Targeting Pathways Implicated in Cholesterol Metabolism for Novel Cancer Therapy
Yi Zhou, Vishakha Sharma, Xiaoyu Li, Rajeev K. Singla, Ankush Kumar, Ashishkumar Kyada, Suhas Ballal, Deepak Nathiya, Apurva Koul, Mohammad Khalid, Monica Gulati, Sandeep Arora, Tapan Behl, Joachim Kavalakatt, Bairong Shen, Anupam Bishayee

TL;DR
This review explores how cholesterol metabolism influences cancer growth and highlights key proteins that could be targeted for new cancer therapies.
Contribution
The paper provides a comprehensive overview of cholesterol metabolism pathways and their role in cancer, identifying potential molecular targets for novel anticancer strategies.
Findings
Dysregulated cholesterol metabolism contributes to tumorigenesis and cancer progression through pathways like SREBP, NPC1, PCSK9, and SOAT1.
Targeting cholesterol-related proteins offers a promising direction for developing metabolism-based cancer therapies.
Preclinical and clinical evidence suggests that high-fat diets may exacerbate cancer development through cholesterol signaling.
Abstract
Cholesterol is known for its function in cell metabolism and lipid transport, but it also serves as a powerful signal that can influence cancer growth. This review seeks to understand the various pathways and mechanisms through which the dysregulation of cholesterol metabolism is implicated in cancer growth and signaling, focusing on key molecular target proteins involved in cholesterol synthesis, transport, and metabolism. Additionally, the review further assesses the potential for targeting these key pathways and proteins as a future strategy of anticancer treatment and therapy. Cholesterol acts as a metabolic cue that reshapes diverse signaling networks, including hedgehog and several sterol-regulated pathways orchestrated by key proteins, including sterol regulatory element-binding protein 2 (SREBP2), sterol O-acyltransferase 1 (SOAT1), Niemann–Pick type C1 (NPC1), and proprotein…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
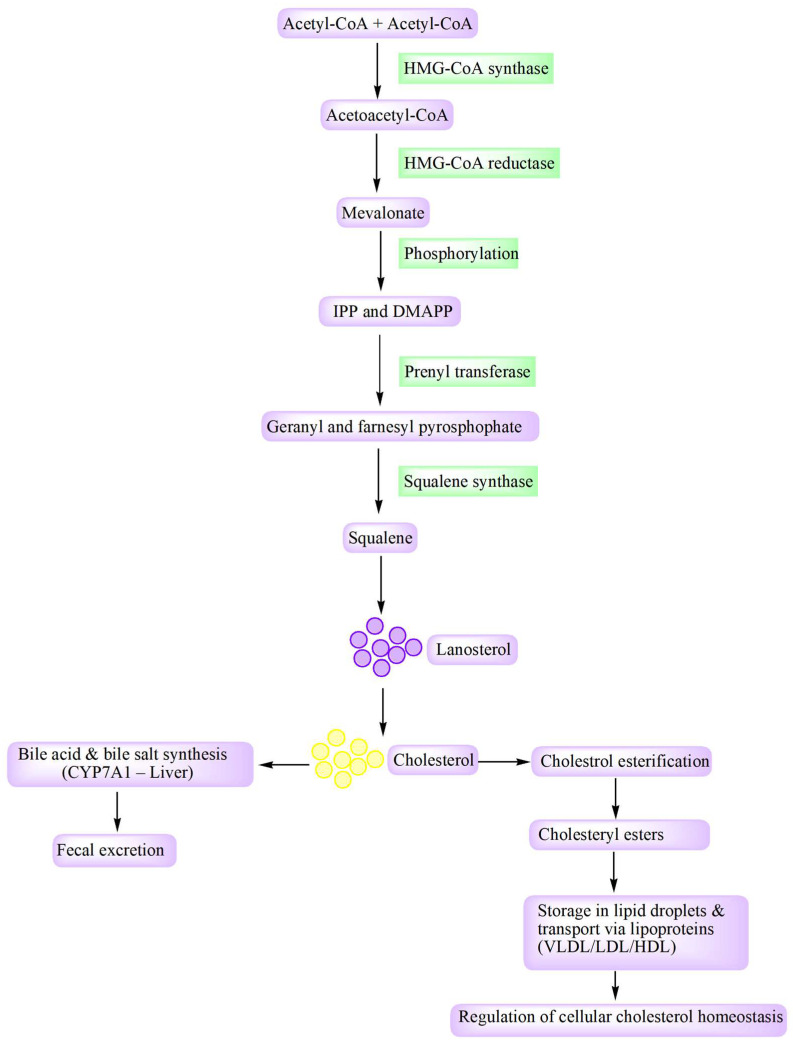 Figure 1
Figure 1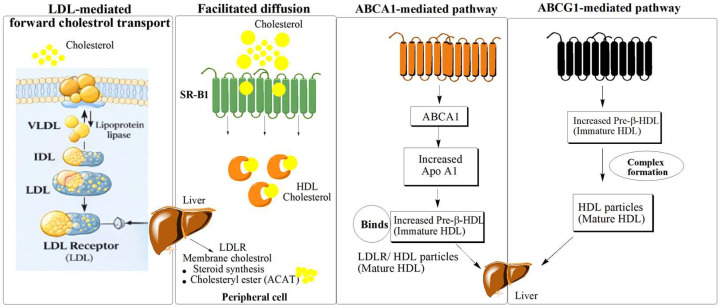 Figure 2
Figure 2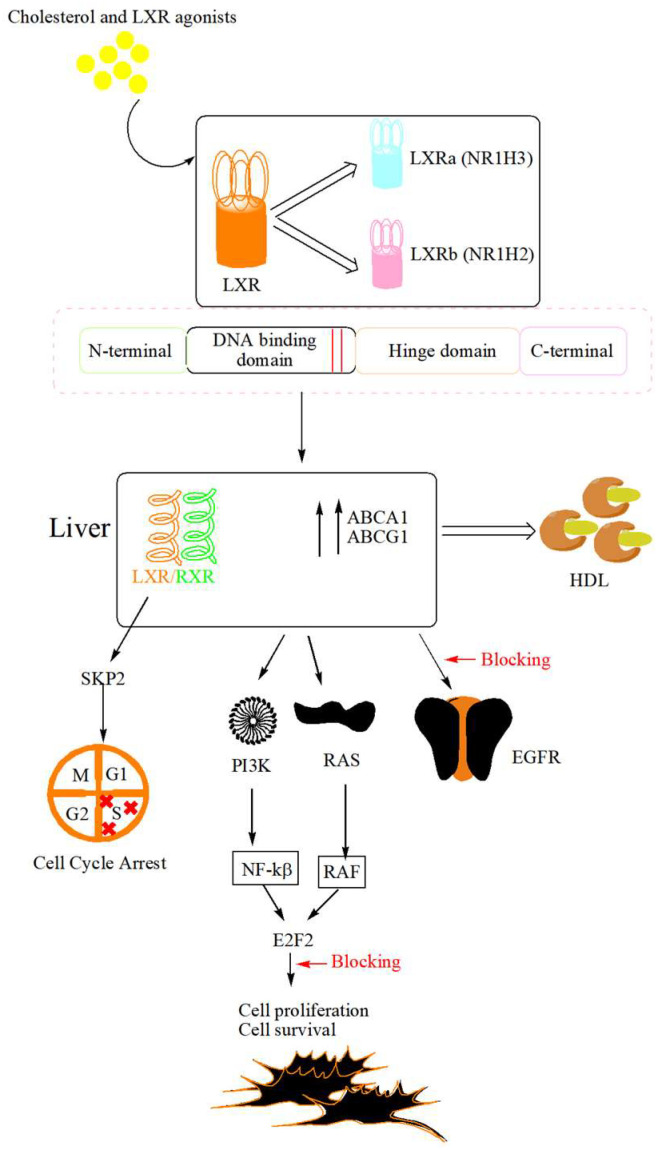 Figure 3
