Simultaneous Study of Circular RNAs and Messenger RNAs in Colorectal Cancer: The Unbalanced Fate of a Couple?
Corentin Levacher, Joanna Delfosse, Camille Charbonnier, Françoise Charbonnier, Mathieu Viennot, Edwige Kasper, Jacques Mauillon, Nathalie Parodi, Stéphanie Baert-Desurmont, Philippe Ruminy, Claude Houdayer

TL;DR
This study explores how circular RNAs and messenger RNAs interact in colorectal cancer patients, finding an imbalance that may contribute to cancer development.
Contribution
The study introduces the SEALigHTS technique to analyze circRNA-mRNA interactions and identifies novel circRNA junctions in colorectal cancer.
Findings
Patients with colorectal cancer had a circRNA/mRNA ratio twice that of healthy controls.
The POLD1 gene produced a circRNA with potential cancer-promoting effects.
The imbalance between circRNAs and mRNAs in CRC is not due to competition between the two.
Abstract
Circular RNAs (circRNAs) can compete with messenger RNAs (mRNAs) from their host gene, which can lead to the downregulation of mRNA. We investigated this potential mechanism of downregulation in colorectal cancer (CRC) predisposition in a large cohort of 712 patients suspected of having hereditary CRC and 249 healthy controls, with a focus on 23 genes associated with CRC. Using the novel SEALigHTS technique (Splice and Expression Analyses by Exon Ligation and High-Throughput Sequencing), we identified 220 circular RNA junctions, including 47 new ones. Patients exhibited a circRNA/mRNA ratio approximately twice that of controls, particularly for the POLD1 gene, which produces a circRNA with potential cancer-promoting effects. The findings imply that the balance between circRNAs and mRNAs is disturbed in CRC, though not due to competition between the two. They also suggest that specific…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
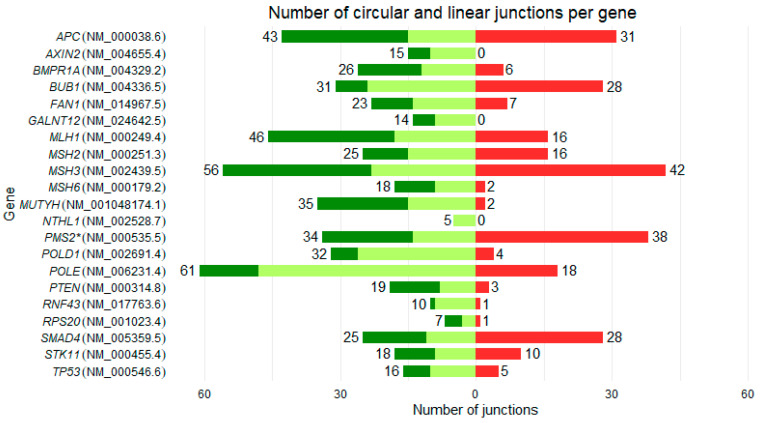 Figure 1
Figure 1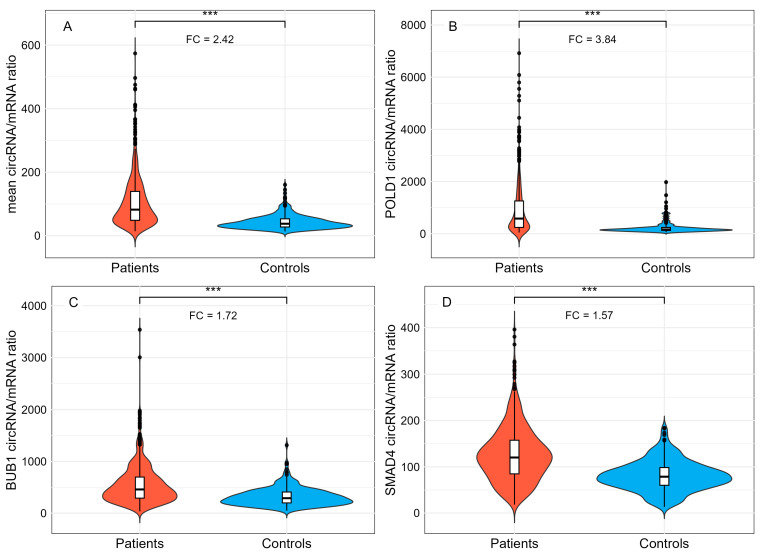 Figure 2
Figure 2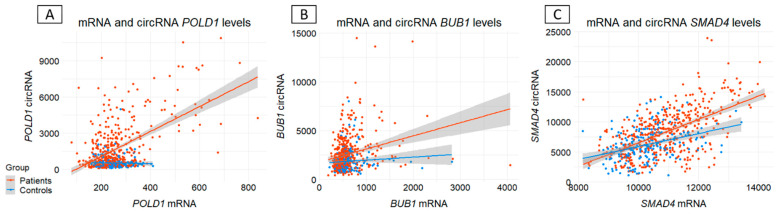 Figure 3
Figure 3| Gene | Reference | Number of Exons | Number of Linear Junctions | Number of Circular Junctions | Number of New Circular RNAs | Circular/Linear Junctions (%) | Circular/Linear UMIs (%) |
