Reduced LOXL3 Expression Disrupts Microtubule Acetylation and Drives TP53-Dependent Cell Fate in Glioblastoma
Talita de Sousa Laurentino, Roseli da Silva Soares, Antônio Marcondes Lerario, Ricardo Cesar Cintra, Suely Kazue Nagahashi Marie, Sueli Mieko Oba-Shinjo

TL;DR
Reducing LOXL3 disrupts microtubules and causes cell death in glioblastoma cells with normal TP53, but leads to cell aging in those with mutated TP53.
Contribution
LOXL3 is identified as a key regulator of microtubule stability and cell fate in glioblastoma, with outcomes dependent on TP53 status.
Findings
Reduced LOXL3 expression lowers α-tubulin acetylation and disrupts cell cycle progression.
TP53 status determines whether LOXL3 knockdown causes apoptosis or senescence.
LOXL3 reduction impairs adhesion and migration in TP53 wild-type glioblastoma cells.
Abstract
Glioblastoma (GBM) is the most aggressive primary brain tumor, marked by molecular heterogeneity and poor clinical prognosis. Lysyl oxidase-like 3 (LOXL3) is frequently upregulated in GBM, but its mechanistic contribution remains insufficiently defined. Here, we investigated the functional role of LOXL3 in GBM using CRISPR-Cas9-mediated LOXL3 knockdown in two genetically distinct GBM cell lines: U87MG (wild-type TP53) and U251 (mutant TP53). Reduced LOXL3 expression markedly reduced α-tubulin acetylation, particularly in U87MG cells, and downregulated genes involved in cell cycle progression and proliferation. Both cell lines exhibited mitotic defects, including delayed cell cycle progression