Figure 3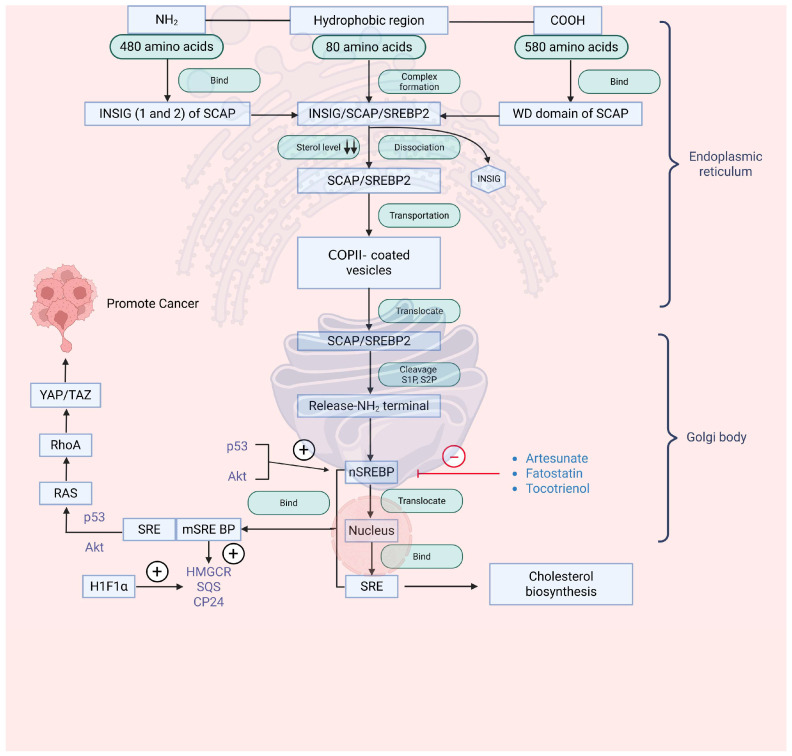 Figure 4
Figure 4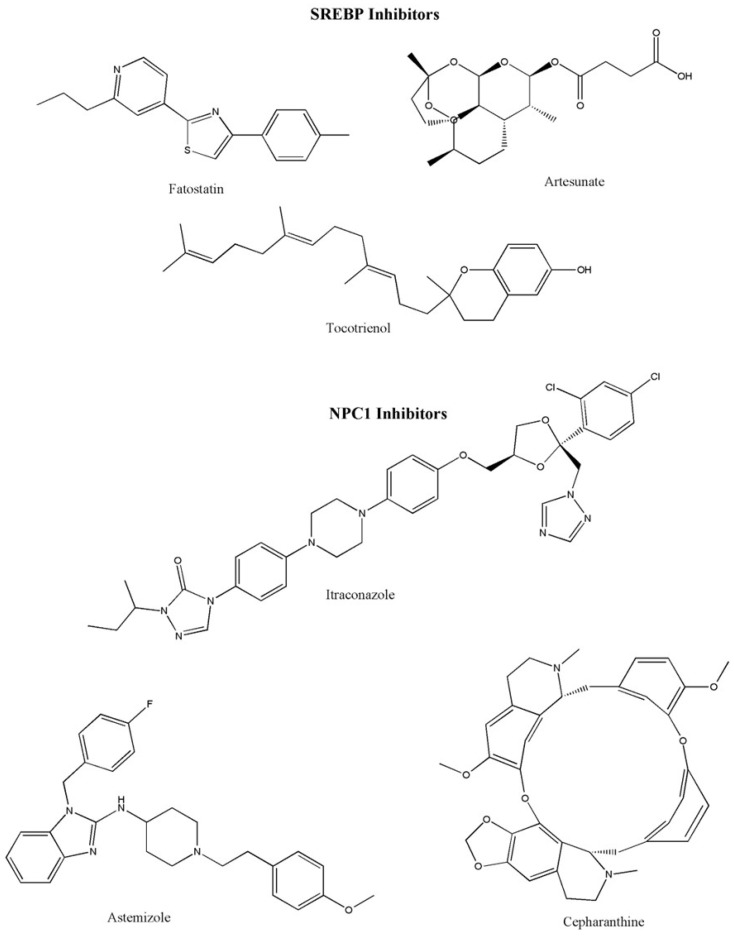 Figure 5
Figure 5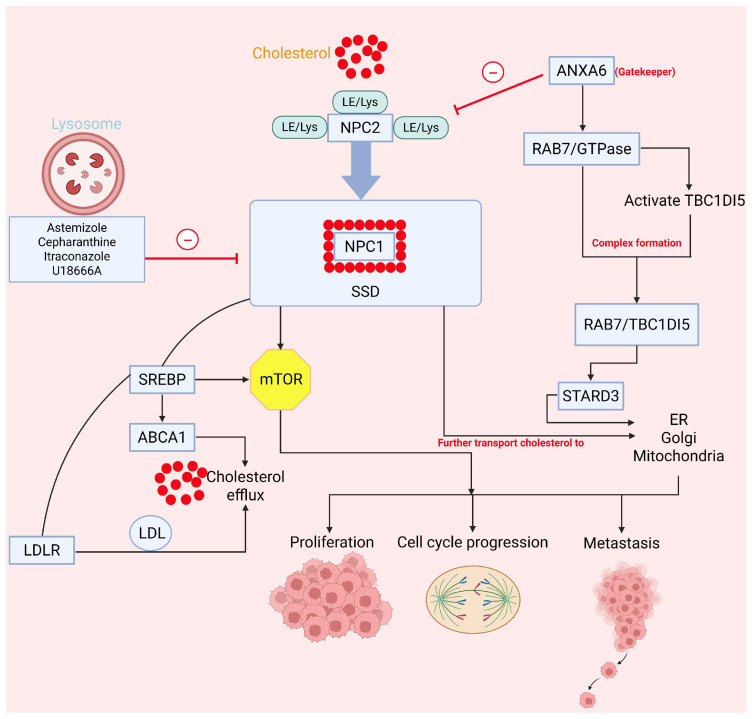 Figure 6
Figure 6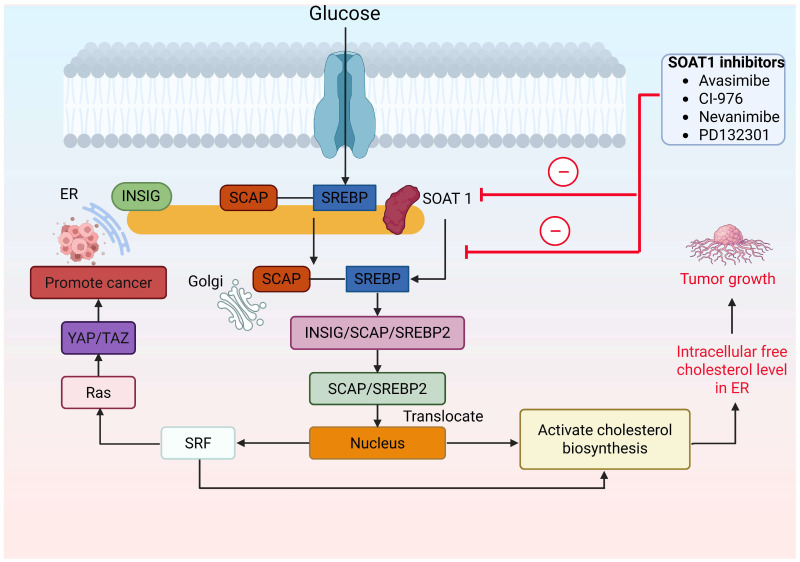 Figure 7
Figure 7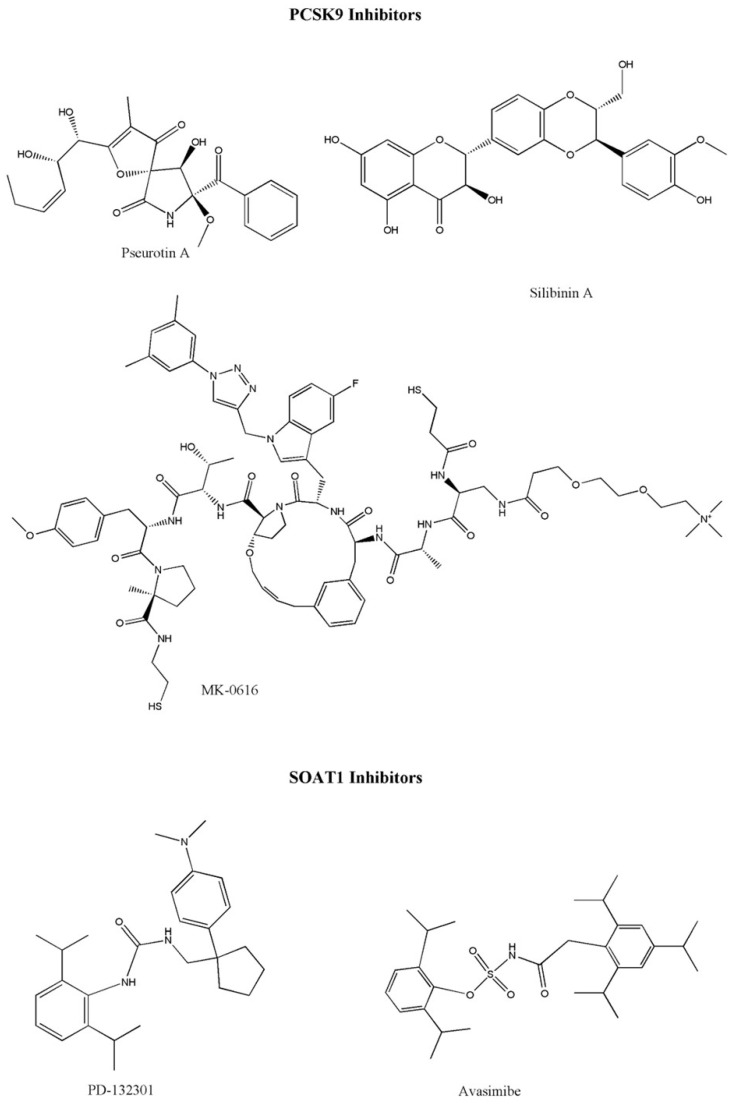 Figure 8
Figure 8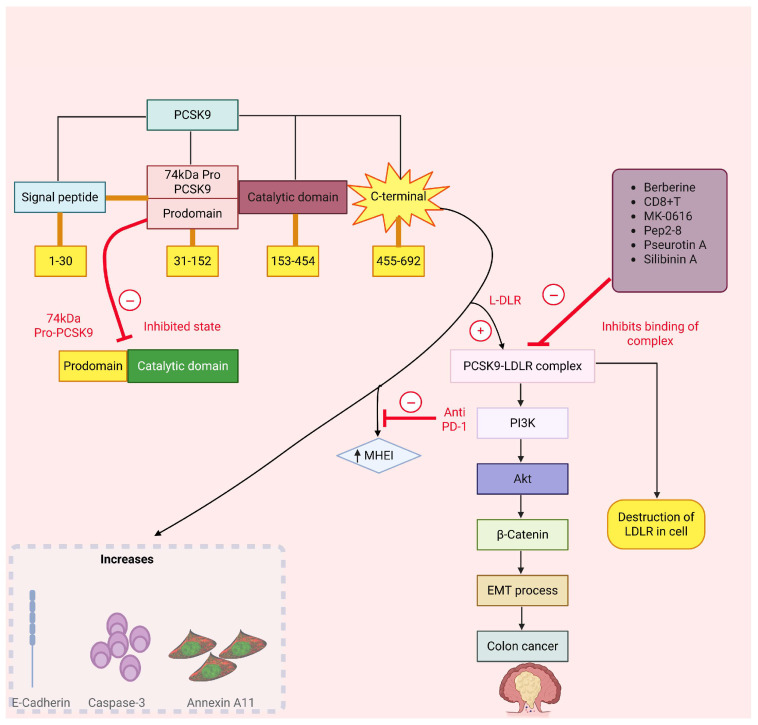 Figure 9
Figure 9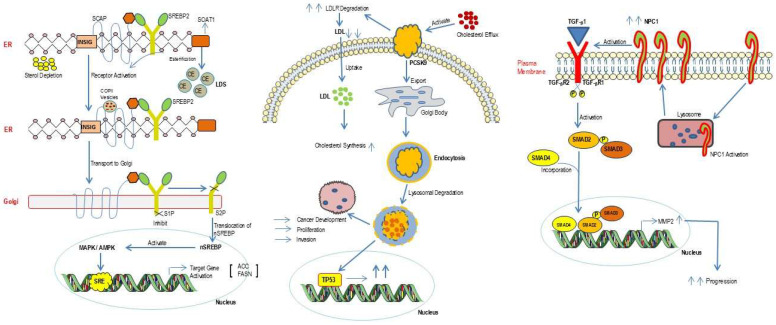 Figure 10
Figure 10- —Sichuan Science and Technology Program
- —National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer, Lipids, and Metabolism · Cholesterol and Lipid Metabolism · Peroxisome Proliferator-Activated Receptors
1. Introduction
Cholesterol is a vital component in building cellular structures and regulates various physiological processes [1,2]. Cholesterol sustains mammalian cellular function by stabilizing membrane organization and acting as a metabolic precursor for signaling molecules such as bile acids and steroid hormones [3,4]. Excessive accumulation of cholesterol in the bloodstream predisposes individuals to atherosclerosis and related cardiovascular disorders [5]. Composed largely of cholesteryl esters at their core, low-density lipoprotein (LDL) and high-density lipoprotein (HDL) facilitate the bidirectional transfer of cholesterol between the liver and peripheral cells [6]. LDL serves as one of the key biochemical mediators in increasing the deposition of cholesterol in arteries, a complex, multifactorial inflammatory process that ultimately drives the pathologies of heart disease and atherosclerosis [7,8]. Beyond its role in cardiovascular disease, LDL-mediated cholesterol transport is recognized as a critical metabolic adaptation in cancer, where rapidly proliferating tumor cells frequently reprogram cholesterol uptake through enhanced LDL receptor-mediated internalization. This elevated intracellular cholesterol pool supports accelerated membrane biogenesis, lipid raft formation, and oncogenic signaling, while reducing the energy cost in de novo cholesterol synthesis, thereby conferring a growth advantage in the tumor microenvironment [9,10].