|---|---|---|---|---|---|---|---|
| POLD1 | 27 | 32 | 4 | 3 | 12.50 | 731.23 | |
| BUB1 | 25 | 31 | 28 | 8 | 90.32 | 492.85 | |
| PMS2 * | 15 | 34 | 38 | 17 | 111.76 | 145.69 | |
| SMAD4 | 12 | 25 | 28 | 6 | 112.00 | 114.25 | |
| POLE | 49 | 61 | 18 | 6 | 29.51 | 61.42 | |
| MSH3 | 24 | 56 | 42 | 1 | 75.00 | 39.01 | |
| RNF43 | 10 | 10 | 1 | 0 | 10.00 | 23.69 | |
| FAN1 | 15 | 23 | 7 | 0 | 30.43 | 20.81 | |
| MLH1 | 19 | 46 | 16 | 6 | 34.78 | 19.67 | |
| MSH2 | 16 | 25 | 16 | 2 | 64.00 | 15.95 | |
| APC | 16 | 43 | 31 | 3 | 72.09 | 12.19 | |
| MUTYH | 16 | 35 | 2 | 1 | 5.71 | 4.05 | |
| STK11 | 10 | 18 | 10 | 4 | 55.56 | 2.93 | |
| BMPR1A | 13 | 26 | 6 | 2 | 23.08 | 2.28 | |
| MSH6 | 10 | 18 | 2 | 2 | 11.11 | 2.24 | |
| TP53 | 11 | 16 | 5 | 2 | 31.25 | 2.15 | |
| PTEN | 9 | 19 | 3 | 0 | 15.79 | 0.18 | |
| RPS20 | 4 | 7 | 1 | 1 | 14.29 | 0.04 | |
| AXIN2 | 11 | 15 | 0 | 0 | 0.00 | 0.00 | |
| GALNT12 | 10 | 14 | 0 | 0 | 0.00 | 0.00 | |
| NTHL1 | 6 | 5 | 0 | 0 | 0.00 | 0.00 |
- —the Region Normandie and the European Regional Development Fund (ERDF)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCircular RNAs in diseases · Cancer-related molecular mechanisms research · MicroRNA in disease regulation
1. Introduction
Known for decades [1,2], the messenger RNA (mRNA) is the centerpiece of the transcriptome. This intermediary between DNA and protein is regulated by various mechanisms, including transcription [3,4,5], splicing [6], stability [7], capping [8], transport [9], and translation [10]. Among the elements involved in this regulation, many types of RNA have been documented. MicroRNAs, as well as pseudogene transcripts and long non-coding RNAs, have been shown to play a role in regulating mRNA [11]. These different elements are grouped together in a network known as competing endogenous RNA (ceRNA), and accumulating evidence points to its preponderant role in various pathologies such as cancer [12], neurodegenerative [13], cardiovascular [14], and autoimmune diseases [15]. Recently, with growing interest, circular RNAs (circRNAs) have been included in this ceRNA network [16].
