Low-Grade Fibromyxoid Sarcoma and Related Subtypes: A Systematic Review and Pooled Analysis of 773 Cases
Gitte G. J. Krebbekx, Elisabeth A. Kleine, C. Dilara Savci-Heijink, Diederik T. Meijer, Donner, Robert Hemke, Floortje G. M. Verspoor

TL;DR
This study reviews 773 cases of low-grade fibromyxoid sarcoma, finding that complete surgical removal is most effective and that the cancer can recur or spread years later.
Contribution
The study provides the largest pooled analysis of LGFMS cases, emphasizing the importance of complete resection and long-term monitoring.
Findings
Complete (R0) surgical resection significantly improves recurrence-free survival compared to incomplete resection.
Nearly 40% of patients experienced either local recurrence or metastasis, highlighting the tumor's potential for late progression.
MUC4 positivity and FUS-CREB3L2 gene fusion are key diagnostic markers for LGFMS.
Abstract
Low-grade fibromyxoid sarcoma (LGFMS) is a rare soft-tissue sarcoma that often appears harmless under the microscope, yet can recur or metastasize many years after surgery. Because of its subtle histological appearance, diagnosis can be difficult and is often confirmed by the presence of MUC4 protein and FUS-CREB3L2 gene fusion. We systematically reviewed all published cases of LGFMS and included four additional patients treated at our institution. Among 773 patients, wide (R0) surgical resection offered the best local control, while chemotherapy and radiotherapy provided little benefit. Nearly one in five patients developed local recurrence or metastasis, sometimes after a long disease-free interval. These findings highlight the need for careful diagnosis, complete resection, and long-term radiological follow-up in all patients with LGFMS. Background: Low-grade fibromyxoid sarcoma…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
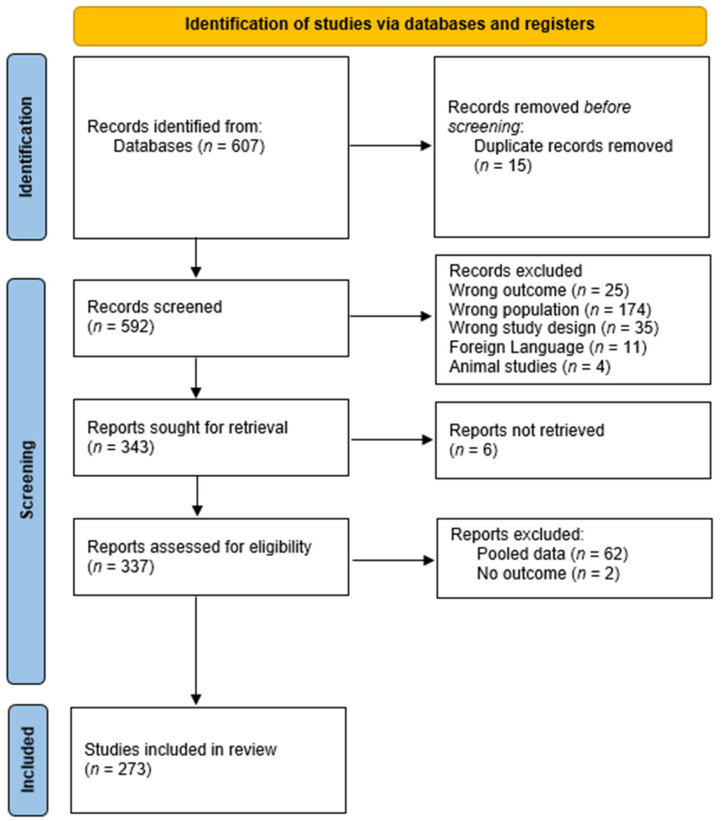 Figure 1
Figure 1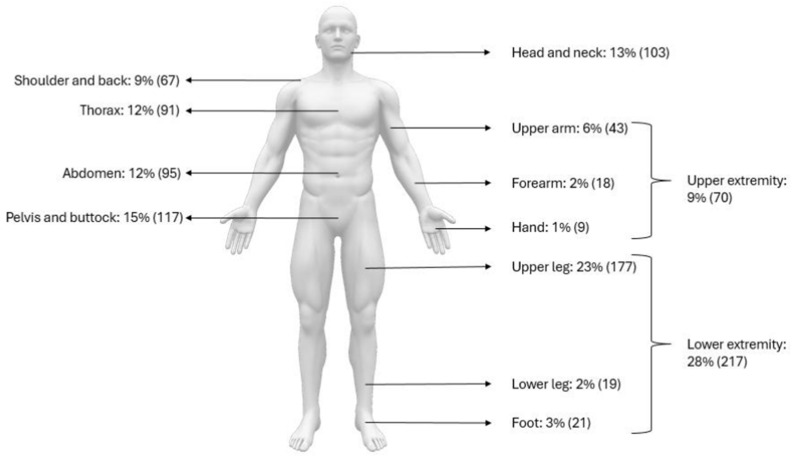 Figure 2
Figure 2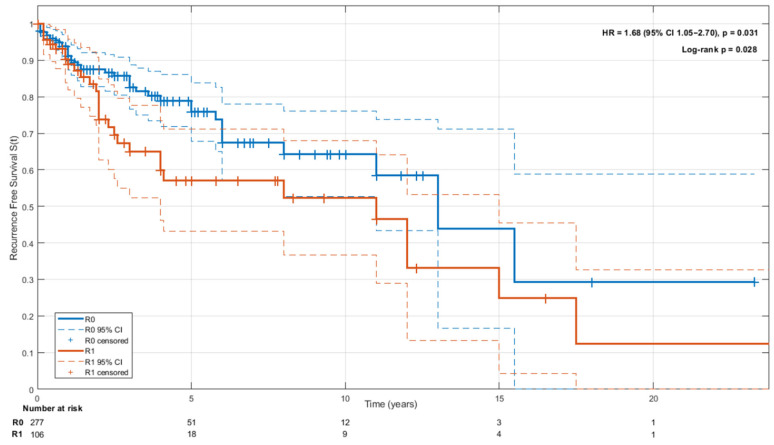 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSarcoma Diagnosis and Treatment · Urologic and reproductive health conditions · Gestational Trophoblastic Disease Studies
1. Introduction
Low-grade fibromyxoid sarcomas (LGFMSs) are rare, slow-growing, and typically painless malignant tumors that arise in the deep soft tissues, accounting for approximately 0.6% of all soft tissue sarcomas [1]. It occurs predominantly in adults between 20 and 50 years of age, with an estimated incidence of 0.2 per million individuals [2]. According to the World Health Organization (WHO, 2020) classification, LGFMS belongs to the group of malignant fibroblastic tumors [3]. The entity was first described by Evans in 1987, who reported two adolescent cases with deceptively benign-appearing lesions, low cellularity, minimal mitotic