and spindle abnormalities; however, cell fate diverged according to TP53 status. U87MG cells, sustained spindle checkpoint activation triggered a p53-dependent spindle checkpoint response…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
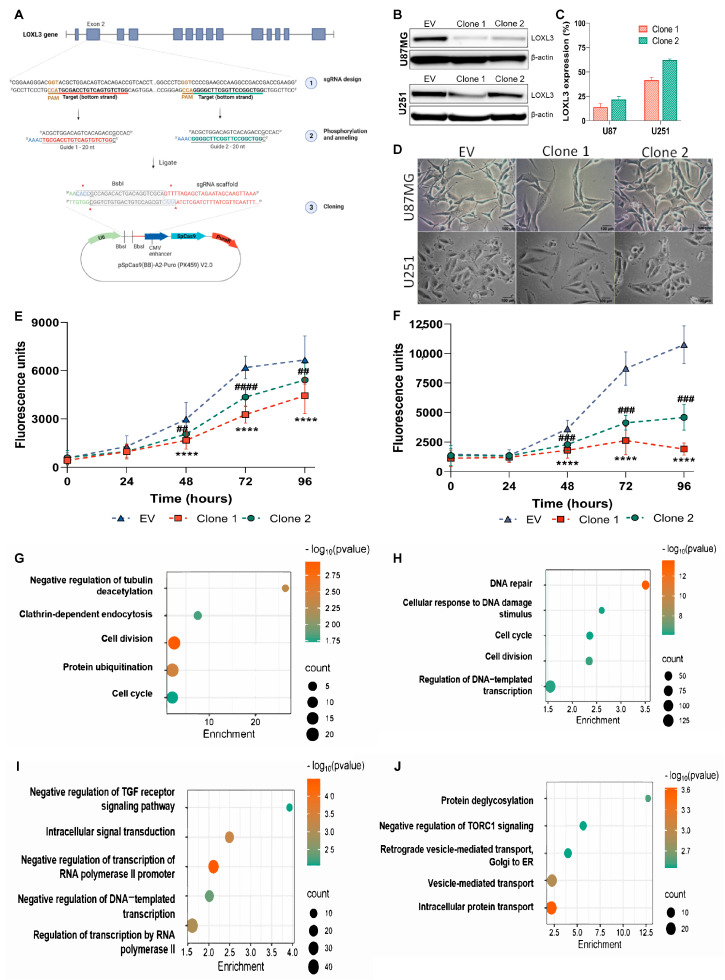 Figure 1
Figure 1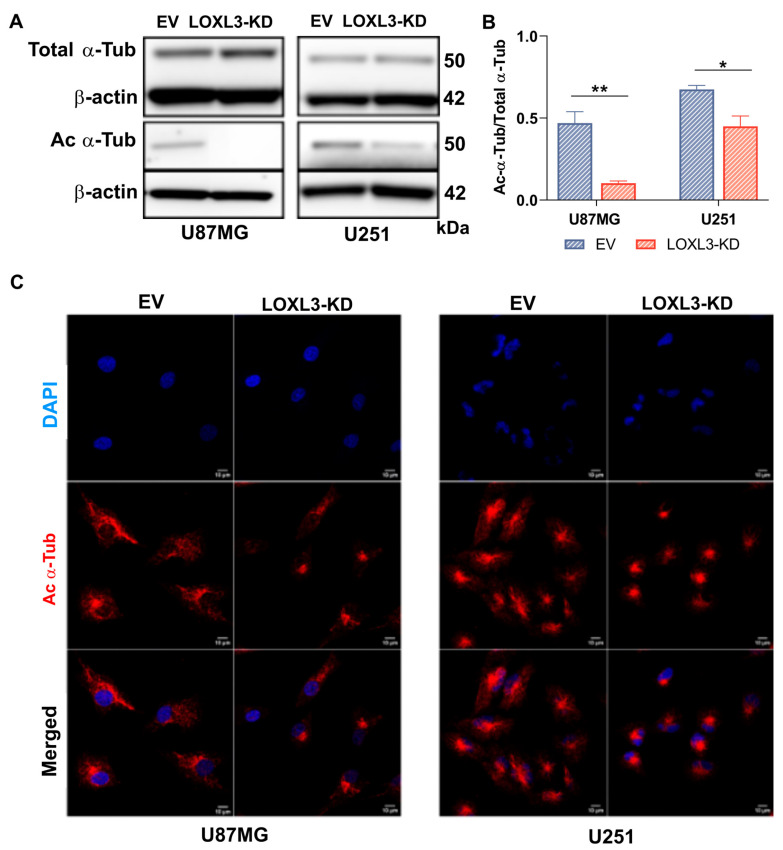 Figure 2
Figure 2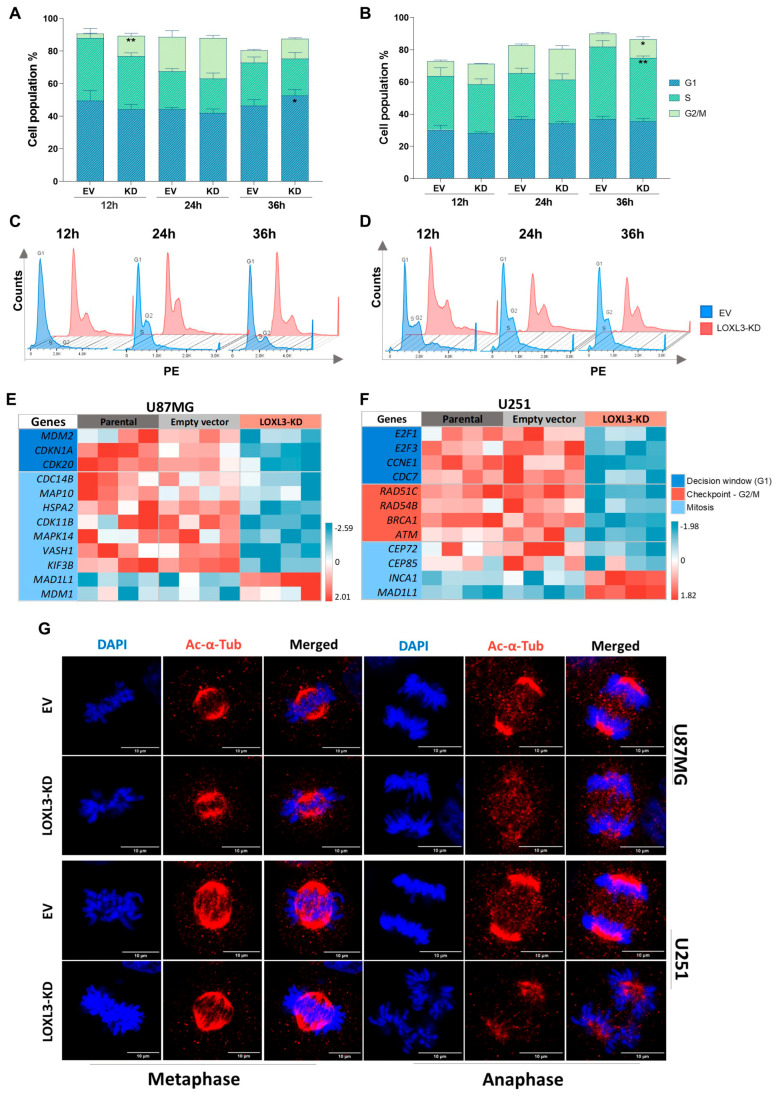 Figure 3
Figure 3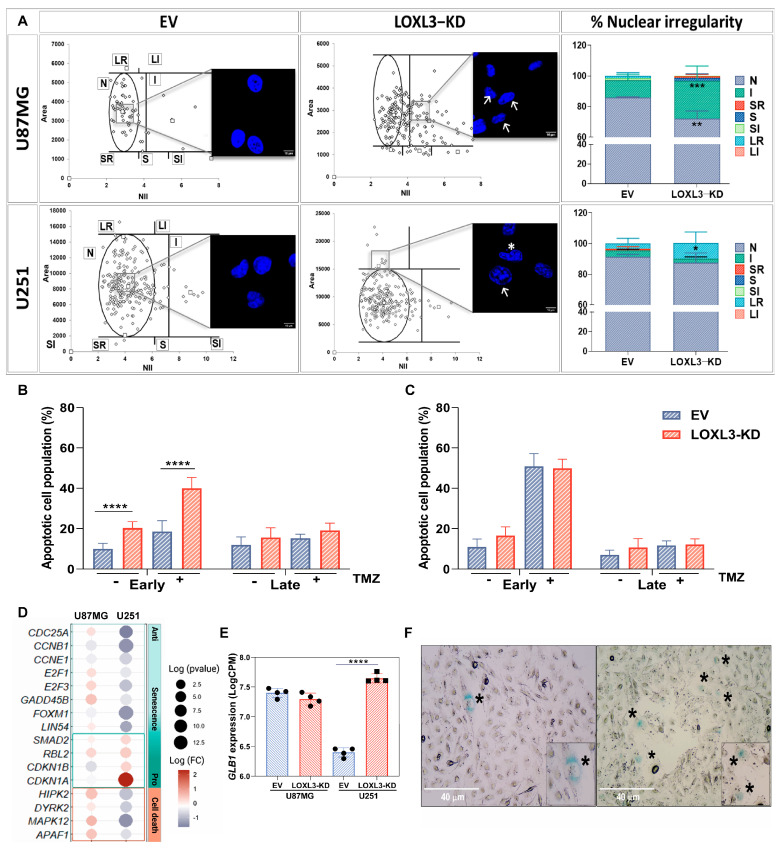 Figure 4
Figure 4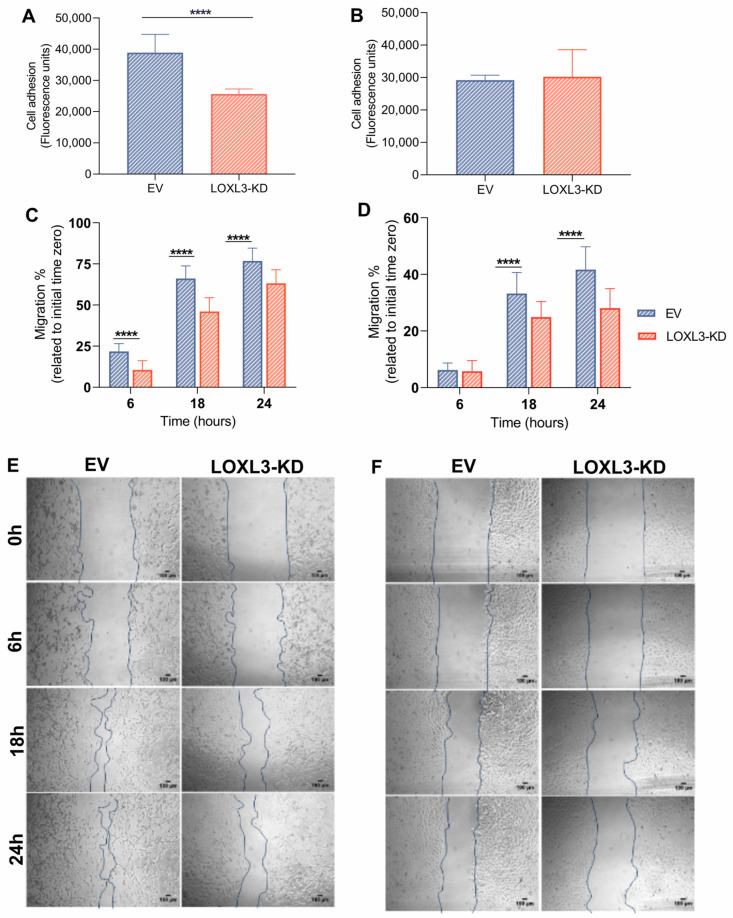 Figure 5
Figure 5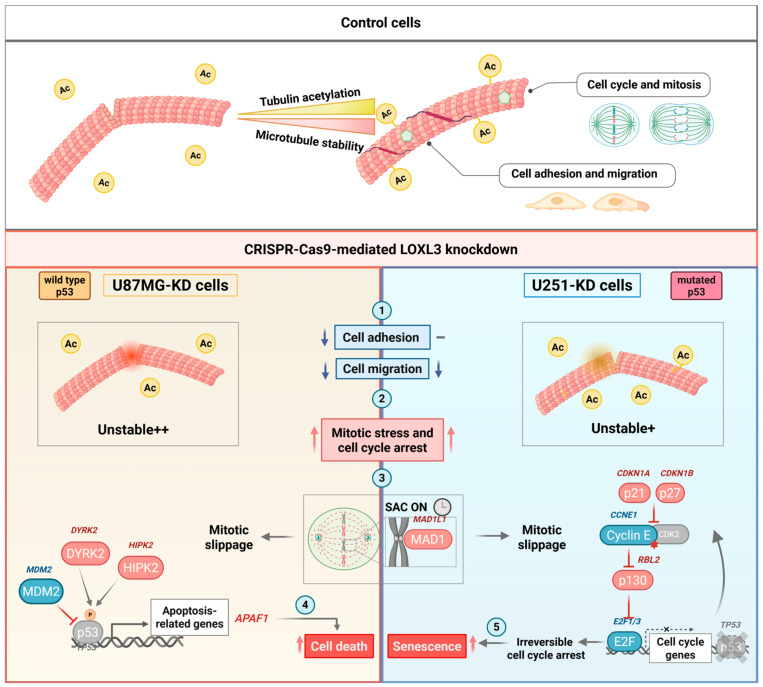 Figure 6
Figure 6- —Conselho Nacional de Pesquisa
- —Sao Paulo Research Foundation
- —Faculdade de Medicina da USP (FMUSP)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial metabolism and enzyme function · Neutrophil, Myeloperoxidase and Oxidative Mechanisms · Atherosclerosis and Cardiovascular Diseases
1. Introduction
Glioblastoma (GBM) is the most common malignant primary tumor of the central nervous system (CNS) in adults, accounting for more than half of high-grade gliomas characterized by a median survival of only 12–15 months, despite multimodal therapy [1]. The intratumoral heterogeneity of GBM is driven by recurrent alterations in major oncogenic pathways, including p53, RB, and PI3K signaling, contributing to therapeutic resistance and limited clinical progress [2].