circRNAs are single-stranded, closed-loop covalent structures, devoid of 5′ caps and 3′ poly-A tails. They result from a process known as backsplicing, whereby a 5′ splice site binds to an upstream 3′ splice site [17]. Interest in circRNAs has increased greatly, mainly because the levels of certain circRNAs have proved important for improved diagnosis, prognosis, and disease monitoring in Alzheimer disease [18], cardiovascular disease [19], and cancer [20]. These observations are often attributed to their functions as microRNA and RNA binding protein sponges [21], but other mechanisms are emerging, e.g., cap-independent translation that enables the generation of peptides involved in different tumorigenesis pathways [22,23], or transcription modulation. This last mechanism is particularly interesting, as it has been shown that circRNAs can regulate the levels of various mRNAs, including those of the parental gene [24,25], and a physiological balance between these two forms of transcript has been demonstrated [26,27]. Consequently, it may be hypothesized that a disruption of this physiological balance represents a novel, hidden mechanism in genetic diseases [28,29,30,31]. The question is particularly relevant in diseases where known mechanisms explain only a fraction of cases; the remainder are referred to as “missing heritability”. Colorectal cancer (CRC) exemplifies this issue, as the majority of non-polyposis familial and early-onset microsatellite stable (MSS) cases have no identified genetic cause [32]. To investigate this hypothesis, we performed a simultaneous analysis of circRNA and mRNA expression in blood samples for 23 CRC predisposition genes in a cohort of 712 CRC patients (i.e., the “missing heritability” cohort) and 249 matched controls, using a novel, dedicated technique named SEALigHTS (Splice and Expression Analyses by exon Ligation and High-Throughput Sequencing) [29,33]. We described the backsplicing landscape for these genes, studied the circRNA–mRNA balance between patients and controls, and searched for a competitive mechanism between circRNA and mRNA expression.
2. Materials and Methods
Patients and controls
The collection of unexplained CRC cases was previously described [32] and includes 1029 patients with a personal and/or familial history suggestive of hereditary predisposition to CRC, with Lynch syndrome and polyposis excluded (Table 1). All patients were recruited by the French network of Cancer Genetics Departments. In addition, 500 healthy volunteers without any personal or familial CRC history among their first-degree relatives were recruited by the Clinical Investigation Center of Rouen University Hospital. Among them, 712 patients and 500 controls had their blood sampled in PAXgene tubes for RNA extraction. To ensure quality, PAXgene tubes were transported within two days after sampling to the investigating center and extracted within 2 weeks. All 712 patients were included in the study. Among 500 healthy controls with available RNA samples, 249 were randomly selected, with a comparable sex distribution and a similar age range to the patient group (Table 2). Written informed consent was obtained from all participants (ethics approval: DC-2013–1759).
Continuous variables are reported as mean ± standard deviation, and categorical variables as number (percentage). Age corresponds to age at blood sampling. RNA concentration refers to total RNA extracted from PAXgene blood samples. Group comparisons were performed using Welch’s t-test for continuous variables and the chi-square test for categorical variables.
RNA extraction
RNA was extracted from peripheral blood using the PAXgene Blood RNA kit (PreAnalytiX, Switzerland), and concentration was determined with the NanoDrop 1000 Spectrophotometer (ThermoFisher Scientific, MA, USA), with a mean of 88 ng/µL (min: 4; max: 446).