activity, yet pulmonary metastases [4]. Histologically, LGFMS consists of bland-appearing spindle cells arranged in whorled or swirling fascicles, often displaying alternating fibrous and myxoid areas. In 1997, Lane et al. introduced the hyalinizing spindle cell tumor with giant rosettes (HSCTGR), which shares similar morphology and clinical behavior [5]. The sclerosing epithelioid fibrosarcoma (SEF), described by Meis-Kindblom in 1995, represents a more aggressive variant with overlapping features, and since 2011, hybrid tumors combining LGFMS, HSCTGR, and SEF patterns have also been recognized [6].
Cytogenetic studies have become essential for distinguishing fibroblastic neoplasms [7]. The most frequent fusion genes involve FUS and EWSR1, both of which encode RNA-binding proteins, and CREB3L1/2, which encode DNA-binding proteins [8,9]. The FUS-CREB3L2 t(7;16) fusion is characteristic and occurs in the vast majority of cases [10], whereas FUS-CREB3L1 t(11;16) and EWSR1-CREB3L1 t(11;22) are less common variants [11]. These translocations generate chimeric transcription factors that are believed to drive tumorigenesis.
Histopathological diagnosis was historically limited to non-specific mesenchymal markers such as vimentin [1]. Since 2012, MUC4 has been identified as a highly sensitive and specific immunohistochemical marker, expressed in nearly all LGFMS [11,12]. MUC4 is a transmembrane glycoprotein involved in cell-signaling and differentiation, normally expressed in endodermal tissue [13]. Misdiagnosis due to sampling errors or limited biopsies may lead to inadvertent, incomplete resections, thereby increasing the risk of local recurrence and requiring re-excision, often with significant functional morbidity [1,14].
Magnetic resonance imaging (MRI) typically demonstrates a heterogeneous lesion with fibrous component showing low signal intensity on both T1- and T2-weighted sequences, and a myxoid component showing high T2 signal intensity and a gyriform pattern or laminated enhancement pattern after gadolinium administration [15,16].
The mainstay of treatment is wide surgical resection, given the risk of both local recurrence and distant metastasis [1]. Adjuvant radiotherapy, chemotherapy, and immunotherapy have shown minimal or no impact on tumor control or survival [2,17,18,19]. Long-term follow-up is therefore essential, although reported data remain limited, with only a minority of patients monitored beyond three years [1]. Despite its deceptively benign histologic appearance, LGFMS carries a significant potential for late recurrence and metastasis, making accurate diagnosis and adequate surgical management critical [1]. Current evidence is derived mainly from numerous case reports and small retrospective series, resulting in fragmented knowledge and a lack of standardized treatment recommendations.