Modern molecular classification has refined these definitions, with the World Health Organization (WHO) 2021 criteria not strictly defining GBM as an isocitrate dehydrogenase (IDH)-wildtype entity [3]. Importantly, this diagnosis is increasingly driven by molecular signatures rather than histology alone. Diffuse astrocytic tumors that appear histologically lower-grade are now classified as GBM, CNS WHO grade 4 if they harbor TERT promoter mutations, EGFR amplification, or +7/−10 chromosome copy number changes [3]. TP53 mutations are foundational to the IDH-mutant astrocytoma lineage, occurring in >90% of cases [4], while the frequency is only 25–30% of IDH-wildtype GBM [5], further underscoring the biological complexity that shapes GBM behavior and clinical outcomes.
Lysyl oxidase-like 3 (LOXL3), a copper-dependent amine oxidase of the lysyl oxidase family, is best known for catalyzing collagen and elastin cross-linking, contributing to extracellular matrix (ECM) stiffness and structural stabilization [6]. Beyond its canonical enzymatic function, LOXL3 participates in intracellular regulatory processes, such as STAT3 modulation through lysine deacetylation [7] and contributions to tumor development, progression, and therapy resistance across multiple malignancies [8,9,10]. In gliomas, LOXL3 expression increases with tumor grade and is higher in low-grade gliomas harboring wild-type IDH, a feature associated with poor prognosis [11]. In GBM, LOXL3 upregulation is associated with poor outcomes, showing a strong positive correlation with genes governing cytoskeletal organization, including tubulin-encoding transcripts, suggesting a functional relationship with microtubule (MT) dynamics [12].