SEALigHTS
Principle
Initially used as a multiplex technique for fusion transcript detection [34] and for measuring gene expression [35], SEALigHTS has been adapted and validated to study and quantify splicing and backsplicing [29]. Briefly, SEALigHTS allows the simultaneous exploration of all exon–exon junctions in a panel of genes of interest, thanks to probes designed at exon extremities. Following reverse transcription and probe hybridization on cDNA, nearby probes are ligated if splicing and/or backsplicing occurs, and the number of ligations is quantified using unique molecular identifiers and high-throughput sequencing. All possible combinations of exons, i.e., splicing and backsplicing, are detectable.
Protocol
Oligonucleotides probes contained (i) specific sequences for each exon (19–30 bases in length to obtain an optimal melting temperature of 70 °C), (ii) Unique Molecular Identifiers (UMI), consisting of 7 random bases, to count the number of ligations, (iii) complementary sequences of universal PCR primers. In accordance with the transcripts present in Ensembl and/or GTex Portal and the literature, 788 probes (Supplementary Table S1) were designed at exon boundaries for the 23 CRC predisposition genes listed by French experts from the Groupe Génétique et Cancer (GGC) [36] (Table 3). For exon ends where a single-nucleotide polymorphism (SNP) is present with a frequency higher than 1% in the Caucasian population, the corresponding probe was designed with a combination of the 2 nucleotides involved to avoid any hybridization problems. If the alternative and canonical splice sites to be explored were close, partial probe overlap occurred. In this case, and when the sequence overlap was greater than 50%, the alternative probe was not selected. To control for DNA contamination, intronic probes were designed for each gene and added to the probe mix (Supplementary Table S1). Following quantification, 14−500 ng of total RNA were converted into cDNA using a SuperScript™ VILO™ cDNA Synthesis kit (Invitrogen, Carlsbad, CA, USA). cDNAs were incubated for 1 h at 60 °C with the mix of 788 oligonucleotide probes (Supplementary Table S1) in 1 × SALSA MLPA buffer (MRC Holland, Amsterdam, The Netherlands). Following hybridization, neighboring probes were ligated using the thermostable SALSA DNA ligase (MRC Holland, Amsterdam, The Netherlands).
Among the 23 genes studied (Table 3), gene expression reported by the GTex Portal varied up to 10,000 times higher for some genes than for others. After an initial trial, it appeared that the high expression of PTEN and RPS20 required a separate amplification mix in combination with the Q5^®^ Hot Start High-Fidelity 2X Master Mix (NEB, Ipswich, MA, USA), which is why 2 independent PCRs were performed using different universal PCR primers. PCR products were purified using AMPure XP magnetic beads (Beckman Coulter, Brea, CA, USA). The library containing PTEN and RPS20 was diluted 1:20 in the library containing the other genes. Sequencing of amplicons was carried out using a NextSeq^®^ system with 75 cycles (Illumina, San Diego, CA, USA).
Sequencing reads, comprising a maximum of 75 bases, including UMIs, left and right probes, and barcodes, were demultiplexed using the barcodes and aligned with probe sequences. Exon junctions were counted for quantification purposes, with the 7 random bases of the UMI allowing 16,384 different combinations of unique molecules. A minimum of 65% mapping of left and right probes was deemed necessary to avoid primer dimers and ensure sufficient ligation and reliable results. A custom Python (v3.11) script, available on request, generated schematic backsplicing and splicing profiles (Supplementary Figure S1).
Bioinformatics and statistical analyses
Ratio circRNAs/mRNAs
Linear mRNAs (splicing) were distinguished from circRNAs (backsplicing) based on the order of the probes. If a probe located at the 3′ boundary of an exon is ligated with a probe located at the 5′ boundary of the following exon, the transcript is linear. Conversely, if this 3′ probe is ligated to a 5′ probe of a preceding exon, the transcript is circular (Supplementary Figure S1). Each circular RNA is a unique molecule, so all circular junctions must be considered, unlike an mRNA molecule composed of successive linear junctions. For each gene, the ratio between circRNAs and mRNAs was obtained by dividing the total number of UMI counts of all circular junctions by the median number of UMI counts of all canonical linear junctions of that gene, i.e., . Importantly, some UMIs attributed to linear junctions may actually derive from circRNAs. This occurs when a circRNA contains multiple exons, allowing probes to hybridize to internal exons. Consequently, the signal may be misinterpreted as linear junctions, affecting mRNA expression calculation. To mitigate this, only linear junction UMIs with no potential influence from circRNAs expressed at more than 10% of the corresponding mRNA were counted for mRNA calculation. A Wilcoxon rank-sum test was used to assess differences between patient and control groups, and p-values were corrected using the Bonferroni method to control the overall type I error rate at 5%.