A comprehensive synthesis of the available literature is thus required to better define the clinical spectrum, improve diagnostic accuracy, and inform evidence-based management. To address this gap, we conducted a systematic review and included a consecutive case series from our own institution. The primary objective was to evaluate current evidence regarding the clinical presentation, diagnostic features, treatment, and outcomes of LGFMS and its related subtypes. As a secondary objective, we compared outcomes between R0 and R1 resections to assess the prognostic relevance of surgical margin status.
2. Method
2.1. Study Design and Overview
This study comprised a systematic literature review including institutional cases. All methods followed the STROBE [20] and PRISMA [21] guidelines, with study quality assessed using the Joanna Briggs Institute (JBI) Checklist for Case Series [22].
The study was registered at Amsterdam UMC (ID: 120491) and PROSPERO (ID: CRD42022310606). The protocol was developed a priori to ensure methodological transparency and reproducibility. Ethical approval for the institutional component was covered under an existing Medical Ethics Review Committee registration (No. 2025.0109).
2.2. Search Strategy and Study Selection
A systematic search of PubMed/MEDLINE and Embase was performed up to September 2025 using the following search terms:
“fibromyxoid sarcoma” OR “LGFMS” OR “Evans tumor” OR “hyalinizing spindle cell tumor with giant rosettes” OR “HSCTGR”.
This search yielded 607 unique records.
Studies were eligible if they reported on LGFMS, HSCTGR, or SEF hybrid tumors and were published in English, French, German, Italian, or Spanish, without date restrictions. Exclusion criteria included review articles, series lacking individual patient data, and studies describing only diagnostic or therapeutic techniques without outcome data.
Two reviewers (GGJK and EAK) independently screened all titles, abstracts, and full texts using Rayyan (Qatar Computing Research Institute, Doha, Qatar) [23]. Discrepancies were resolved by consensus. Reasons for exclusion were documented at each stage.
2.3. Institutional Cases
In addition to the literature review, four consecutive patients with histologically confirmed LGFMS treated at Amsterdam UMC between 2018 and 2025 were included. Clinical, radiological, and histopathological data were extracted retrospectively from the institutional database. All patients provided informed consent for data use and publication. Institutional cases were identified from the Musculoskeletal Tumor (MUST) registry and are described in summary here; detailed case descriptions are provided in Supplementary Materials S1.
To preserve methodological consistency, these cases were analyzed together with the literature-derived cohort as part of the pooled dataset.
2.4. Quality Assessment
The methodological quality of all cohorts with ≥5 cases was independently assessed by two reviewers using the JBI Critical Appraisal Checklists for case series and observational studies [24,25]. Smaller case reports were not formally scored due to the inherent risk of bias and design heterogeneity. Each JBI was rated as “yes” (low risk of bias), “no” (high risk of bias), or “unclear/not applicable”. No automation or IA-based tools were used in this process. Any disagreement between reviewers was resolved by consensus discussion.
2.5. Data Extraction and Synthesis
The primary objective of this study was to evaluate the clinical presentation, diagnostic characteristics, treatment strategies, outcomes, and follow-up of LGFMS and related subtypes. The secondary objective was to compare recurrence-free survival (RFS) between patients who underwent R0 and R1 resections.
Data were manually extracted into a structured database. Extracted variables included: demographics (age, sex), tumor features (anatomical site, depth, size, symptoms, imaging characteristics), histopathology and immunohistochemistry features (MUC4 expression, cellularity, mitotic index), cytogenetics (FUS-CREB3L2, FUS-CREB3L1, and EWSR1-CREB3L1 fusions), treatment (initial diagnosis, inadvertent resections, resection margins, adjuvant therapy), outcomes (postoperative complications, recurrence, metastasis, and follow-up duration.
Tumor size was primarily extracted from pathology reports; if unavailable, measurements from imaging were used. Resection margins were standardized according to the AJCC 7th edition (R0–R2) [26]. Diagnoses were categorized as LGFMS, HSCTGR, SEF hybrid, or unclassified fibromyxoid sarcoma (UFMS). When relevant data were missing or aggregated, corresponding authors were contacted to obtain individual-level information.
2.6. Statistical Analysis
All statistical analyses were conducted using IBM SPSS Statistics, version 26 (IBM Corp., Armonk, NY, USA). Categorical variables (e.g., resection margin, histopathological subtype) were analyzed using Pearson’s Chi-square test, following verification of test assumptions. Continuous variables (e.g., age, tumor size) were compared using the Mann–Whitney U test or independent-samples t-test, depending on data distribution. Descriptive statistics (frequencies, medians, and interquartile ranges) summarized demographic, pathological, and treatment characteristics.