MTs are the main structural component of the eukaryotic cytoskeleton and undergo continuous cycles of polymerization and depolymerization, a process termed dynamic instability. These processes are regulated by MT-associated proteins (MAPs) that bind to and stabilize the MT lattice [13]. These interactions are modulated by post-translational modifications of tubulin [14], with acetylation being particularly significant. Acetylation of α-tubulin, primarily catalyzed α-tubulin N-acetyltransferase 1 (α-TAT1), enhances MT stability by reducing breakage susceptibility, prolonging MT lifespan, and preventing structural damage [15,16]. Conversely, deacetylation is mediated by histone deacetylases HDAC5/6 and sirtuin 2 [17]. MT acetylation supports processes essential to cell survival, including mitotic spindle architecture, intracellular transport, and regulation of cell morphology, adhesion, and motility [13,16].
These observations raise the possibility that LOXL3 promotes GBM progression through the regulation of MT integrity. However, the mechanistic contribution of LOXL3 to cytoskeletal dynamics and cell fate in GBM remains largely undefined. Therefore, we investigated the functional role of LOXL3 in two genetically distinct GBM cell lines, U87MG (wild-type TP53) and U251 (mutant TP53), focusing on MT acetylation, cell cycle progression, ant tumor cell behavior. Our findings reveal that reduced LOXL3 expression disrupts MT homeostasis and proliferation, while TP53 status determines whether cells undergo apoptosis or senescence. These results identify LOXL3 as a key regulator of GBM cell aggressiveness and support its potential as a molecular target for genotype-informed therapeutic strategies.
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
Human GBM cell lines U87MG and U251 were obtained from American Type Culture Collection and authenticated by short tandem repeat (STR) profiling using GenePrint 10 System (Promega, Fitchburg, WI, USA). U87MG cells harbor wild-type TP53, whereas U251 cells carry TP53 mutations [18]. This classification follows the WHO’s 2016 system [19]. Cells were cultured in DMEM supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin (Thermo Fisher Scientific, Waltham, MA, USA), and 10% heat-inactivated fetal bovine serum (FBS; Cultilab, Campinas, SP, Brazil), at 37 °C in a 5% CO_2_. Mycoplasma contamination was routinely tested.
2.2. CRISPR-Cas9-Mediated LOXL3 Reduction
Two single-guide (sgRNAs) targeting exon 2 of LOXL3 (NM_032603.4) were designed using an online platform [20]. sgRNA sequences targeting the coding exon 2 (Figure S1A) were cloned into the pSpCas9(BB)-2A-Puro V2.0 (PX459; Addgene, Cambridge, MA, USA) as described previously [21]. U87MG and U251 cells were transfected with 2 μg of plasmids (including empty vector [EV] controls), using FuGENE (Promega), and selected with puromycin (2.5 and 5 μg/mL, for U87MG and U251 cells, respectively, 72 h). Single-cell clones were isolated by limiting dilution.
2.3. DNA and RNA Extraction
Genomic DNA and total RNA were extracted using the AllPrep DNA/RNA Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. Concentration and purity were determined by spectrophotometry. Samples with A260/A280 ratio greater than 1.8 were considered to have satisfactory purity.
2.4. Clone Validation
PCR amplification of the CRISPR-Cas9 LOXL3 target region was performed with 100 ng of DNA in GoTaq Green Master Mix buffer containing 3 mM MgCl_2_, 1 U of GoTaq DNA polymerase (Promega), 10 µM primers, 2.5 µM dNTPs in a final volume of 25 µL. Amplicons were visualized on 2% agarose gel electrophoresis. Primer sequences are provided in Figure S1A. PCR products were subcloned into the pGEM-T Easy Vector (Promega), transformed into competent bacteria. PCR products were purified using Agencourt AMPure XP beads (Beckman Coulter Biosciences, Indianapolis, IN, USA) and sequenced using BigDye Terminator v3.1 Kit on an ABI 3500 Genetic Analyzer (Thermo Fisher Scientific). Sequences were aligned to the LOXL3 reference from GenBank.
2.5. Western Blotting
Cells were lysed in RIPA buffer (50 mM Tris-HCl 454, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA) supplemented with protease inhibitors (Sigma–Aldrich, St. Louis, MO, USA). Protein concentration was determined by the Bradford assay (Thermo Fisher Scientific). Equal amounts of protein (20 μg) were separated on 4–12% SDS-PAGE gels (NuPAGE, Thermo Fisher Scientific) and transferred to PVDF membranes (iBLOT, Thermo Fisher Scientific). Membranes were blocked with 5% non-fat milk and incubated overnight at 4 °C with primary Abs against LOXL3 (1:1000, rabbit; Aviva, San Diego, CA, USA), acetylated α-tubulin (1:1000, mouse; Sigma–Aldrich), α-tubulin (1:8000, mouse; Sigma–Aldrich), and β-actin (mouse, Sigma–Aldrich; 1:20,000). After washing, HRP-conjugated secondary Abs (1:1000; Sigma–Aldrich) were applied for 1 h at room temperature. Signal was detected using the Clarity Western ECL substrate (Bio-Rad, Hercules, CA, USA) and imaged on ImageQuant LAS4000 (GE Healthcare, Pittsburgh, PA, USA). Densitometry was performed using ImageJ v1.53 (National Institutes of Health, Bethesda, MD, USA). Two independent biological replicates were analyzed.
2.6. Immunofluorescence Microscopy
Cells grown on glass coverslips were fixed with 4% paraformaldehyde (15 min), permeabilized with 0.1% Triton X-100 (10 min), and blocked with 4% goat serum (30 min). Overnight incubation at 4 °C with anti-acetylated α-tubulin Ab (1:200; Sigma–Aldrich) was followed by Alexa Fluor 568-conjugated secondary Ab (1:400; Thermo Fisher Scientific). Nuclei were counterstained with DAPI (Thermo Fisher Scientific). Images were acquired by confocal microscopy (Zeiss 510 LSM META and 780-NLO; Carl Zeiss Microscopy, Thornwood, NY, USA) and analyzed with ImageJ v1.53.
