fatty acid–FAs derived respiration, as FAs entry into mitochondria is blocked. In contrast, OCR measured under vehicle conditions represents total mitochondrial respiration. The difference between vehicle- and etomoxir-treated conditions was therefore used as an estimate of FAO-dependent respiration, with a larger difference indicating higher reliance on FAO. After the inhibition of FAO with Etomoxir, CTL fibroblasts exhibited a more pronounced reduction in OCR compared to patient-derived fibroblasts. This suggests that patient-derived fibroblasts are less dependent on FAs for oxidative respiration, as evidenced by the smaller difference in OCR between Etomoxir-treated and untreated cells (Figure 4f). To evaluate lipid-based ATP production, maximal lipid respiratory capacity, and lipid spare respiratory capacity, we measured the difference in OCR between Etomoxir-treated and untreated fibroblasts under conditions of palmitic acid surplus. This approach ensures that the availability of FAs does not mask the results, allowing us to accurately assess the capacity of the mitochondria to metabolize FAs. As it is shown in Figure 4g, lipidic ATP production may be diminished in GRN+/− fibroblasts, although this analysis did not reach statistical significance due to the notable variability of CTL fibroblasts. However, a significant impairment in Lipidic maximal respiration capacity and Lipidic spare respiratory capacity was distinctly evident in GRN+/− fibroblasts (p < 0.05), attributed to a concurrent reduction in spare respiration (p < 0.05), when compared to CTL fibroblasts (Figure 4g, detail in Figure S14b).
To confirm that FAs transport is intact and that the reduced effect of Etomoxir is not due to impaired FAs transport into the mitochondria, we tracked Red-C12, a saturated FAs analog with a fluorophore with an overall length approximately equivalent to that of an 18-carbon FA, in both CTL and GRN+/− fibroblasts. We observed a similar accumulation of Red-C12 within the mitochondria of GRN+/− fibroblasts, as indicated by Red-C12 staining in Mitotracker-positive structures (Figure S15). This suggests that FAs transport into the mitochondria is not compromised.
Our data indicate that GRN haploinsufficiency is associated with reduced mitochondrial FAO, as control cells exhibit higher FAO levels compared to GRN+/− fibroblasts. This reduction is accompanied by impaired mitochondrial respiratory performance, consistent with altered mitochondrial metabolism. This suggests dysfunctional mitochondrial respiration that may increase the likelihood of lipid accumulation.
3.5. GRN Insufficiency Causes Impaired Lipid Metabolism in Skin Fibroblasts from FTD-GRN Patients
Accordingly, our focus shifted towards intracellular organelles termed LDs, which act as reservoirs for stored lipids destined for future utilization. Analysis of LDs by TEM images unveiled a significant rise in these structures in GRN+/− fibroblasts compared to CTL fibroblasts (p < 0.05), often located proximal to or in direct contact with the endoplasmic reticulum, and characterized by an electron-lucent core encompassed by an electron-dense periphery (Figure 5a, detail in Figures S16 and S17). This pattern is consistent in GRN+/− fibroblasts where the incorporation of newly synthesized TAGs which are more electron-dense, into pre-existing LDs enriched with cholesterol esters which are electron-lucent [37], results in liquid-to-liquid phase separation of these two lipid components.
This discovery prompted us to delve into how the regulation of LD dynamics is affected in GRN+/− fibroblasts and to elucidate whether treatment with rhPGRN can exert a modulatory influence on this process. To monitor the recent incorporation of esterified FAs into LDs, we implemented a pulse-chase assay using Red-C12 and BD493, which incorporates into LD-specific neutral lipids (Figure 5b) [23]. Under CM condition GRN+/− fibroblasts demonstrated elevated levels of BD493 staining in comparison to CTLs (p < 0.05), as indicated in Figure 5c (uncropped images in Figure S18). This observation is in accordance with our TEM imaging findings (Figure 5a) and is further supported by a larger portion of Red-C12 being localized within LDs in GRN+/− fibroblasts in comparison to CTLs (p < 0.05) (Figure 5c). The elevated LD levels observed in PGRN haploinsufficient fibroblasts suggest lipid dysfunction in these mutant cells.
The dynamics of LDs are tightly regulated, especially under conditions of nutrient deprivation. During such periods, FAs can be mobilized from membrane-bound organelles through autophagy and lysosomal lipolysis, thereby augmenting the LD pool. Conversely, FAs can be released from LDs themselves by lysosomal (lipophagy) and cytoplasmic lipolysis, facilitating their transfer into mitochondria [20,23].