Both clinical and experimental observations suggest that alterations in cholesterol metabolism play a significant role in cancer development [11,12,13,14,15]. Growing research indicates that cholesterol metabolism intricately shapes diverse aspects of tumor behavior by reprogramming oncogenic pathways, influencing ferroptosis, and altering the tumor microenvironment [16,17]. Preclinical data suggest that disruption of cholesterol metabolic pathways, including synthesis and uptake, restrains oncogenic transformation and tumor proliferation [18]. Despite increasing interest, the current state of literature broadly discusses cholesterol metabolism in cancer without thoroughly examining the specific signaling pathways and regulatory proteins that orchestrate these processes [19,20]. This review focuses on unraveling the central regulatory roles of key molecular pathways in cholesterol metabolism, with particular attention to SREBP2, SOAT1, NPC1, and PCSK9. These pathways pathologically impact lipid raft formation, membrane receptor signaling, and immune cell function, which contribute to tumor growth and metastasis. By synthesizing recent preclinical and clinical findings, we highlight the therapeutic potential of targeting these pathways for cancer therapy.
2. Literature Search Strategy
2.1. Database and Time Frame
We searched PubMed and the Web of Science Core Collection from database inception to September 2025 to identify studies on cholesterol metabolism pathways as therapeutic targets in cancer.
2.2. Search Concepts and Keywords
Search terms were developed using controlled vocabulary, such as Medical Subject Headings (MeSHs) and free-text keywords. For cholesterol metabolism and lipid homeostasis, we used the following keywords: “cholesterol metabolism,” “cholesterol biosynthesis,” “mevalonate pathway,” “cholesterol efflux,” “cholesterol transport,” “lipid raft,” and “lipoprotein*”. The cancer-, treatment-, and target-related keywords were “cancer”, “tumor/tumour”, “neoplasm”, “malignan”, “carcinoma”, “therapy”, “therapeutic target”, “SREBP2”, “SOAT1/ACAT1”, “NPC1”, “PCSK9”, “LXR”, “LDLR”, “HMGCR”, “statin”, “itraconazole”, “U18666A”, “SOAT inhibitor”, and “PCSK9 inhibitor”.
2.3. Eligibility Criteria