mRNA and circRNAs expression
For each sample and junction, the UMI counts for each junction were normalized according to library size, i.e., . For each gene and sample, mRNA expression was calculated as the mean of normalized “circular-free” canonical linear junctions. In contrast, as circRNAs represent distinct transcripts, expression per gene was obtained by summing the UMIs of normalized circular junctions.
Circular RNAs characterization
To characterize the full circRNA sequences, divergent primers were designed (Supplementary Figure S2A,B). From 500 ng of RNA, cDNA was generated using the SuperScript™ VILO™ cDNA Synthesis kit (Invitrogen, Carlsbad, CA, USA), followed by PCR with the Q5^®^ Hot Start High-Fidelity 2X Master Mix kit (NEB, Ipswich, MA, USA). Gel-purified PCR products were sequenced using PCR primers and the BigDye v3 kit (Applied Biosystems, South San Francisco, CA, USA) and analyzed by capillary electrophoresis on a “3500 Genetic Analyzer” (Applied Biosystems, South San Francisco, CA, USA).
3. Results
3.1. General Considerations
Of the 961 samples analyzed, 657 (68.4%), i.e., 433 patients and 224 controls, met the quality criteria (65% mapping), with a mean UMI count of 1,183,735, ensuring homogeneous samples and results, as shown by principal component analysis (PCA) (Supplementary Figure S3 and Supplementary Table S2).
GREM1 and EPCAM genes are poorly expressed in whole blood. As the number of UMIs is gene expression-dependent, we were unable to detect all canonical junctions; therefore, GREM1 and EPCAM were excluded from further analyses.
For the 21 genes analyzed, 559 linear junctions (canonical and alternative) were identified (Supplementary Table S1), with diversity in their numbers between genes (Figure 1 and Table 3). Although the number of linear junctions depends on the number of exons, the number of expressed alternative junctions differed between genes, e.g., APC and MSH2 both have 16 exons, but 43 and 25 total linear junctions were expressed in blood, respectively. As the probes hybridize on both PMS2 and its pseudogene PMS2CL, their respective mRNAs and circRNAs cannot be distinguished. Therefore, PMS2 was not considered in the analyses.
3.2. Backsplicing Landscape
Of the 20 genes studied (PMS2 excluded), SEALigHTS detected 220 circular junctions (Supplementary Table S1), of which 47 (21.36%) were novel (Table 3), not reported in RJunBase [37], a database compiling data on linear junctions, circular junctions, and fusion transcripts. A wide diversity in the number and expression level of circRNAs (Table 3 and Figure 1) was observed. Three genes (AXIN2, GALNT12, and NTHL1) did not show circular junctions. For the remaining 17, the median number of circular junctions per gene was 7 and ranged from 1 to 42 for RPS20 and MSH3, respectively. The number of circular junctions was not linked to the number of linear junctions; e.g., MUTYH had 35 linear junctions and only two circular junctions, unlike SMAD4, which had 25 and 28 respectively (Table 3 and Figure 1). While circRNAs were expressed at low levels for most genes (17/20), POLD1, BUB1, and SMAD4 had circRNA levels similar or higher than their mRNA molecules. The relative expression of circRNA to mRNA varied between genes and was not related to the number of circRNAs produced by the gene. For example, POLD1 had the highest circRNA/mRNA expression level at 731.23%, i.e., a relative abundance of 731 circRNAs per 100 mRNA molecules, with only four circRNAs produced, whereas APC had a ratio of 12.19% with 31 circRNAs produced (Table 3).