As expected, our observations revealed an increase in LD formation under STV conditions, a phenomenon that was similar in both CTL and GRN+/− fibroblasts. This was evident through both increased BD493 staining of LDs and, importantly, the vast majority of Red-C12 being localized within LDs in GRN+/− fibroblasts compared to CTLs (p < 0.05) (Figure 5c). The observed reduction in differences under nutrient deprivation could be attributed to either a saturation point in the cells’ ability to store LDs, or a reduced biogenesis of LDs through autophagolysosome-driven processes in GRN+/− fibroblasts. However, considering that lipid dynamics shift towards LDs accumulation and increased FAs metabolism under nutrient scarcity, the accumulation of LDs observed under CM condition in GRN haploinsufficient fibroblasts resembles the response observed during nutrient deprivation [23]. This finding suggests that PGRN-deficient cells may exhibit features that more closely resemble a STV-like phenotype.
To determine whether the observed alterations in LD dynamics within GRN+/− fibroblasts were directly attributable to GRN insufficiency, we treated both CTL and GRN+/− fibroblasts with rhPGRN at 500 ng/mL for 2 h. As depicted in Figure 5d, the 2 h rhPGRN treatment in GRN+/− fibroblasts showed a significant reduction in LD accumulation when compared to GRN+/− NT fibroblasts (p < 0.05), effectively restoring up to CTL fibroblast levels. It is noteworthy that this effect was specific to GRN+/− fibroblasts, as rhPGRN treatment did not modify LD levels within CTL fibroblasts (Figure 5d, uncropped images in Figure S19). rhPGRN treatment in GRN+/− fibroblasts reduced the proportion of Red-C12 in LDs when compared to GRN+/− NT fibroblasts (p < 0.05), bringing levels to those observed in CTLs under CM conditions (Figure 5d). However, this treatment showed no effect under nutrient-deprived conditions (Figure 5d), likely because the strong impact of the STV condition masked the effect of rhPGRN. In any case, under CM conditions, LD dynamics appear to be directly influenced by PGRN leading to lipid dysfunction in GRN haploinsufficient fibroblasts.
4. Discussion
This study investigated the cellular mechanisms underlying FTD in GRN c.709-1G>A mutation carriers. GRN insufficiency was found to result in a complex interplay of cellular alterations in GRN+/− fibroblasts, providing new insights into the disease mechanisms.
Comparison of fibroblasts from FTD-GRN patients and healthy CTLs revealed several key pathological hallmarks, similar to those observed in the brains of FTD patients. We confirmed reduced GRN protein and mRNA levels in GRN+/− fibroblasts, consistent with the haploinsufficiency seen in GRN-associated FTD [29,30]. We also observed impaired lysosomal acidification and dysregulated autophagic flux, likely contributing to the accumulation of storage material. This phenomenon is well-documented in the cerebral cortex of FTD-GRN patients [33] and is consistent with reports of lysosomal abnormalities and the accumulation of storage material in various experimental models, including animal studies [38,39] and patient-derived extraneuronal cell cultures, such as fibroblasts [14]. Although fibroblasts are peripheral cells, they share key PGRN-related pathways with neurons, including expression of several membrane receptors and reliance on conserved endocytic and lysosomal mechanisms. In both cell types, PGRN deficiency leads to lysosomal dysfunction, a central feature of GRN-associated neurodegeneration, making fibroblasts a relevant system to study core cellular consequences of PGRN haploinsufficiency. At the same time, we acknowledge that fibroblasts do not fully capture the specialized features and functional demands of neurons. Taken together, these findings reinforce the relevance of patient-derived fibroblasts as a suitable initial model for investigating specific cellular mechanisms associated with GRN haploinsufficiency, while also highlighting the need for complementary studies in CNS-derived cells and in vivo models to fully address disease complexity.
Mitochondrial dysfunction is a hallmark of various neurodegenerative diseases, including FTD [40]. Although only a few studies have specifically linked mitochondrial alterations to FTD-GRN [41,42]. In this study, we observed mitochondrial swelling and impaired respiratory function in GRN+/− fibroblasts, in line with previous reports suggesting that PGRN contributes to mitochondrial homeostasis in podocytes by regulating mitochondrial biogenesis, via the PGRN–Sirt1–PGC-1α/FoxO1 signaling pathway, and mitophagy [43]. These findings suggest that affected cells may struggle to meet elevated energy demands during stress conditions requiring increased ATP production. Furthermore, impaired mitochondrial function could lead to the accumulation of dysfunctional mitochondria, lipids, and reactive oxygen species, thereby exacerbating cellular damage.