We included original studies (preclinical and clinical) and high-quality reviews that examined cholesterol metabolic regulation in cancer biology or therapy, with priority given to evidence involving SREBP2, SOAT1, NPC1, and PCSK9 and related effectors. We excluded conference abstracts without full text, non-cancer studies not informing oncologic mechanisms, and articles lacking relevance to cholesterol pathway modulation or cancer outcomes.
2.4. Study Selection and Synthesis
The titles and abstracts of publications were screened first, followed by full-text assessment. Reference lists of key articles were hand-searched to capture additional relevant studies. Evidence was synthesized narratively, emphasizing mechanistic links and translational/therapeutic implications.
3. Molecular Regulation of Cholesterol Metabolism: Key Pathways and Effectors
3.1. Cholesterol Biosynthesis Pathway
Cholesterol serves as a vital lipid constituent within the membranes of mammalian cells [21,22,23]. The cellular regulation of cholesterol levels involves a tightly coordinated network overseeing its synthesis, import, export, conversion, esterification, and movement across intracellular compartments [24,25,26]. Around 80% of the total cholesterol required by humans is typically biosynthesized within the body, while the remaining 20% is obtained from dietary sources [27,28]. The biosynthesis pathway of cholesterol is depicted in Figure 1, where we can observe that the cholesterol biosynthesis initiator is acetyl coenzyme A (acetyl-CoA) [29]. Cholesterol biosynthesis begins with the condensation of two acetyl-CoA molecules catalyzed by 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) synthase, producing acetoacetyl-CoA. Reduction of this intermediate by HMG-CoA reductase gives rise to mevalonate, which undergoes sequential phosphorylation to generate isopentyl and dimethylallyl pyrophosphate. These activated isoprenoids are subsequently linked by prenyl transferase to form geranyl and farnesyl pyrophosphate intermediates. Further, squalene synthase converts these products into squalene (intermediate), a triterpene composed of six isoprene units. Through epoxidation of squalene, it gets converted to lanosterol, which further leads to the synthesis of cholesterol and other sterols [30,31].