Following circRNA detection, we aimed to describe the entire sequence of the most frequent circRNAs (Supplementary Table S3), i.e., circPOLD1(3-2), circBUB1(19-10), circBUB1(9-2), circBUB1(9-6), circSMAD4(8-6), circSMAD4(10-5), and circSMAD4(8-5), and found alternative backsplicing events in two cases. Sequencing the circRNA joining exons 3 to 2 of POLD1 (circPOLD1(3-2)) characterized a circPOLD1(3-2p) (Supplementary Figure S2C), i.e., with use of an alternative splice site in exon 2, reported in RJunBase for both linear and circular transcripts. For the circRNA joining exons 10 to 5 of SMAD4 (circSMAD4(10-5)), sequence analysis revealed a full-length backsplice transcript and an alternative one with exons 6 and 7 skipped (Supplementary Figure S2D). As these alternative splicing events are also present in mRNAs, this suggests that the same (back)splicing machinery produces both messenger and circular RNAs, increasing circRNA diversity.
3.3. Ratio circRNA/mRNA Between CRC Patients and Controls
The ratio was calculated for the 17 genes harboring circular junctions (Supplementary Table S2). We observed a significant 2.42-fold increase in the mean circRNA/mRNA ratio in patients compared to controls (105 vs. 43; (Wilcoxon test, p < 10^−16^), (Figure 2A and Supplementary Figure S4). This result was confirmed independently for POLD1, BUB1, and SMAD4, which exhibited the highest circRNA/mRNA ratios. Hierarchical clustering confirmed these findings and did not reveal confounding factors (Supplementary Figure S5). The increase in the circRNA/mRNA ratio was predominantly driven by POLD1 (Figure 2B), with a fold change of 3.84, rather than by BUB1 and SMAD4 (fold changes of 1.72 and 1.57, respectively) (Figure 2C,D). We identified 31 outlier patients, i.e., patients with a circRNA/mRNA ratio above mean +/−2 DS. Among these patients, 11 exhibited outlier ratios for all three genes (POLD1, BUB1, and SMAD4), while 12 and 8 patients displayed outlier ratios for two and one of these genes, respectively. To explore potential genotype–phenotype correlations, we investigated whether this subgroup of 31 patients was associated with specific clinical phenotypes. No significant association was found between this subgroup and the presence of adenocarcinoma (p = 0.43; Fisher’s exact test), adenoma (p = 0.75; Pearson’s chi-squared test), familial cancer (p = 0.51; Pearson’s chi-squared test), early sporadic cancer (p = 0.41; Fisher’s exact test), or multiple primary tumors (p = 0.64; Fisher’s exact test). Age and sex were tested and did not appear to be confounding factors (t-test p-value = 0.887 and Pearson’s chi-squared test: p-value: 0.55, respectively).
To shed light on a possible mechanism, we then investigated whether this circRNA/mRNA increase was linked to a decrease or increase in mRNA or circRNA levels, respectively. Regression analysis between circRNA and mRNA expression showed that this imbalance probably reflected increased circRNA production or accumulation rather than decreased POLD1, BUB1, and SMAD4 expression, thereby suggesting independent regulation (Figure 3).
4. Discussion
CircRNAs are now established as key players in human pathology, but original data remain scarce. We investigated whether disruption to the coregulation of mRNA–circRNA could represent a novel disease mechanism in a cohort of 712 unexplained CRC cases and 249 matched controls. While our data do not support a competitive regulatory mechanism, they reveal a significant imbalance in the mRNA–circRNA couple in blood between CRC patients and controls.
These results were obtained using SEALigHTS, a novel targeted high-throughput approach that, to our knowledge, is the only technology enabling the simultaneous quantitative analysis of mRNAs and circRNAs, including from FFPE-compatible material. By relying on probe hybridization at exon boundaries and UMI-based quantification prior to amplification, SEALigHTS circumvents the major limitations of conventional RNA-seq strategies for circRNA detection [38], without requiring RNA enrichment or specific sample treatment [39].