Reflecting broader metabolic dysregulation, PGRN-haploinsufficient fibroblasts also displayed increased accumulation of LDs, indicative of impaired lipid homeostasis. Abnormal LD levels are commonly observed in neuronal cells from patients with neurodegenerative disorders, including FTD [44,45]. Notably, brain lipid composition in both humans and mice with PGRN deficiency exhibits disease-specific alterations [46]. Although LDs are not inherently toxic, mounting evidence suggests they may exert detrimental effects over time [47]. In LSDs, secondary lipid-based storage products are frequently observed and are believed to contribute directly to disease pathogenesis [48,49]. Similarly, lipofuscin accumulation in the central nervous system, once seen as a passive aging marker, is now understood as an active mechanism to sequester misfolded proteins. However, its buildup may ultimately worsen neurodegeneration by promoting the spread of toxic protein species [15]. In addition, changes in membrane composition driven by lipid imbalances can compromise membrane integrity and disrupt cellular signaling. The interaction between lipids and reactive oxygen species can generate peroxidated lipids, which are cytotoxic and can trigger cell death [50]. Lipid signaling has also been implicated in the regulation of neuroinflammation [51]. Taken together, these findings suggest that PGRN-deficient cells exhibit profound disruptions in lipid metabolism, potentially leading to altered membrane composition and impaired cell signaling. Combined with the accumulation of dysfunctional mitochondria, these defects may synergistically impair energy homeostasis and exacerbate the accumulation and propagation of toxic lipid and protein species, ultimately contributing to disease progression.
The relationship between lysosomal, mitochondrial, and lipid dysfunction is complex and not fully understood. Consistent with the observations, rhPGRN treatment produced the most rapid and pronounced effects on lysosomal function, followed by significant effects on lipid metabolism and, ultimately, mitochondrial function. The temporal sequence of effects may suggest, but does not establish, a model in which lysosomal recovery precedes improvements in downstream metabolic parameters, while alternative causal relationships remain possible. It is important to consider that extracellular rhPGRN can act through at least two complementary mechanisms: receptor-mediated signaling at the cell surface and intracellular uptake followed by lysosomal processing. Fibroblasts express sortilin, and extracellular PGRN is known to bind sortilin and prosaposin, undergo rapid endocytosis, and traffic to lysosomes where it is processed into mature granulin peptides [52]. In parallel, extracellular PGRN can directly signal through membrane receptors such as EphA2, Notch, TLR9, and TNFR, modulating cellular responses independently of internalization [9]. Our data indicate a strong link between PGRN function, lysosomal activity, and lipid metabolism. Notably, the lipid droplet accumulation and mitochondrial impairment observed in GRN+/− cells closely resemble the phenotype of control cells under nutrient deprivation. This suggests that lysosomal failure in FTD-GRN may produce a phenotypic resemblance to a ‘starvation-like’ metabolic state—characterized by lipid mobilization and reduced respiratory capacity—even in the presence of nutrients. In this context, the connection between GRN haploinsufficiency and mitochondrial dysfunction appears to be part of a broader bioenergetic shift secondary to lysosomal insufficiency. Further experiments targeting canonical nutrient-sensing pathways, such as AMPK and mTORC1, will be needed to fully elucidate the mechanistic basis of this relationship. The time-dependent effects observed in our study may therefore reflect an interplay between extracellular signaling and intracellular processing that ultimately converges at the lysosome to restore cellular nutrient homeostasis. Lysosomal dysfunction has been closely associated with both mitochondrial and lipid abnormalities in various neurodegenerative diseases and multiple LSDs [17,53,54]. In particular, studies on a specific form of neuronal NCL6 have reinforced the connection between lysosomal failure and defects in these two organelles [49,55]. However, similar findings have not yet been reported in NCL11, which is caused by GRN mutations—likely due to the limited number of identified cases and the lack of in-depth studies [56]. Lysosomes regulate lipid transport and biogenesis both directly, through the processing of lipids via saposins [57], and indirectly, via mechanisms involving the mTORC1 [58]. They also influence mitochondrial quality control through mitophagy. However, in the context of marked LD accumulation, it is plausible that lipid imbalance may lead to membrane composition alterations, which could in turn impair or aggravate mitochondrial homeostasis [59]. Our data indicates a close association between PGRN function, lysosomal activity, and lipid metabolism, whereas the relationship between GRN haploinsufficiency and mitochondrial dysfunction appears less direct. While lysosomal alterations may represent an important contributing factor to the observed lipid and mitochondrial changes, we cannot exclude parallel, bidirectional, or alternative pathways linking these processes. Further mechanistic studies will be required to clarify the hierarchy and causal relationships among lysosomal, lipid, and mitochondrial dysfunctions.