Recurrence-free survival (RFS) was analyzed using Kaplan–Meier survival curves, comparing R0 and R1 resections; statistical significance was defined as p < 0.05. Survival plots were generated in MATLAB R2023b (MathWorks, Natick, MA, USA).
3. Results
3.1. Study Selection and Characteristics
The systematic search identified 607 unique studies, of which 273 met the inclusion criteria and were included in the pooled analysis (Supplementary Materials S2) After screening and full-text review, 333 articles were excluded due to duplication, foreign language, wrong outcome, wrong population or study design, lack of individual patient data, or insufficient outcome information (Figure 1). In total, 773 individual patients were analyzed, including four additional institutional cases from Amsterdam UMC (Supplementary Materials S1). Most included studies were retrospective single-center case series or isolated case reports (244 small case series (n < 5) and 30 larger case series), with publication years ranging from 1987 to 2025. The median follow-up across all series was 3.0 years (IQR 1.0–6.1) (Table 1).
3.2. Critical Appraisal
Across all 11 JBI domains, an average of 88% of checklist items were rated as low risk of bias (“yes”), 9% as unclear (“?”), and 3% as high risk of bias (“no” = red). Nearly all studies (97%) clearly defined inclusion criteria, exposure variables, and outcome measures. Study populations were generally representative, with most cohorts derived from institutional or national sarcoma registries and based on pathologically confirmed diagnoses of LGFMS or related subtypes. Diagnostic confirmation using MUC4 immunopositivity and/or FUS-CREB3L2 fusion was reported in 93% of studies.
Outcomes were assessed using valid and reproducible criteria in approximately 90% of studies. Potential confounders were identified and addressed in 75%, while 25% provided insufficient information or lacked statistical adjustment. Adequate follow-up to detect recurrence or metastasis was reported in 85% of studies, and loss to follow-up was minimal or appropriately accounted for in 83%. Only three studies (7%) demonstrated incomplete follow-up reporting.
Overall, 26 of 30 cohorts (87%) fulfilled at least nine of eleven JBI criteria, indicating high methodological robustness across the included evidence base. Minor limitations mainly related to incomplete control for confounding or lack of detailed statistical analysis due to small sample sizes. Importantly, no studies were excluded based on the JBI assessment, as all met acceptable quality standards for pooled synthesis. Detailed JBI domain scoring is provided in Supplementary Materials S3.
3.3. Patient Demographics and Tumor Distribution
The median age at diagnosis was 35 years (IQR 21–49, range: neonatal [27] to 97 years [28]), with an equal sex distribution (50% females, n = 389). Tumors were predominantly deep-seated (80%, n = 475), and located in the lower extremity (43%, n = 334), most frequently in the thigh (23%, n = 177), followed by the upper extremity (18%, n = 138), pelvis/sacrum/buttock (15%, n = 117), thorax (12%, n = 91), and abdomen (12%, n = 95). Superficial lesions were uncommon (8%) and mainly affected the chest wall and abdominal wall. Reported tumor size (70%, n = 547) in maximum diameter was median 6.1 cm (IQR 3.8–10.0, range 0.7 cm–50 cm [28,29]). Detailed information and patient demographics and tumor distribution Table 1 and Figure 2.
3.4. Histopathology, Immunohistochemistry, and Cytogenetics
Histopathological evaluation in the included studies was performed on both fresh-frozen tissue and formalin-fixed, paraffin-embedded (FFPE) samples, frequently obtained from external institutions. Tumors typically demonstrated the classic biphasic architecture, composed of bland spindle cells arranged in whorled or swirling fascicles within alternating fibrous and myxoid zones, often with low cellularity, minimal atypia, and absent necrosis.
Immunohistochemical data were available for 428 patients (62%). The non-specific mesenchymal marker vimentin was positive in 198 cases (46%) and negative in one case [16] (0.5%), confirming its limited diagnostic specificity. MUC4, the key diagnostic marker for LGFMS, was analyzed in 428 cases and showed strong cytoplasmic positivity in 243 (57%), while only eight cases (3%) were negative. Additional immunostains, including β-catenin, CD34, calponin, caldesmon, desmin, EMA, ERG, keratin, p53, p63, S100, α-SMA, and SOX10, were inconsistently reported and largely negative across series. Ki-67 nuclear labeling indices were generally low (<5%), consistent with the indolent proliferative activity of LGFMS.
Cytogenetic testing was performed in 318 patients (41%), of whom 268 (84%) demonstrated a detectable gene rearrangement. The FUS gene was frequently altered, showing rearrangement or translocation in 263 cases (83%). The predominant fusion was FUS-CREB3L2 (47%, n = 148). A smaller subset showed EWSR1 rearrangements (8%, n = 24), with only one case confirming an EWSR1–CREB3L2 translocation [30]. In 31 cases, the FUS gene showed no rearrangement, and no other alternative fusions were analyzed.