2.7. RNA Sequencing and Bioinformatics Analysis
RNA-seq libraries were generated from quadruplicates of parental, control EV, and LOXL3-KD cells using QuantSeq 3′mRNA-Seq Library Prep Kit-FWD for Illumina (Lexogen, Vienna, Austria). Library size distribution was assessed with TapeStation 4200 (Agilent Technologies, Santa Clara, CA, USA). Pooled DNA libraries were sequenced on a NextSeq 500 (Illumina, San Diego, CA, USA) to a mean depth of 5 million single-end 75-bp reads per sample at SELA Facility Core, School of Medicine, University of Sao Paulo. Data (GEO accession number GSE288138) were processed using FASTQC for quality control [22], STAR for alignment to hg38 [23], and featureCounts for read quantification [24]. Counts per million (CPM) were normalized using edgeR v2 [25]. Differential expression was assessed using limma v3.50.3 following log_2_ CPM transformation [26]. Differentially expressed genes (DEGs; genes differentially expressed in LOXL3-KD cells relative to controls) (|log_2_FC| ≥ 0.5; adj. p ≤ 0.05) were analyzed using the Database for Annotation, Visualization and Integrated Discovery (DAVID, 2021 update) and the Gene Ontology enrichment (GO, biological process). Heatmaps were generated from z-score-transformed log_2_ CPM values. See Tables S1 and S2 for DEFs and enrichment data.
2.8. Cell Viability Assay
Cells (1 × 10^3^/well) were seeded in 96-well plates. Cell viability was assessed daily for 4 days using PrestoBlue Cell Viability Reagent (Thermo Fisher Scientific), with fluorescence recorded at 540/560 nm (GloMax-96 Microplate Reader, Promega). Background fluorescence from medium-only wells was subtracted. Each condition was tested in octuplicate in two independent experiments.
2.9. Cell Cycle Analysis
Cells (5 × 10^3^/well) were serum-starved overnight to synchronize cycling and re-stimulated with complete medium. At 12, 24, and 36 h, cells were fixed with 70% ethanol, washed, and stored at 4 °C. Samples were treated with RNase A (30 µg/mL; Sigma-Aldrich) and stained with propidium iodide (PI; 50 µg/mL). DNA content was analyzed by flow cytometry (FACS Canto II; BD Biosciences, San Jose, CA, USA). Cell cycle profiles were evaluated in triplicate from two independent experiments using FlowJo v10.
2.10. Nuclear Morphometric Analysis (NMA)
Cells were fixed and stained with DAPI, and nuclei were imaged using EVOS M5000 fluorescence microscopy (Thermo Fisher Scientific). Nuclear area, perimeter, and irregularity index were quantified using ImageJ v1.53. Nuclei were classified as normal, large, irregular, and small, with subdivisions into regular and irregular groups, as previously described [27]. Control EV (n = 64, n = 108) and LOXL3-KD (n = 198, n = 233) from U87MG and U251 cells were analyzed across two independent experiments.
2.11. Cell Adhesion Assay
Cells (5 × 10^4^/well) were seeded into 96-well plates and allowed to adhere for 3 h. Non-adherent cells were removed by three PBS washes. Attached cells were quantified using the PrestoBlue assay. Experiments were performed in octuplicate across two independent replicates.
2.12. Cell Migration Assay
Migration was evaluated using a wound-healing assay. Cells (8 × 10^4^/well) were seeded in 48-well plates and grown to 70–80% confluence. A scratch was made with a sterile pipette tip, debris was removed, and cells were cultured in DMEM with 1% FBS. Images were taken at 0, 6, 18, and 24 h using a phase-contrast microscope. Assays were performed in octuplicate across two independent experiments. Wound closures were quantified with ImageJ.
2.13. Apoptosis Assay
Cells (5 × 10^4^/well) were seeded in six-well plates (quadruplicate, two experiments) and treated with temozolomide (TMZ, 0.5 mM) or vehicle (DMSO). After 4 days, apoptosis was assessed using Annexin V-FITC/PI staining (Dead Cell Apoptosis Kit, Thermo Fisher Scientific) and analyzed by flow cytometry (30,000 events/sample; FACS Canto II; BD Biosciences). Populations were classified as viable, early apoptotic, late apoptotic, or necrotic, and data were analyzed using FlowJo v10.
2.14. Senescence Assay
Senescence-associated-β-galactosidase (SA-β-gal) activity was detected in cells (5 × 10^4^/well) cultured in six-well plates containing DMEM with 10% FBS. Cells were fixed with formaldehyde and glutaraldehyde (5 min, room temperature) and incubated with X-gal solution (1 mg/mL) overnight at 37 °C, as described previously [28]. Images were acquired by phase-contrast microscopy.
2.15. Statistical Analyses
Statistical analyses were executed using SPSS v20.0 (IBM Corporation, Armonk, NY, USA) and GraphPad Prism v8 (GraphPad Software, San Diego, CA, USA). Comparisons among multiple groups for tubulin expression, cell viability, cell cycle, cell adhesion, cell migration, apoptosis, and nuclear morphology were conducted using two-way analysis of variance (ANOVA) with Tukey’s post hoc test. Gene expression data were analyzed using one-way ANOVA with Tukey’s post hoc test or Student’s t-test. Z-score-normalized values were utilized to generate heatmaps. Data are presented as mean ± SD unless indicated otherwise. Statistical significance was set at p ≤ 0.05.