3.2. Liver X Receptors and Cholesterol Efflux: Central Mechanisms Governing Lipid Homeostasis
Cholesterol efflux represents a key regulatory pathway that expels excess intracellular cholesterol, safeguarding lipid homeostasis and ensuring proper cellular physiology [12]. Cholesterol efflux prevents cholesterol accumulation in arteries, working against plaque formation and the development of atherosclerosis [32]. During cholesterol efflux, an acceptor molecule is required to transport the cholesterol [33]. HDL acts as an acceptor of cholesterol, transporting the compound to the liver through four mechanisms [34,35]: aqueous diffusion, the ATP-binding cassette transporter A1 (ABCA1)-mediated pathway, the ATP-binding cassette transporter G1 (ABCG1)-mediated pathway, and facilitated diffusion (Figure 2). In aqueous diffusion, the cholesterol diffuses from the plasma membrane to HDL through the aqueous medium [36,37]. Lecithin–cholesterol acyltransferase (LCAT) functions as a key enzyme in reverse cholesterol transport (RCT), mediating the transfer and esterification of free cholesterol within HDL particles. After this chemical complex formation, cholesteryl esters are transferred to different lipoproteins of the body in the serum. This step is part of a physiological process, RCT. In RCT, excess cholesterol is cleared from peripheral cells and directed to the liver for biliary excretion. Dysregulation of this pathway causes cholesterol retention within cells, a condition linked to increased cancer cell proliferation, resistance to apoptosis, and metastatic progression [38]. Facilitated diffusion of cholesterol is carried out by regulation of a homo-oligomeric glycoprotein known as scavenger receptor class B, type 1 (SR-B1) [21], which is a type of HDL receptor that transfers the HDL-cholesterol to the liver through a hydrophobic channel of the SR-B1 receptor. Larger HDL particles bind tightly with SR-B1 than small ones and promote more free cholesterol efflux because even after saturation, SR-B1 receptor continue to reorganize cholesterol into the cell.
ABCA1 and aqueous diffusion are two major pathways to efflux 70% of total cholesterol prominently from the cell membrane of macrophages to pre-formed HDL particles [39], while the rest of the removal is carried out by other proteins [40]. ABCA1 is a gene that is known for the presence of four domains, two of which are nucleotide-binding domains (NBD), while the other two are six-helix transmembrane domains (TMD) [41]. Chen and the team indicated nine models to clarify the possible mechanism of cholesterol efflux by the ABCA1 pathway, and we discuss two models here as a mechanism elucidation [42]. The earliest conceptual framework describing this process is the molecular efflux, often referred to as the sequential model. This model states that the ABCA1 gene enhances the production of lipid-enriched microdomains, which further provides an active site for apo A1 [43]. The lipidation process of apo A1 gives rise to phospholipid-rich pre-β1 HDL particles that efficiently mobilize cholesterol from various intracellular and membrane regions [44]. The membrane solubilization model proposes that α-helical domains of apo A1 insert into cellular lipid bilayers, triggering apo A1-lipid clustering and partial membrane dissolution, which ultimately yields discoidal HDL particles [45]. The process culminates in the coupled release of membrane-derived cholesterol and phospholipids toward HDL particles, facilitating lipid redistribution and efflux [21]. Through its transporter function, ABCA1 drives the efflux of cholesterol originating from the breakdown of lipoproteins and its subsequent accumulation in the plasma membrane. The clearance of endosomal cholesterol relies on ABCA1-dependent retro-endocytosis of apo A1. Complementing this mechanism, ABCG1 provides an additional pathway that enhances cholesterol efflux and maintains lipid equilibrium [40]. ABCG1, a member of the ABC transporter family, functions in cholesterol export. Structurally, it possesses one NBD and TMD, characteristics that define it as a half transporter [46]. ABCG1 resides on the endoplasmic reticulum (ER) and participates in redistributing intracellular sterols, transferring cholesterol and oxysterols from the ER to the plasma membrane. At the cell surface, it channels cholesterol toward HDL and other lipid acceptors. The principal signaling pathways influenced by cholesterol dynamics are summarized in Table 1.
Mechanisms governing cholesterol efflux play a pivotal role in sustaining cancer cells’ viability and proliferation by maintaining intracellular cholesterol balance [54]. Multiple regulatory proteins, including ABCG1 [55], SR-B1 [56], LCAT, and apolipoprotein E (APOE), participate in this process, ultimately facilitating the organization of lipid raft microdomains essential for signal transduction.
Liver X receptors (LXRs), functioning as ligand-activated transcription factors within the nuclear receptor superfamily, are expressed as two isoforms—LXRα (nuclear receptor subfamily 1 group H member; NR1H3) and LXRβ (nuclear receptor subfamily 1 group H member 2; NR1H2) [57]. The principal role of LXRs is to maintain the cellular and systemic balance of cholesterol and lipid metabolism [58]. LXRα is mostly expressed in the liver, kidneys, and small intestine, with around 77–80% compared to the rest of the places like the adrenal, brain, heart, spleen, and testes [59,60]. The architecture of LXRs includes four distinct domains: the N-terminal activated function-1 (AF-1), a central DNA-binding domain, a hydrophobic ligand-binding pocket, and a C-terminal activation domain responsible for transcriptional regulation [61]. The LXR forms a heterodimer with the retinoid X receptor (RXR) that interacts with liver X-responsive elements (LXRE) within target gene promoters to modulate transcription in a ligand-dependent manner. In the unliganded state, the LXR/RXR complex associates with corepressor proteins, thereby suppressing gene expression. Upon binding of oxysterol ligands, conformational rearrangements within the receptor lead to the dissociation of corepressors and the recruitment of coactivator complexes. This transition initiates the transcription of downstream genes, including ABCA1 and ABCG1, which mediate cholesterol efflux to HDL [62,63]. The structural organization of LXR and its functional role in cancer are depicted in Figure 3.
The contributory role of LXRs in tumor progression is related to various types of activation and inhibition processes. Stimulation of LXR signaling results in a blockade of cell-cycle advancement from G1 to S phase, primarily through the reduced expression of S phase kinase-associated protein-2 (SKP2) [64,65]. The role of E2F transcription factor 2 (E2F2) has also been observed in estrogen receptor (ER)-positive breast cancer cell lines [66,67], showing constant downregulation of E2F2 after exposure to LXR agonists via inhibition of proliferation. LXR-mediated inhibition of cholesterol synthesis in prostate cancer systems, including both xenografted nude mice and in vitro cultures, affects the Ak strain transforming (Akt/protein kinase B), resulting in downstream dysregulation of signaling networks essential for cellular viability [47]. LXR activation in pancreatic ductal adenocarcinoma models interferes with growth factor signaling, prominently attenuating epidermal growth factor receptor (EGFR)-dependent pathways [52]. Furthermore, the transcriptional activation of melanoma cell secretion of APOE by LXR leads to inhibition of angiogenesis [53].