Molecular diagnostics were primarily conducted by fluorescence in situ hybridization (FISH) to detect FUS gene rearrangements, while selected series used reverse transcriptase polymerase chain reaction (RT-PCR) or targeted next-generation sequencing (NGS). The Archer FusionPlex Sarcoma Panel was applied in several contemporary studies to identify FUS–CREB3L2 fusion transcripts among a panel of soft-tissue tumor–associated genes [31].
3.5. Clinical Presentation and Diagnostic Accuracy
Clinical symptoms were reported for 257 patients (33%). The majority presented with a slow-growing mass (27%, n = 69) or, less frequently, a rapidly enlarging lesion (9%, n = 24). Pain was absent in most cases (29%, n = 75), while 19% (n = 48) reported localized pain or discomfort. Median symptom duration before diagnosis was 1 year (IQR 0.5–4.0) (Table 1), although the longest reported symptom duration reached 34 years [32].
An initial diagnosis was documented in 203 patients (26%). Correct identification by imaging or biopsy was achieved in 54% of cases as LGFMS (n = 110) and in 38% as low-grade sarcomas (n = 77); the remaining cases were frequently misdiagnosed as benign lesions (47%, n = 95).
Misclassification occurred after biopsy (n = 37, including 26 FNA), imaging alone (n = 14), clinical assessment (n = 11), or frozen-section analysis (n = 2).
A total of 101 patients (32%) underwent an unplanned (“WHOOPS”) resection, defined as inadvertent excision without prior diagnosis or oncological planning. Of the 29 cases in which re-excision was reported, 25 (86%) achieved secondary R0 margins upon definitive surgery.
Following resection and histopathological review, 602 tumors (78%) were confirmed as LGFMS, 96 (12%) as HSCTGR, 44 (6%) as LGFMS/SEF hybrid, 10 (1%) as HSCTGR/SEF hybrid, 4 (<1%) as pure SEF variants, and 18 (2%) as unclassified fibromyxoid sarcomas (UFMSs).
3.6. Radiological Features
Imaging findings were reported for 273 patients, of whom 25 (14%) received a diagnosis based on imaging alone. Among these, 13 cases (52%) were misclassified as benign. The imaging modalities used included CT in 127 patients (47%), MRI in 109 (40%), ultrasound in 44 (16%), and FDG–PET/CT in 15 (6%).
Lesions typically appeared as well-defined, heterogeneous soft-tissue masses hypo- to isodense on CT, hypoechogenic on ultrasound. On MRI, tumors demonstrated mixed fibrous and myxoid signal patterns, with low T1 and high T2 signal intensity, and heterogeneous post-contrast enhancement. Occasional calcifications, hemorrhagic areas, or adjacent bone involvement were described. FDG–PET/CT findings were variable, showing low to moderate metabolic activity consistent with the tumor’s low-grade biology, although rare hypermetabolic cases were reported.
3.7. Treatment and Adjuvant Therapy
Surgical excision was the primary treatment modality in 98% (n = 760) of patients, 2% (n = 13) had unresectable or metastatic disease [2,30,31,33,34]. Resection margins were reported as R0 in 36% (n = 280), R1 in 15% (n = 113), R2 in 3% (n = 19), and unknown in 47% (n = 362) (Table 1). Intraoperative or postoperative complications were reported in 25 patients (3%). The severity of postoperative events varied from minor wound infections [35] to major systemic complications such as pulmonary embolism [36]. Some patients required emergency reoperations for procedure-related obstructions or vascular events, including bowel resection [37], ureteric stenting [38], nephrectomy [39], and above-knee amputation following thrombosis of the external iliac artery [40].
Adjuvant therapy was administered in 17% of patients (n = 128), predominantly radiotherapy 10% (n = 79) and chemotherapy (6%, n = 49). Reported agents included cyclophosphamide plus fecitabine [18], docetaxel [41], adriamycin [41], trabectedin [2], pazopanib [41,42], sunitinib [19] and anastrozole [41]. One patient experienced a coma secondary to Sunitinib therapy, which resolved after discontinuation of the drug [21].
3.8. Oncological Outcomes and Follow-Up
Follow-up status was available for 71% of patients (n = 545). At last follow-up, 422 (55%) had no evidence of disease (NED), 79 (10%) were alive with disease (AWD), 41 (5%) had died of disease (DOD), and 4 (0.5%) had died of other causes (DOOD) [43,44,45].
Thirty patients (4%) were lost to follow-up, while 198 (26%) had no follow-up information reported.