Our study provides new insights into (i) the mechanism of backsplicing, (ii) the landscape of circRNAs for 20 CRC genes, and (iii) the involvement of circRNAs from these 20 genes in CRC. These different aspects are discussed successively. Linear splicing and backsplicing of exons are mediated by the spliceosome and occur simultaneously during the transcription of most human genes. Briefly, two main models for exonic circRNA biogenesis coexist. In the first model, known as “lariat-driven circularization” or “exon skipping”, skipping of one or more exons from the pre-mRNA results in a spliced mRNA and a lariat containing the exon(s) that will be circularized. In the second model, “intron-pairing-driven circularization” [17], the presence of intronic repeated inverted complementary sequences, such as Alu sequences, enables the pairing of two introns that border exons that will be circularized. These sequences promote the spatial proximity of the 5′ splice site junction to the upstream 3′ site [40], leading to the formation of a circular transcript. Although both models may explain our results, we counted 220 circular junctions and 138 alternative linear splicing junctions (i.e., exon skipping), which for the most part were not related to circRNAs. For example, BUB1 has 28 circular junctions but only seven alternative linear splicing junctions. Consequently, based on these observations, the “intron-pairing-driven circularization” model is more likely to be involved.
Albeit counterintuitive, the number of circRNAs does not increase with the number of exons or introns (Table 3). This might be explained by independent regulation and/or by the fact that circRNA biogenesis depends on several parameters, such as the size of flanking introns and the presence of Alu sequences (see the “intron-pairing-driven circularization” model), transcription speed [41], notably through interaction with DNA forming circRNA:DNA hybrids (circR loops) [42], and splice site strength [26]. circPOLD1(3-2), circBUB1(9-2), and circBUB1(4-3) were more highly expressed than their parental mRNAs, but the vast majority of circRNAs (217/220, 94%) represented less than 10% of the corresponding mRNA (Supplementary Table S1).
These results confirm that backsplicing is less efficient than splicing [43] and suggest a transcriptional backsplicing background, as already described for linear splicing. More surprisingly, at the gene level, overall circRNA expression relative to the corresponding mRNAs is often greater than 10% (9/20 genes; Table 3). The values observed for POLD1, BUB1, and SMAD4, five times higher in the patients’ outlier group for POLD1 (Figure 2), can hardly be considered background noise and may have biological relevance (Table 3). This suggests that each gene has its own circRNA repertoire, with different and finely regulated but uncoupled circRNA/mRNA expression levels [44,45]. Although a competitive mechanism has been ruled out, we demonstrate that this couple may be unbalanced in certain pathological contexts [20,31]. Few large, high-throughput studies have reported circRNA/mRNA ratios, and, to our knowledge, none have done so in blood. Recently, a large integrative RNA-sequencing analysis of circRNA profiles in colorectal cancer demonstrated that circRNA downregulation was associated with decreased expression of circRNA host genes [46]. We also observed a similar trend in circRNA and mRNA levels, but in the direction of increased expression.
Patients suspected of hereditary predisposition to colorectal cancer exhibited a 2.42-fold higher circRNA/mRNA ratio compared to controls, an imbalance largely driven by POLD1, with a 3.84-fold change. No correlation with phenotype was identified for the subgroup of outlier patients with the highest circRNA/mRNA ratios. Our results suggest increased production or impaired clearance of POLD1 circRNA rather than reduced POLD1 expression, consistent with the known contribution of POLD1 to hereditary CRC predisposition, which is related not to decreased expression but to missense pathogenic variants in the exonuclease domain [47]. Interestingly, circPOLD1(3,2) has recently been shown to play a functional role in tumorigenesis. In a recent study, circPOLD1(3,2) expression increased with lesion severity in cervical cancer and promoted oncogenic signaling through interactions with RNA-binding proteins such as YBX1, leading to activation of pathways involved in cell proliferation and tumor progression [48]. These findings support the biological relevance of circPOLD1(3,2) and reinforce the significance of the imbalance observed in our study.