4. Correlation Between Cholesterol Metabolism and Cancer Progression
4.1. SREBP as a Potential Target
SREBP-2 is a transcription factor required for maintaining cellular cholesterol balance [68]. It belongs to the SREBP family, which encompasses various isoforms, such as SREBP-1a and SREBP-1c [69,70], and acts as a major regulator of cholesterol metabolism in cellular homeostasis [1,71]. SREBP-2 consists of 1141 amino acids, an NH2 domain, a hydrophobic region in the middle, and a carboxyl group (COOH) at the terminal point [72]. The amine (NH2) domain at the terminal position contains DNA binding site known as the Bhlh-Zip motif. By binding to SP1 and NF-Y transcription factors, this regulatory element/motif contributes to the modulation of target gene expression. The middle region, comprising 80 amino acids, consists of a hydrophilic loop extending to the ER. Translocation and localization processes are carried out by the COOH domain. This COOH domain binds to WD40 (tryptophan (W) and aspartic acid (D)) of SREBP cleavage-activating protein (SCAP). In the ER, insulin-induced gene (INSIG), SCAP, and SREBP2 form a complex. Under low sterol levels, INSIG dissociates, allowing the SCAP/SREBP2 complex to be transported to COPII-coated vesicles from the ER to the Golgi apparatus. In the Golgi, site-1 and site-2 proteases (S1P and S2P) cleave SREBP2, releasing the active NH2-terminal fragment (nSREBP2). Following its activation, the processed fragment translocates to the nucleus and engages sterol regulatory elements (SREs) to enhance cholesterol biosynthetic activity. Concurrently, oncogenic networks, including RAS, RhoA, and YAP/TAZ signaling, facilitate malignant progression, as depicted in Figure 4.
SREBP2 expression is frequently altered in diverse tumors, notably in cancers of the breast, prostate, pancreas, and liver. The promotion of prostate cancer metastasis is associated with an aberrant lipogenic program dependent on SREBP, mainly when promyelocytic leukemia and the phosphatase and tensin homolog gene are simultaneously inactivated. This interplay helps in elucidating mechanisms for prostate cancer progression, possessing insights into potential targets for managing this cancer [73]. The significant correlation between elevated SREBP2 expression and unfavorable clinical outcomes in prostate cancer highlights the potential significance of SREBP2 as a prognostic indicator. Accumulating research indicates that heightened SREBP2 expression observed in advanced prostate cancer implicates this transcription factor in the regulation of tumor growth and progression [74]. The involvement of SREBP in prostate cancer progression is largely attributed to their ability to activate transcriptional programs governed by c-Myc and Akt [74].
Despite the clinical success of lapatinib and trastuzumab as HER2-targeted treatments, resistance, whether intrinsic or acquired, emerges in a significant subset of HER2-positive breast cancers [75,76,77,78]. Efforts to overcome this resistance have centered on modulating the mevalonate pathway, aiming to inhibit mechanistic target of rapamycin complex 1 (mTORC1) and SREBP2-mediated stimulation of the YAP/TAZ (Yes-associated protein/WW domain-containing transcription regulator 1) axis [79]. Zhang and co-workers [48] elucidated a novel mechanism in which the interaction between SREBP2 and transcription factor CP2 (TFCP2) activates HMGCR expression by inhibiting TFCP2-mediated senescence in prostate cancer cells [48]. ABCA9, another cholesterol transporter, has been found to inhibit the translocation of ER to the nucleus via Phosphatidylinositol 3-kinase (PI3K)-Akt-Forkhead Box O1 (FOXO1) pathway in breast cancer cells. ABCA9 operates as a cholesterol transporter in the ER, where it restrains sterol biosynthesis by attenuating SREBP2 activity. Reestablishing ABCA9 function markedly impedes the growth of breast cancer cells [49]. Pharmacological or genetic inhibition of eukaryotic elongation factor 2 kinase (EEF2K) hampers cholesterol biosynthesis by attenuating SREBP2-mediated signaling, and effect that can be mitigated by external cholesterol supply. This highlights the potential importance of targeting EEF2K to modulate cholesterol metabolism in cancer. Selective inhibition of EEF2K reduces tumor formation by downregulating SREBP2-mediated transcription of cholesterol-biosynthetic genes [50]. Attachment of X-box binding protein 1 (XBP1) to SREBP2 enhances 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) activity, fostering cancer progression. Hence, simultaneous inhibition of SREBP2 and its co-regulatory elements may provide a valuable therapeutic avenue against cholesterol-driven tumorigenesis [80].
Similarly, the drug artesunate (used for the treatment of malaria) is shown to inhibit the localization of SREBP2 by downregulating isopentenyl pyrophosphate (IPP) and glutathione peroxidase 4 (GPX4) in myeloma cells [81]. γ-Tocotrienol is another drug that decreases the level of triacylglycerol and cholesterol in prostate cells and rat hepatocytes [82,83]. These results emphasize that focusing on SREBP-2 and the mevalonate pathways can become a promising approach in the field of cancer therapy. Various SREBP1 inhibitors are displayed in Figure 5.
4.2. Niemann–Pick Type C1 as a Potential Target
The lysosomal membrane protein NPC1 governs the efflux of cholesterol and sphingolipids from the lysosomal lumen to the cytoplasm [84,85]. Through this trafficking function, NPC1 is essential for maintaining cholesterol equilibrium and preventing intracellular lipid overload. The NPC1 gene mutation causes various genetic disorders and also leads to the formation of cancer [86].
When cholesterol binds to late endosomes/lysosomes (LE/Lys), NPC2 transfers cholesterol to NPC1. Engagement of cholesterol with the sterol-sensing domain of NPC1 initiates mTOR signaling [87]. Activated SREBP, in turn, upregulates ABCA1 to enhance cholesterol import through LDL [88]. The exported lysosomal cholesterol is subsequently trafficked to the Golgi apparatus for distribution.
In response to reduced cellular cholesterol, steroidogenic acute regulatory-related lipid transfer domain-3 (STARD3) acts as a molecular shuttle, directing cholesterol trafficking from the ER to the lysosomal–endosome system [89], leading to activation of Ras-related protein Rab-7 (RAB7)/guanosine triphosphatase (GTPase) by forming the RAB7/TBC1D15 complex [90,91], which ultimately promotes metastasis.