The overall median follow-up was 3.0 years (IQR 1.0–6.1). During follow-up, local recurrence occurred in 143 patients (19%), lung metastases in 62 (11%), and other distant metastases in 54 (10%). The median time to first recurrence was 3.1 (IQR 1.3–7) years, with the latest recurrence reported after 30 years [46]. The median time to local recurrence was 2.8 years (IQR 0.8–6.0), to lung metastasis 4.0 years (IQR 0.0–11.3), and to other metastases 2.3 years (IQR 0.0–9.3). Most patients (15%, n = 83) experienced a single recurrence, whereas 51 (9%) patients developed multiple recurrences, and 9 patients (2%) had ≥5 recurrences. Remarkedly, one patient had 17 recurrences [45] (Table 1). Local control and survival were not consistently improved by adjuvant therapy: no evidence of disease was observed in 5 patients (10%) treated with chemotherapy and in 21 (27%) of those treated with radiotherapy, compared with 442 patients overall (55%). Death of disease occurred in 4 patients (8%) after chemotherapy and in 5 (6%) after radiotherapy, compared with 41 patients (5%) overall. Local control and survival were better in superficial compared with deep tumors. No evidence of disease was observed in 258 patients (54%) with deep tumors and in 82 (67%) with superficial tumors, compared with 422 patients overall (55%). Death of disease occurred in 33 patients (7%) with a deep tumor and in 2 (2%) with a superficial tumor, compared with 41 patients (5%) overall.
The four institutional patients underwent wide or marginal resection without adjuvant treatment and remained free of disease recurrence or metastasis after a median follow-up of 3.1 years (range 1.1–5.1).
No significant differences were found between pathology-proven and unproven cases in survival, local recurrence, lung metastasis and distant metastases outcomes, allowing combined analysis of all R0 and R1 resection margin groups (Supplementary Materials S4).
The median follow-up time for R0 resections was 2.1 years (IQR 1.3–5.2) and for R1 resections was 2.9 (IQR 1.0–7.5) years. Overall survival was higher after R0 resection (96%, n = 226) than after R1 resection (89%, n = 69; p = 0.02). Local recurrence occurred less frequently following R0 resection (9%, n = 24) than R1 resection (24%, n = 27; p < 0.001). No significant differences were observed in the incidence of lung metastases (4% vs. 6%; p = 0.42) or other distant metastases (5% vs. 6%; p = 0.81). (Table 2)
The Kaplan–Meier survival analysis (Figure 3) demonstrated significantly improved recurrence-free survival R0 compared to R1 resections. Median RFS was longer in the R0 group, with a hazard ratio (HR) of 1.68 (95% CI 11.05–2.70, p = 0.03). The log-rank test confirmed a significant difference between the two groups (p = 0.03). The 5-, 10-, and 15-year recurrence-free survival (RFS) rates were 57% (95% CI, 43–67%), 52% (36–68%), and 25% (4–49%) for patients with R1 resections, and 78% (71–83%), 64% (53–76%), and 29% (0–59%) for those with R0 resections, respectively.
Follow-up recommendations varied substantially between studies. Suggested surveillance modalities included whole-body imaging [27], MRI [47,48,49], CT [50,51,52,53,54], chest imaging [55,56,57,58,59], or combinations of local and distant imaging techniques [40,60,61]. The recommended surveillance duration ranged from several decades [32,55] to lifelong follow-up [59,62]. Proposed imaging intervals varied from every 3 months to every 6 months or annually
4. Discussion
Low-grade fibromyxoid sarcomas (LGFMSs) and their related subtypes occur across various locations, predominantly in young adults. These deep-seated soft tissue tumors usually present as painless, slowly enlarging lesions, often reaching considerable size before detection. Approximately 90% of tumors were deep-seated, a proportion slightly higher than previously reported (83%). Superficial tumors appear to have a more favorable prognosis, likely due to earlier recognition, smaller size, and facilitated excision, as demonstrated by Billing et al. [63]. Histopathological confirmation is based on morphology in combination with MUC4 immunopositivity and characteristic FUS–CREB3L2 or, less frequently, EWSR1-CREB3L1 translocations.
4.1. Diagnostic Challenges and Biopsy Technique
Diagnostic delay is common, resulting from the tumor’s indolent course and deceptively benign morphology. Fine-needle aspiration (FNA) frequently produced non-diagnostic or misleading results, leading to inadvertent resections and repeat surgery. Core-needle or open biopsy provides a higher diagnostic yield and should be preferred to ensure accurate molecular and immunohistochemical analysis before resection. The most frequent false-negative benign diagnoses included desmoid-type fibromatosis, nodular fasciitis, and peripheral nerve-sheath tumors, while false-negative malignant diagnoses often included solitary fibrous tumor and malignant fibrous histiocytoma [28,64,65,66].