3. Results
3.1. CRISPR-Cas9-Mediated LOXL3 Reduction in Expression in GBM Cell Lines
CRISPR–Cas9 editing was employed to generate partial LOXL3-knockout (LOXL3-KD) clones in U87MG (wild-type TP53) and U251GBM (mutant TP53) cell lines to investigate the functional role of LOXL3 in GBM. Two sgRNAs targeting exon 2 of LOXL3 were cloned into the pSpCas9(BB) vector (Figure 1A) and transfected into cells. Following clonal selection, Sanger sequencing confirmed the introduction of heterozygous genomic alterations in all clones, including a nucleotide conversion (U87MG clone 1) and deletions of variable length (U87MG clone 2; U251 clones 1 and 2). Notably, none of the clones exhibited a complete knockout, and all retained one wild-type allele (Figure S1B).
Western blotting analysis identified clones exhibiting the most substantial LOXL3 protein reduction, designated as clones 1 and 2 for each cell line (Figure 1B). Densitometric quantification showed LOXL3 expression decreases to 13.9% and 21.8% of control in U87MG clones 1 and 2, and to 41.6% and 62.1% in U251 clones 1 and 2, respectively (Figure 1C). LOXL3-KD cells displayed visible enlarged morphology, which was most pronounced in clones with stronger LOXL3 suppression (Figure 1D).
LOXL3 reduced expression impaired cell proliferation in both cell lines, with clone 1 demonstrating the greatest effect. In U87MG clone 1 cell viability was reduced 1.7-, 1.8-, and 1.5-fold at 48, 72 and 96 h compared with control cells (Figure 1E). U251 clone 1 exhibited an even stronger phenotype with viability decreases of 2, 3.14, and 5.6-fold at the same time points (Figure 1F). Based on these results, subsequent analyses were conducted using clone 1 from each cell line, hereafter referred to as LOXL3-KD.
Transcriptomic profiling identified 921 DEGs in U87MG cells and 1974 DEGs in LOXL3-KD U251 cells (|log_2_FC| ≥ 0.5; adjusted p ≤ 0.05), including 520 and 1074 downregulated genes, and 401 and 900 upregulated genes, respectively (Table S1). GO enrichment indicated significant repression of pathways associated with cell cycle regulation (GO:0007049) and cell division (GO:0051301) in both cell lines (Figure 1G–J; Table S2). Additionally, LOXL3-KD U87MG cells showed reduced enrichment of negative regulation of the tubulin acetylation (GO:1904428), whereas LOXL3-KD U251 cells exhibited suppression of DNA repair (GO:0006281) and cellular response to DNA damage stimulus (GO:0006974). These data demonstrate successful LOXL3 suppression in both GBM cell lines and reveal a shared transcriptional signature linked to impaired proliferation and cell cycle progression, along with cell line-specific vulnerabilities associated with microtubule stability or DNA damage responses. Our results also revealed that LOXL3 is essential for DNA repair and cell division, positioning LOXL3 as a target for new therapies of IDH-wildtype GBM and IDH-mutant astrocytomas, WHO grade 4.
3.2. LOXL3 Partial Knockout Reduces α-Tubulin Acetylation
Based on transcriptomic enrichment linking LOXL3 to microtubule regulation, we next assessed α-tubulin acetylation. Western blotting revealed a marked reduction in acetylated α-tubulin levels in LOXL3-KD cells from both lines, with a substantially stronger decrease in U87MG cells (4.6-fold) than in U251 cells (1.5-fold) (Figure 2A,B). Immunofluorescence confirmed reduced acetylated microtubules in both models (Figure 2C). These results establish that LOXL3 positively regulates microtubule acetylation in GBM cells, establishing the LOXL3 axis as a requirement for microtubule stability and DNA damage response.
3.3. Reduced LOXL3 Expression Delays Cell Cycle Progression and Disrupts Mitosis
Flow cytometry analysis following synchronization demonstrated that LOXL3 partial knockout impaired cell cycle progression in both cell lines but with distinct checkpoint responses. LOXL3-KD U87MG cells displayed transient G2/M accumulation at 12 h followed by G1 arrest at 36 h, whereas LOXL3-KD U251 cells predominantly accumulated in S and G2/M phases at 36 h (Figure 3A–D).
Consistently, transcriptomic profiling revealed a broad downregulation of cell-cycle-related genes in both cell lines (Figure 3E,F). In U87MG cells, downregulation of genes involved in p53 regulation (MDM2, CDKN1A, MAPK14), mitotic exit and G1 initiation (HSPA2, CDC14B), and microtubule-associated proteins (MAP10), together with spindle assembly checkpoint (SAC) activation (MAD1L1 upregulation) and negative regulation of centriole duplication (MDM1 upregulation), suggested sustained mitotic surveillance. In contrast, U251 cells showed reduced expression of G1/S controllers (E2F1, E2F3, CCNE1, CDC7), DNA repair genes (RAD51C, RAD54B, BRCA1, ATM), and mitotic centrosome regulators (CEP72, CEP85), together with upregulation of INCA1 (CDK inhibitor) and MAD1L1, implying checkpoint failure and genomic stress. Immunofluorescence imaging further confirmed mitotic spindle defects in both models, with U251 LOLX3-KD cells displaying pronounced spindle disorganization and increased multipolarity (Figure 3G). Together, these observations provide evidence of essential role of LOXL3 for mitotic fidelity and that its reduced expression triggers a TP53-dependent fate.
3.4. Reduced LOXL3 Expression Alters Nuclear Morphology and Differentially Modulates Cell Fate
NMA identified distinct phenotypes in LOXL3-KD populations: U87MG cells showed irregular and fragmented nuclei, consistent with mitotic catastrophe, whereas U251 cells displayed enlarged, round nuclei, characteristic of senescence (Figure 4A). Apoptosis assays showed significantly increased early apoptotic cell fractions in LOXL3-KD U87MG cells under basal conditions, which were further enhanced by TMZ treatment (Figure 4B). By contrast, LOXL3-KD U251 cells did not display increased apoptosis, with or without TMZ treatment (Figure 4C).