Overall, the circRNA/mRNA imbalance observed in CRC patients suggests that the circRNAs studied play a role in a subset of CRC cases, either as a cause or a consequence of other underlying defects. CircRNAs are known to be finely regulated in a cell-type dependent manner [49]; for example, they are highly expressed in muscle, and tumor studies have shown that circRNA levels are lower in tumors than in surrounding healthy tissue [50]. Thus, the observed imbalance may also reflect changes in cellular composition following tumorigenesis or chemotherapy and may serve as a biomarker associated with cancer onset and progression [20]. Given the phenotype of the cohort studied, i.e., patients suspected of hereditary predisposition to colon cancer, the small subgroup of 31 outlier patients is of particular interest and warrants further investigation, including cosegregation analyses in families, circRNA–mRNA tumor analyses, and the search for newly formed peptides to elucidate novel mechanisms.
5. Conclusions
Overall, splicing and backsplicing analyses using SEALigHTS highlight the RNA architecture of individual genes and provide no evidence for a competitive mechanism or coordinated regulation between circular and linear transcripts. The observed imbalance is largely driven by a single circRNA with oncogenic potential (circPOLD1(3,2)), suggesting that specific circRNAs may play a pivotal role in disease susceptibility. These findings open new avenues for investigating circRNAs in blood as potential biomarkers or functional contributors to cancer predisposition.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Brenner S. Jacob F. Meselson M. An Unstable Intermediate Carrying Information from Genes to Ribosomes for Protein Synthesis Nature 196119057658110.1038/190576 a 020446365 · doi ↗ · pubmed ↗
- 2Gros F. Hiatt H. Gilbert W. Kurland C.G. Risebrough R.W. Watson J.D. Unstable Ribonucleic Acid Revealed by Pulse Labelling of Escherichia Coli Nature 196119058158510.1038/190581 a 013708983 · doi ↗ · pubmed ↗
- 3Bentley D.L. Coupling m RNA Processing with Transcription in Time and Space Nat. Rev. Genet.20141516317510.1038/nrg 366224514444 PMC 4304646 · doi ↗ · pubmed ↗
- 4Anvar S.Y. Allard G. Tseng E. Sheynkman G.M. de Klerk E. Vermaat M. Yin R.H. Johansson H.E. Ariyurek Y. den Dunnen J.T. Full-Length m RNA Sequencing Uncovers a Widespread Coupling between Transcription Initiation and m RNA Processing Genome Biol.2018194610.1186/s 13059-018-1418-029598823 PMC 5877393 · doi ↗ · pubmed ↗
- 5Rodríguez-Molina J.B. West S. Passmore L.A. Knowing When to Stop: Transcription Termination on Protein-Coding Genes by Eukaryotic RNAPII Mol. Cell 20238340441510.1016/j.molcel.2022.12.02136634677 PMC 7614299 · doi ↗ · pubmed ↗
- 6Mironov A. Petrova M. Margasyuk S. Vlasenok M. Mironov A.A. Skvortsov D. Pervouchine D.D. Tissue-Specific Regulation of Gene Expression via Unproductive Splicing Nucleic Acids Res.2023513055306610.1093/nar/gkad 16136912101 PMC 10123112 · doi ↗ · pubmed ↗
- 7Passmore L.A. Coller J. Roles of m RNA Poly(A) Tails in Regulation of Eukaryotic Gene Expression Nat. Rev. Mol. Cell Biol.2022239310610.1038/s 41580-021-00417-y 34594027 PMC 7614307 · doi ↗ · pubmed ↗
- 8Aregger M. Cowling V.H. Regulation of m RNA Capping in the Cell Cycle RNA Biol.201714111410.1080/15476286.2016.125154027791484 PMC 5270520 · doi ↗ · pubmed ↗