The researchers identified myeloid zinc Finger 1 (MZF1) as a controller of NPC1 expression induced by p95ErbB2, suggesting that ErbB2 signaling activation and NPC1 are essential in the cholesterol metabolism of cancer cells [92]. The link between MZF1 and NPC1 was reinforced through quantitative imaging-based cytometry (QIBC) analysis, revealing a notable decrease in cytosolic NPC1 levels in MZF1-knockdown cells or cells depleted of MZF1. According to Gene Ontology Biological Process (GOBP) enrichment, p95ErbB2-expressing cells display elevated expression of genes involved in cholesterol efflux, suggesting altered cellular cholesterol handling and homeostasis, potentially linked to changes in cholesterol transport [92]. The consequential hyper-activation of mTORC1 upon cholesterol accumulation due to NPC1 inhibition suggests a complex regulatory relationship that warrants further investigation into the underlying molecular mechanisms and potential implications for cellular functions [93]. In the presence of NPC1/2, lysosome-associated membrane protein (LAMP) serves as a crucial membrane component in lysosomes, which causes exocytosis, chaperone-mediated autophagy, or mTORC1 signaling [94]. Long and colleagues elucidated the binding site and mechanism by which itraconazole inhibits NPC1’s cholesterol-transport function [51]. They found, using cryo-electron microscopy (cryo-EM), that the 3D structure of NPC1 bound to itraconazole revealed the drug occupying the luminal cavity adjacent to the sterol-sensing domain (SSD), thereby blocking cholesterol transport; this was further confirmed by functional assays. The inhibitory mechanism closely resembles that of NPC1 blockade observed in the Hedgehog signaling pathway [51]. U18666A (Figure 5) is another small-molecule NPC1 inhibitor that suppresses the proliferation of triple-negative breast cancer cells and shows a synergistic effect with paclitaxel [86]. Cepharanthine and astemizole are additional NPC1 inhibitors that further disrupt cholesterol trafficking [95]. Figure 6 illustrates the crosstalk between cholesterol metabolism and cancer driven by NPC1 activity.
4.3. Sterol-Acyltransferase 1 in Cholesterol Metabolism and Cancer
The enzyme SOAT1 plays a pivotal role in regulating cholesterol metabolism [96] through its function in cholesterol esterification, a fundamental step for the proper storage and intracellular distribution of cholesterol [97]. It is located in the ER and in tissues such as the liver and adipose. Alterations in SOAT1 induce a negative impact on cholesterol levels and the initiation of many physiological processes, such as gene mutation [98]. Free cholesterol in the ER controls SREBP2. When ER cholesterol is high (enhanced when SOAT1 is inhibited), INSIG binds the SCAP-SREBP2 complex to form an ER-retained INSIG-SCAP-SREBP2 complex, preventing SCAP from escorting SREBP2 to the Golgi and thus blocking SREBP activation. When the concentration of ER cholesterol falls, INSIG dissociates and SCAP ferries SREBP2 to the Golgi for proteolytic activation (Figure 7) [99]. Additionally, it activates the SRF, RAS, and YD/TAZ proteins, which promote cancer progression. There are several SOAT inhibitors that decrease the cholesterol accumulation. PD-132301 is one of the SOAT1 inhibitors that reduces the accumulation of cholesterol ester with a half-maximal inhibitory concentration (IC50) of 37 nM. In vivo studies showed that the introduction of PPPA derivatives to Apoe^−^/^−^ mice resulted in a significant reduction of total plasma cholesterol concentration of 57.9 ± 9.3% compared to the control group [100]. Nevanimibe hydrochloride is another SOAT1 inhibitor, which is given orally (1.6–158.5 mg/kg/day) with limited efficacy at a specific target [101]. Figure 8 shows the chemical structure of SOAT1 inhibitors. SOAT1 plays a crucial role in the mevalonate pathway, exerting negative feedback on unesterified cholesterol through p53 mutations in pancreatic cancer [96]. Xu and colleagues [102] reported that cholesterol activates the Wnt/PCP-YAP signaling pathway via SOAT1 in colon cancer. They also observed that cholesterol depletion lowers HMGCR levels, which in turn suppresses YAP activity, indicating YAP is dependent on cellular cholesterol levels.
4.4. PCSK9 as a Potential Target
The enzyme PCSK9 contributes to hypercholesterolemia by modulating the activity of key lipid-regulatory genes, including LDLR and ApoB [103]. The biosynthesis of PCSK9 yields a 74 kDa proenzyme comprising a signal peptide (residues 1–30), an N-terminal prodomain (31–152), a catalytic core (153–454), and a C-terminal cysteine-rich region (455–692). In the ER, PCSK9 undergoes autocatalytic cleavage; the cleaved prodomain remains tightly bound as an intramolecular chaperone/inhibitor, and the secreted protein is catalytically inactive. The extracellular form of PCSK9 serves as a ligand for the LDL receptor (LDLR) by binding its EGF-A domain through the catalytic site. The C-terminal region directs the PCSK9-LDLR complex to endolysosomal compartments for degradation, diminishing surface levels of LDLR and VLDLR. Downstream, activation of the PCSK9-LDLR signaling cascade promotes PI3K-Akt-β-catenin pathway activity, induces epithelial–mesenchymal transition (EMT), and accelerates colon tumor progression, accompanied by alterations in caspase-3, Annexin A11, and E-cadherin expression. Small molecules and peptides (e.g., pseurotin A, silibinin A, MK-0616, Pep2–8, and berberine) can disrupt PCSK9-LDLR binding and prevent receptor destruction (Figure 9) [104,105,106]. Preclinical investigations revealed that phytochemical eugenol inhibits LDL by blocking PCSK9 through LOX1 inhibition [107,108]. Pseurotin A is one of the inhibitors that bind with PCSK9 in the interface pocket necessary for interaction with LDLR [108]. In vitro, pseurotin A reduces PCSK9 and boosts LDLR in hormone-dependent breast cancer cells, suggesting its potential as a dual inhibitor for hormone-dependent breast malignancies [107]. Figure 8 shows the chemical structure of various PCSK9 inhibitors. Currently, evolocumab and alirocumab are two PCSK9 inhibitor antibodies used to decrease lipid levels. These inhibitors exhibit adequate bioavailability (85% and 72%, respectively). Wong and co-workers [109] identified PCSK9 as a crucial determinant of cholesterol homeostasis and colorectal tumor progression under adenomatous polyposis coli (APC) and Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations. In such tumors, PCSK9 downregulates cholesterol import while upregulating endogenous synthesis and elevating geranylgeranyl pyrophosphate (GGPP) levels, collectively enhancing KRAS/MEK/ERK signaling cascades. ERK1 and ERK2 were the most upregulated genes among the top 24 kinases. Various findings revealed that KRAS also activates p-MEK/p-ERK. MK-0616 is an orally available PCSK9 inhibitor that enhances the degradation of LDLR by binding with Toll-like receptors and other proteins [110,111]. It exerts its action by binding with the LDL-receptor and could act as a novel target in cancer. In a clinical trial with 380 treated participants, MK-0616 (6–30 mg) produced significant week-8 LDL-c reductions compared to placebo (−41.2%, −55.7%, −59.1%, −60.9%; all p < 0.001) with adverse event rates comparable to placebo (39.5–43.4% vs. 44.0%) [112].