Transcriptomic profiles supported these divergent fates. LOXL3-KD U87MG cells upregulated p53-dependent pro-apoptotic mediators (HIPK2, DYRK2, MAPK12, APAF1), whereas LOXL3-KD U251 cells downregulated anti-senescence drivers (CDC25A, CCNB1) and upregulated senescence-associated genes (RBL2, CDKN1A, CDKN1B) (Figure 4D). Correspondingly, GLB1 (β-galactosidase) expression was significantly elevated in LOXL3-KD U251 cells but unchanged in U87MG cells (Figure 4E). Moreover, SA-β-gal activity confirmed increased senescence in LOXL3-KD U251 cells (Figure 4F). These findings elucidate a bifurcation of terminal cellular fates, revealing LOXL3 attenuation triggers apoptosis in TP53-competent cells and irreversible senescence in TP53-deficient cells. This establishes LOXL3 inhibition as a universal therapeutic axis that bypasses the TP53 mutational status.
3.5. LOXL3 Partial Knockout Impairs Cell Adhesion and Migration
Given the reduced microtubule acetylation in LOXL3-KD cells, we evaluated cytoskeleton-dependent behaviors. LOXL3-KD U87MG cells exhibited impaired adhesion (1.5-fold reduction; Figure 5A) and decreased migration across time (11.1%, 20.1%, and 13.6% reductions at 6, 18, and 24 h, Figure 5C,E). In contrast, U251 LOXL3-KD cells displayed no significant loss of adhesion (Figure 5B) and only modest impairments in migration at later points (8.3% and 13.6% at 18 and 24 h; Figure 5D,F). Analysis of cytoskeleton-related genes (GO:0005856) revealed 17 and 33 DEGs in LOXL3-KD U87MG and U251 cell lines, respectively. However, the ECM gene set (GO:0044420) yielded only minimal differential expression (one gene in U87MG; three genes in U251) (Figure S2). These findings suggest that LOXL3-regulated microtubule acetylation contributes to adhesion and motility, with a more substantial impact observed in U87MG cells, where acetylation loss is greater. Targeting LOXL3 opens a clear vulnerability to disrupt GBM’s invasive ability.
4. Discussion
4.1. LOXL3 as a Driver of GBM Aggressiveness
LOXL3 upregulation has been implicated in tumorigenesis and tumor progression, including viability, adhesion and invasion in GBM [11,29]. This study of CRISPR-Cas9-mediated knockout in two genetically distinct GBM cell lines, U87MG (wild-type TP53) and U251 (mutant TP53) cells, confirmed these associations, producing increased cell size, reduced viability, and impaired proliferation, consistent with our previous transient silencing study [12]. While TP53 status profoundly influences cellular stress responses and cell fate decisions, it is not expected to directly affect the molecular efficiency of LOXL3 knockdown. Differences in TP53 status do not block the catalytic ability of Cas9 to induce DNA breaks; rather, TP53 acts as a downstream modulator of the cellular phenotypic response to genomic stress rather than an upstream determinant of the molecular efficiency of CRISPR-Cas9-mediated gene editing [30]. Therefore, TP53 status is likely to modulate the cellular interpretation of LOXL3 reduced expression—determining the severity of the growth arrest or apoptotic response—rather than the primary degree of gene silencing achieved [31].
Reductions in viability following LOXL3 silencing were also reported in melanoma cells, where knockdown via shRNA induced genomic instability, mitotic defects, and death by apoptosis [32]. Only heterozygous clones were obtained, all retaining one wild-type allele and partial reduction in LOXL3 protein expression, suggesting selective pressure against complete LOXL3 loss. This observation aligns with an in vivo study in mice, where homozygous deletion of Lolx3 resulted in perinatal lethality, highlighting the essential role of LOXL3 in cellular survival [33].
4.2. Microtubule Acetylation and Cell Cycle Control
Mechanistically, reduced LOXL3 expression impaired α-tubulin acetylation, destabilized MT, and activated SAC. Tubulin acetylation is known to enhance MT flexibility, protect against structural damage, and support polymerization [13,34]. Transcriptomic analyses revealed downregulation of MT-stabilizing genes (MAP10, HSPA2, CDC14B) [35,36,37] in U87MG cells, suggesting that impaired MT acetylation disrupts G2/M arrest and subsequent G1 arrest. U251 cells showed altered G1/S regulators (E2F1, E2F3, CCNE1, CDC7) [38,39] and reduced homologous recombination DNA repair gene expression (RAD51C, RAD54B, BRCA1, ATM) [40,41,42,43]. These changes indicate compromised genomic stability and disrupted cell cycle progression, underscoring the role of LOXL3 in maintaining MT integrity and mitotic fidelity.
4.3. SAC Activation and Spindle Defects
Both LOXL3-KD cell lines exhibited SAC activation, a surveillance mechanism that delays mitosis until all chromosomes are correctly attached to MTs [44]. MT disruption is known to activate and prolong SAC, resulting in mitotic arrest [45]. In both cell lines, MAD1L1, encoding the SAC component MAD1, was upregulated. In addition, LOXL3-KD U87MG cells upregulated MDM1, an MT-binding protein that negatively regulates centriole duplication [46]. On the other hand, U251 cells displayed multipolar mitosis, consistent with excessive SAC activity and p53-independent mitotic slippage [47]. These findings highlight the importance of LOXL3 in spindle organization and checkpoint regulation.
4.4. Divergent Cell Fate Outcomes
GBM cell fate outcome diverged according to TP53 status. In LOXL3-KD U87MG cells, prolonged mitotic arrest triggered mitotic catastrophe and apoptosis via caspase activation [48], enhanced by TMZ, with transcriptomic evidence of p53-dependent pro-apoptotic signaling (HIPK2, DYRK2, APAF1) [49,50], alongside downregulating CDKN1A and MDM2 [51]. In contrast, LOXL3-KD U251 cells underwent senescence [52], characterized by flattened morphology, β-galactosidase activity [53], and upregulation of CDKN1A/CDKN1B (p21/p27), consistent with p53-independent CDK inhibition [54,55].