A wide spectrum of imaging modalities was used for diagnosis and follow-up, including ultrasound, radiography, CT, PET-CT, and MRI. Although MRI and CT best delineate fibromyxoid tumors [15], imaging findings remain non-specific, and diagnosis should never rely solely on radiologic assessment. The combination of tissue biopsy with molecular testing remains the diagnostic gold standard.
4.2. Histopathology and Cytogenetics
Consistent with prior literature [67], our review confirmed MUC4 positivity in 96% of tested cases and FUS–CREB3L2 rearrangement in 47%. These rates are slightly higher than earlier series, reflecting improvements in molecular testing over time. The cytogenetic profile in our cohort—with 90% FUS and 6% EWSR1 rearrangements—was slightly higher than reported in previous studies. The French Sarcoma Group [10] identified FUS–CREB translocations in 81% of 59 patients, including several non-LGFMS sarcomas (four pure SEF cases), while the smaller cohort of Matsuyama et al. [68] (n = 16) demonstrated 84% positivity for FUS–CREB3L2.
EWSR1 rearrangements were rare, typically confined to pure SEF (90%) [11] and, in a few additional cases, observed in LGFMS/SEF hybrid tumors [27,35,36,69,70] and LGFMS/SEF hybrid tumors [33,34,37,38,39]. Beyond these canonical fusions, several novel rearrangements have been described (e.g., KMT2A–YAP1, HEY1–NCOA2, YAP1–TFE3), though their biological relevance remains uncertain. Together, these data emphasize that LGFMS represents a molecular and morphological spectrum, rather than a single uniform entity.
Several non-specific immunohistochemical markers, including α-SMA, EMA, S100, BCL2, CD34, and vimentin, were variably expressed, reflecting overlap with other fibroblastic and spindle-cell sarcomas [10]. Tumors with giant rosettes occasionally showed additional positivity for CD68, CD57, and NSE [5,71,72]. Ki-67 indices were low (<10%), supporting the indolent nature of LGFMS.
Given this non-specific profile, MUC4 remains the diagnostic gold standard, with ~100% sensitivity and 70% specificity. In this review, 96% of tested cases were MUC4 positive, confirming its reliability as the most robust immunohistochemical discriminator for LGFMS and related subtypes.
Beyond the canonical FUS– and EWSR1–CREB fusions, a number of rare or novel gene rearrangements and somatic mutations (e.g., t(7;18;16) [73], t(7/10;16) [61], t(4;18) [74], KMT2A-YAP1 [75], KMT2D-PRRX1 [75], HEY1-NCOA2 [42], YAP1-TFE3 [76], MDM2 [77], DDIT3 [77], APC [78], SOX9 [79], TFE3 [79]) have been identified in recent years. Although their clinical significance is unclear, these emerging alterations may refine the molecular classification and prognostic stratification of LGFMS in the future.
4.3. Surgical Management and Adjuvant Therapy
Surgical resection remains the cornerstone of curative treatment. Our analysis demonstrated that R0 resections are associated with significantly improved recurrence-free survival and overall survival compared with R1 resections, whereas differences in metastatic risk were not significant. Adjuvant radiotherapy or chemotherapy was rarely administered and showed no proven survival benefit, consistent with prior reports.
The TRASTS phase I–II trial [80], evaluating trabectedin with radiotherapy for advanced soft-tissue sarcomas, reported promising local control but included no LGFMS cases. The potential role of combined (neo)adjuvant regimens therefore remains unproven and should be investigated in prospective, multicenter studies.
4.4. Oncological Outcomes and Prognosis
The median follow-up of 3 years in the present review likely underestimates late recurrences and metastases, which can occur decades after surgery, as reflected by the median times to first, local, lung, and other distant recurrence of 3.1, 2.8, 4.0, and 2.3 years, respectively. Local recurrence was observed in 19% of patients and metastasis in 21%, rates consistent with the systematic review by Tang et al. (2010) [1], who reported 29% local and 18% pulmonary recurrence. In contrast, the historic cohort of Evans et al. [45], with follow-up up to 70 years, documented recurrence in 64% and metastasis in 45% of patients, illustrating how extended surveillance dramatically alters perceived outcomes.
In our pooled analysis, 8% of patients died of disease, compared with 3% in Tang’s series and 42% in Evans’. These discrepancies likely reflect shorter observation periods, differences in diagnostic certainty, and earlier inclusion of pathologically unconfirmed cases in older studies.
Overall, the prognosis of LGFMS is generally favorable but unpredictable. Even patients with initially low-grade histology may develop late recurrences or metastases after prolonged disease-free intervals. Since malignant transformation is exceedingly rare and typically symptomatic, a patient-centered, symptom-driven follow-up strategy, with thorough patient education at discharge, is preferred over routine lifelong radiologic surveillance.