5. Cholesterol Metabolism and Cancer Immunotherapy
Cholesterol metabolism has recently emerged as a critical regulator of antitumor immune responses, particularly in T cell-based immunotherapies. Emerging evidence indicates that cholesterol metabolism plays a crucial role in shaping antitumor immune responses and immunotherapy efficacy [113]. Beyond tumor-intrinsic effects, altered cholesterol homeostasis profoundly influences immune cell function within the tumor microenvironment. Recent studies have shown that excessive cholesterol accumulation in T cells can drive cellular exhaustion and impair cytotoxic activity, whereas controlled modulation of cholesterol flux enhances immune responsiveness. In this context, Su et al. [114] demonstrated that inhibition of cholesterol esterification through acetyl-CoA acetyltransferase-1 (ACAT1) knockdown markedly potentiates CD19-directed CAR-T cell therapy. ACAT1 inhibition increased free cholesterol levels in the plasma membrane, promoting T cell receptor clustering, improving immunological synapse formation, and enhancing downstream signaling. As a result, metabolically reprogrammed CAR-T cells exhibited increased proliferation, cytokine secretion (IFN-γ), degranulation, and superior tumor cell killing in lymphoma models [114].
Additional evidence further suggests that cholesterol metabolism interacts with immune checkpoint regulation, antigen presentation, and macrophage polarization. Dysregulated cholesterol synthesis and efflux pathways within the tumor microenvironment can suppress effector T cell activity while promoting immunosuppressive phenotypes such as exhausted T cells and tumor-associated macrophages. Targeting key nodes of cholesterol metabolism, such as ACAT1, SREBP signaling, or cholesterol transporters, has therefore emerged as a promising approach to strengthen antitumor immunity and enhance the therapeutic efficacy of immune checkpoint inhibitors and adoptive cell therapies. Collectively, these findings underscore cholesterol metabolic reprogramming as a novel and actionable axis for improving cancer immunotherapy outcomes [115,116,117,118].
6. Conclusions
Collectively, the interplay between cholesterol metabolism and cancer progression encompasses several key molecular pathways, including SREBP, NPC1, PCSK9, and SOAT1 (Figure 10). SREBP2 orchestrates lipid biosynthetic processes that foster tumor growth; SOAT1 mediates cholesterol esterification within tumor cells; NPC1 regulates intracellular cholesterol transport; and PCSK9 promotes LDL receptor degradation, resulting in elevated LDL-cholesterol levels. Perturbation of these regulatory axes contributes significantly to oncogenic advancement. Elucidating the mechanistic roles of these proteins not only enhances our understanding of tumor metabolism but also identifies cholesterol regulatory axes as compelling therapeutic targets. Accordingly, Table 2 briefly summarizes the therapeutic strategies for targeting cholesterol metabolism.
Accumulating clinical and experimental evidence underscores the pivotal role of altered cholesterol metabolism in oncogenesis. Deciphering the molecular interrelationships among cholesterol-regulating proteins offers a strong foundation for developing targeted metabolic interventions. In this context, drug repurposing represents a particularly attractive strategy, enabling the identification of anticancer applications for clinically approved lipid-lowering agents. Nevertheless, further in-depth studies are required to develop novel inhibitors, define new mechanisms of action, and clarify the role of cholesterol efflux-associated proteins in cancer initiation, progression, and therapeutic resistance.
Future Perspectives
Future progress in this field will depend on addressing several critical knowledge gaps. These include elucidating the context-dependent functions of cholesterol-regulating proteins across distinct cancer subtypes and tumor microenvironments; developing highly selective modulators of SREBP2, SOAT1, NPC1, and PCSK9 with favorable safety profiles; and exploring comprehensive treatment strategies that combine cholesterol-targeting approaches with immunotherapy or iron death inducers, in order to overcome the resistance mechanisms. In parallel, systematic drug repurposing methods and patient stratification strategies based on biomarkers are crucial for advancing the application of precision oncology. Ultimately, large-scale and well-designed clinical trials are needed to translate the mechanistic insights into effective, patient-specific therapies. Collectively, continuous interdisciplinary collaboration and innovation can establish cholesterol metabolism as a transformative treatment frontier in oncology, offering new opportunities for improving patient outcomes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Luo J. Yang H. Song B.L. Mechanisms and regulation of cholesterol homeostasis Nat. Rev. Mol. Cell Biol.20202122524510.1038/s 41580-019-0190-731848472 · doi ↗ · pubmed ↗
- 2Martin-Perez M. Urdiroz-Urricelqui U. Bigas C. Benitah S.A. The role of lipids in cancer progression and metastasis Cell Metab.2022341675169910.1016/j.cmet.2022.09.02336261043 · doi ↗ · pubmed ↗
- 3Riscal R. Skuli N. Simon M.C. Even Cancer Cells Watch Their Cholesterol!Mol. Cell 20197622023110.1016/j.molcel.2019.09.00831586545 PMC 7225778 · doi ↗ · pubmed ↗
- 4Ikonen E. Cellular cholesterol trafficking and compartmentalization Nat. Rev. Mol. Cell Biol.2008912513810.1038/nrm 233618216769 · doi ↗ · pubmed ↗
- 5Carson J.A.S. Lichtenstein A.H. Anderson C.A.M. Appel L.J. Kris-Etherton P.M. Meyer K.A. Petersen K. Polonsky T. Van Horn L. Dietary Cholesterol and Cardiovascular Risk: A Science Advisory From the American Heart Association Circulation 2020141 e 39e 5310.1161/CIR.000000000000074331838890 · doi ↗ · pubmed ↗
- 6Duran E.K. Aday A.W. Cook N.R. Buring J.E. Ridker P.M. Pradhan A.D. Triglyceride-Rich Lipoprotein Cholesterol, Small Dense LDL Cholesterol, and Incident Cardiovascular Disease J. Am. Coll. Cardiol.2020752122213510.1016/j.jacc.2020.02.05932354380 PMC 8064770 · doi ↗ · pubmed ↗
- 7Das P. Ingole N. Lipoproteins and Their Effects on the Cardiovascular System Cureus 202315 e 4886510.7759/cureus.4886538106760 PMC 10724412 · doi ↗ · pubmed ↗
- 8Groenen A.G. Halmos B. Tall A.R. Westerterp M. Cholesterol efflux pathways, inflammation, and atherosclerosis Crit. Rev. Biochem. Mol. Biol.20215642643910.1080/10409238.2021.192521734182846 PMC 9007272 · doi ↗ · pubmed ↗