4.5. Impact on Adhesion and Migration
Partial LOXL3 knockout impaired adhesion and migration, with a stronger effect in U87MG cells, consistent with their higher knockout efficiency and greater loss of MT acetylation. These observations align with our previous study [12]. Given the role of MT acetylation in focal adhesion and motility of astrocytes [56], and LOXL3 expression correlation with CTNNB1 (β-catenin) and SNAIL pathway activation in low-grade astrocytomas [11] and hepatocellular carcinoma [8], these results suggest that LOXL3 supports GBM invasiveness through MT-dependent cytoskeletal remodeling. Whether the canonical role of LOXL3 ECM crosslinking contributes to the observed phenotypes remains an open question. Nonetheless, our data suggest that the primary impact of LOXL3 in GBM is centered on the maintenance of intracellular structure integrity. While the expression of ECM genes remained unchanged across both cell lines, the internal cellular architecture was profoundly compromised following LOXL3 reduction. This cytoskeleton destabilization was characterized by a decrease in α-tubulin acetylation and the concomitant downregulation of key cytoskeletal regulatory genes in both U87MG and U251 cells. Consequently, LOXL3 appears to play a non-redundant role in stabilizing the GBM cytoskeleton. This function is distinct from that of other lysyl oxidase family members, particularly LOX, which is described as the predominant driver of ECM stiffening in various malignancies [57] and exhibits the strongest correlation with the ECM genes in GBM [11].
4.6. Therapeutic Implications
Collectively, our findings establish LOXL3 as a critical regulator of MT acetylation, mitotic fidelity, and cell fate in GBM. Reduced LOXL3 expression destabilizes MT, activates SAC, and drives TP53-dependent outcomes—apoptosis in wild-type TP53 cells and senescence in TP53-mutant cells—while reducing invasiveness. This phenotype parallels the effects of benzimidazole carbamate compounds and other MT-targeting drugs, which induce G2/M arrest and mitotic catastrophe in GBM cells, leading to variable fates such as mitotic death, slippage, or senescence [58]. Therefore, the balance between mitotic catastrophe/apoptosis and senescence is dictated by p53 functionality, as summarized in Figure 6. Given that TP53 mutations occur in approximately 25–30% of GBM, IDH-wildtype [5] and up to 90% of astrocytoma, IDH-mutant, grade 4 cases [4], LOXL3 inhibition may be therapeutically exploited in both genetic contexts. Future studies should validate these findings in orthotopic and patient-derived models, clarify enzymatic versus non-enzymatic contributions, and explore synergy with TMZ, microtubule-targeting drugs that inhibit acetylation or senolytic agents.
5. Conclusions
LOXL3 emerges as a multifunctional contributor to GBM aggressiveness, integrating structural and regulatory roles in MT dynamics, mitotic surveillance, and invasive capacity. By converging on pathways of MT destabilization and mitotic catastrophe, LOXL3 inhibition offers a mechanistic bridge to existing MT-targeting strategies, while its TP53-dependent divergence in cellular response underscores the importance of genomic context in therapeutic exploitation, supporting LOXL3 as a promising molecular target for genotype-informed combinational strategies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Price M. Ballard C. Benedetti J. Kruchko C. Barnholtz-Sloan J. Ostrom Q. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2018–2022 Neuro Oncol.202527 iv 1iv 6610.1093/neuonc/noaf 19441092086 PMC 12527013 · doi ↗ · pubmed ↗
- 2Brennan C. Verhaak R. Mc Kenna A. Campos B. Noushmehr H. Salama S. Zheng S. Chakravarty D. Sanborn J. Berman S. The Somatic Genomic Landscape of Glioblastoma Cell 201315546247710.1016/j.cell.2013.09.03424120142 PMC 3910500 · doi ↗ · pubmed ↗
- 3Louis D. Perry A. Wesseling P. Brat D. Cree I. Figarella-Branger D. Hawkins C. Ng H. Pfister S. Reifenberger G. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary Neuro Oncol.2021231231125110.1093/neuonc/noab 10634185076 PMC 8328013 · doi ↗ · pubmed ↗
- 4Wong Q.H. Li K.K. Wang W.W. Malta T.M. Noushmehr H. Grabovska Y. Jones C. Chan A.K. Kwan J.S. Huang Q.J. Molecular landscape of IDH-mutant primary astrocytoma Grade IV/glioblastomas Mod. Pathol.2021341245126010.1038/s 41379-021-00778-x 33692446 · doi ↗ · pubmed ↗
- 5Esperante D. Galicia K.D. Rivas-Cuervo K.G. Cacho-Díaz B. Trejo-Becerril C. Taja-Chayeb L. Aboud O. Carlos-Escalante J.A. Wegman-Ostrosky T. TP 53 oncogenic variants as prognostic factors in individuals with glioblastoma: A systematic review and meta-analysis Front. Neurol.202415149024610.3389/fneur.2024.149024639744115 PMC 11688405 · doi ↗ · pubmed ↗
- 6Laurentino T. Soares R. Marie S. Oba-Shinjo S. LOXL 3 Function Beyond Amino Oxidase and Role in Pathologies, Including Cancer Int. J. Mol. Sci.201920358710.3390/ijms 2014358731340433 PMC 6678131 · doi ↗ · pubmed ↗
- 7Ma L. Huang C. Wang X. Xin D. Wang L. Zou Q. Zhang Y. Tan M. Wang Y. Zhao T. Lysyl Oxidase 3 Is a Dual-Specificity Enzyme Involved in STAT 3 Deacetylation and Deacetylimination Modulation Mol. Cell 20176529630910.1016/j.molcel.2016.12.00228065600 · doi ↗ · pubmed ↗
- 8Li R. Shang R. Li S. Ren Y. Shen L. Yang L. Chen S. Chen X. Li J. Xu M. LOXL 3-promoted hepatocellular carcinoma progression via promotion of Snail 1/USP 4-mediated epithelial-mesenchymal transition Environ. Toxicol.2022372540255110.1002/tox.2361735841383 · doi ↗ · pubmed ↗