4.5. Study Limitations
This review is limited by the heterogeneity of included studies, retrospective designs, and variable data completeness—inevitable in rare diseases. Missing data, particularly on follow-up duration and imaging, may have introduced selection and reporting bias. Because of the small number of patients in key subgroups, we were unable to perform reliable comparative analyses for chemotherapy, radiotherapy, or superficial versus deep tumors. Publication bias is also likely, as case reports tend to highlight atypical or aggressive presentations. Efforts were made to minimize duplication by cross-checking large institutional cohorts, but overlapping cases cannot be entirely excluded.
The JBI critical appraisal confirmed overall high methodological quality but recurrent weaknesses in case selection, follow-up completeness, and confounder control.
Despite these limitations, this review represents the largest pooled cohort of LGFMS and subtypes to date, integrating clinical, histopathological, and molecular findings across more than 700 patients.
4.6. Clinical Implications
The present analysis underscores the importance of early multidisciplinary evaluation in specialized sarcoma centers, with attention to biopsy quality, margin control, and long-term surveillance. Diagnostic awareness among clinicians and radiologists can substantially reduce inadvertent resections and diagnostic delay.
Given the rarity of LGFMS, international collaboration is essential to establish standardized diagnostic criteria, develop evidence-based follow-up protocols, and explore the role of novel systemic or targeted therapies.
5. Conclusions
LGFMS and its subtypes are rare, indolent soft-tissue sarcomas primarily affecting young adults. Although overall survival is favorable, local and distant recurrences remain common and may occur decades after surgery. Wide (R0) resection provides the best local control, whereas (neo)adjuvant treatments lack proven efficacy.
Recommendations
Diagnosis should rely on histology and molecular confirmation obtained through core-needle or open biopsy.Primary treatment is surgery with the aim of achieving R0 margins.Adjuvant therapies may be considered only within clinical trial settings or for unresectable/metastatic disease.Follow-up is advised, initially biannually MRI for five years, and then annually MRI up to 10 years, followed by careful patient education at discharge.
Future prospective multicenter studies are required to determine optimal systemic treatment and define the true long-term prognosis of this rare sarcoma spectrum. Long-term effects of these cases are therefore recommended.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Tang Z. Zhou Z.-H. Lv C.-T. Qin L.-Y. Wang Y. Tian G. Luo X.-L. Zhu Q. Xu X.-G. Low-Grade Fibromyxoid Sarcoma: Clinical Study and Case Report J. Oral Maxillofac. Surg.20106887388410.1016/j.joms.2009.04.13620307771 · doi ↗ · pubmed ↗
- 2Maretty-Nielsen K. Baerentzen S. Keller J. Dyrop H.B. Safwat A. Low-Grade Fibromyxoid Sarcoma: Incidence, Treatment Strategy of Metastases, and Clinical Significance of the FUS Gene Sarcoma 2013201325628010.1155/2013/25628023818812 PMC 3683502 · doi ↗ · pubmed ↗
- 3WHO Classification of Tumours Editorial Board Chapter 1 Soft Tissue Tumours Soft Tissue and Bone Tumours 5th ed.IARC Lyon, France 2020112
- 4Evans H.L. Low-grade fibromyxoid sarcoma. A report of two metastasizing neoplasms having a deceptively benign appearance Am. J. Clin. Pathol.19878861561910.1093/ajcp/88.5.6153673943 · doi ↗ · pubmed ↗
- 5Lane K.L. Shannon R.J. Weiss S.W. Hyalinizing spindle cell tumor with giant rosettes: A distinctive tumor closely resembling low-grade fibromyxoid sarcoma Am. J. Surg. Pathol.1997211481148810.1097/00000478-199712000-000119414192 · doi ↗ · pubmed ↗
- 6Rekhi B. Deshmukh M. Jambhekar N.A. Low-grade fibromyxoid sarcoma: A clinicopathologic study of 18 cases, including histopathologic relationship with sclerosing epithelioid fibrosarcoma in a subset of cases Ann. Diagn. Pathol.20111530331110.1016/j.anndiagpath.2011.02.00521550274 · doi ↗ · pubmed ↗
- 7Storlazzi C.T. Mertens F. Nascimento A. Isaksson M. Wejde J. BrosjöO. Mandahl N. Panagopoulos I. Fusion of the FUS and BBF 2H 7 genes in low grade fibromyxoid sarcoma Hum. Mol. Genet.2003122349235810.1093/hmg/ddg 23712915480 · doi ↗ · pubmed ↗
- 8National Center for Biotechnology Information, U.S. National Library of Medicine Available online: http://www.ncbi.nlm.nih.gov(accessed on 1 June 2018)