Lysosomal dysfunction in GRN+/− fibroblasts is associated with disruptions in lipid and mitochondrial homeostasis, resembling, in part, the phenotype observed under control starvation conditions, such as lipid droplet accumulation and impaired mitochondrial membrane potential. These disturbances reflect key features of lysosomal storage and neurodegenerative diseases and may contribute to disease-relevant cellular stress [60,61]. While lysosomal impairment, mitochondrial dysfunction and LD accumulation could potentially influence processes related to cellular aging or senescence, we did not directly assess canonical senescence markers (e.g., SA-β-gal, p16, or p21), and therefore these aspects remain a hypothesis for future investigation. Exploring how lysosomal dysfunction intersects with lipid and mitochondrial pathways, as well as potential senescence-like responses, represents a particularly interesting avenue for further research.
In conclusion, this study highlights the multifaceted cellular mechanisms underlying FTD-GRN pathology. The observed lysosomal dysfunction, altered autophagy, mitochondrial abnormalities, and impairments in lipid metabolism collectively contribute to the disease’s complexity. The data indicates that mitochondrial swelling and LD accumulation may play a role in the pathology of FTD-GRN. Together with established evidence of lysosomal dysfunction and autophagy alterations, our observations suggest a shift in the metabolic phenotype associated with GRN haploinsufficiency. This work opens several avenues for future research into FTD-GRN pathology and paves the way for identifying novel biomarkers to assess therapeutic efficacy and develop potential treatments. However, acknowledging the limited sample size, further research is needed to fully elucidate FTD-GRN pathogenesis and advance effective therapies.
5. Conclusions
In addition to impairing lysosomal function, GRN haploinsufficiency disrupts cellular metabolism, affecting both mitochondrial and lipid homeostasis. These metabolic alterations resulting from PGRN deficiency provide valuable insights into the fundamental pathological processes driving disease progression. Furthermore, acknowledging the limited sample size, these findings are hypothesis-generating and highlight pathways that warrant further investigation in neuronal and in vivo models, potentially guiding future research into therapeutic strategies, biomarkers, or other disease-relevant interventions for FTD-GRN.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bang J. Spina S. Miller B.L. Frontotemporal Dementia Lancet 20153861672168210.1016/S 0140-6736(15)00461-426595641 PMC 5970949 · doi ↗ · pubmed ↗
- 2Rohrer J.D. Guerreiro R. Vandrovcova J. Uphill J. Reiman D. Beck J. Isaacs A.M. Authier A. Ferrari R. Fox N.C. The Heritability and Genetics of Frontotemporal Lobar Degeneration Neurology 2009731451145610.1212/WNL.0b 013e 3181 bf 997a 19884572 PMC 2779007 · doi ↗ · pubmed ↗
- 3Rademakers R. Neumann M. Mackenzie I.R. Advances in Understanding the Molecular Basis of Frontotemporal Dementia Nat. Rev. Neurol.20128423434 Erratum in Nat. Rev. Neurol. 2013, 9, 240.10.1038/nrneurol.2012.11722732773 PMC 3629543 · doi ↗ · pubmed ↗
- 4Gijselinck I. Van Broeckhoven C. Cruts M. Granulin Mutations Associated with Frontotemporal Lobar Degeneration and Related Disorders: An Update Hum. Mutat.2008291373138610.1002/humu.2078518543312 · doi ↗ · pubmed ↗
- 5López de Munain A. Alzualde A. Gorostidi A. Otaegui D. Ruiz-Martínez J. Indakoetxea B. Ferrer I. Pérez-Tur J. Sáenz A. Bergareche A. Mutations in Progranulin Gene: Clinical, Pathological, and Ribonucleic Acid Expression Findings Biol. Psychiatry 20086394695210.1016/j.biopsych.2007.08.01517950702 · doi ↗ · pubmed ↗
- 6De Muynck L. Van Damme P. Cellular Effects of Progranulin in Health and Disease J. Mol. Neurosci.20114554956010.1007/s 12031-011-9553-z 21611805 · doi ↗ · pubmed ↗
- 7Cenik B. Sephton C.F. Kutluk Cenik B. Herz J. Yu G. Progranulin: A Proteolytically Processed Protein at the Crossroads of Inflammation and Neurodegeneration J. Biol. Chem.2012287322983230610.1074/jbc.R 112.39917022859297 PMC 3463300 · doi ↗ · pubmed ↗
- 8Kao A.W. Mc Kay A. Singh P.P. Brunet A. Huang E.J. Progranulin, Lysosomal Regulation and Neurodegenerative Disease Nat. Rev. Neurosci.20171832533310.1038/nrn.2017.3628435163 PMC 6040832 · doi ↗ · pubmed ↗