Comparative Mitogenomics of Channa pyrophthalmus Unveils Orogeny-Driven Speciation and Lineage-Specific Adaptive Evolution in Snakeheads
Qing Luo, Jiafeng Liu, Jiajun Liu, Mi Ou, Shuzhan Fei, Haiyang Liu, Xincheng Zhang, Jian Zhao

TL;DR
The Fire and Ice Snakehead's mitochondrial genome reveals it split from relatives 7 million years ago due to mountain uplift and evolved conservatively in its mountain stream habitat.
Contribution
This study provides the first complete mitochondrial genome of Channa pyrophthalmus and links geological events to lineage-specific adaptive evolution in snakeheads.
Findings
C. pyrophthalmus diverged from C. gachua around 7.1 million years ago, coinciding with the uplift of the Indo-Burman Ranges.
The Fire and Ice Snakehead shows conservative evolution with purifying selection, contrasting with positive selection in giant snakehead lineages.
Mitochondrial structural variation in the control region and accelerated evolution in ATP8 correlate with ecological and body size diversification.
Abstract
Snakeheads are iconic freshwater predatory fishes in Asia, but their classification is often difficult due to similar appearances. Recently, a new species, the Fire and Ice Snakehead (Channa pyrophthalmus), was discovered in the Tenasserim Region of Myanmar. In this study, we decoded its complete mitochondrial genome to understand its evolutionary history. Our analysis confirms that this species is genetically distinct and split from its closest relatives approximately 7 million years ago, likely isolated by ancient mountains. Interestingly, unlike the widespread Striped Snakehead (Channa striata) which evolved rapidly to adapt to diverse environments, the Fire and Ice Snakehead shows a conservative evolutionary strategy, maintaining a stable genome suited to its specific mountain stream habitat. This study highlights the importance of the Tenasserim region as a biodiversity shelter and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
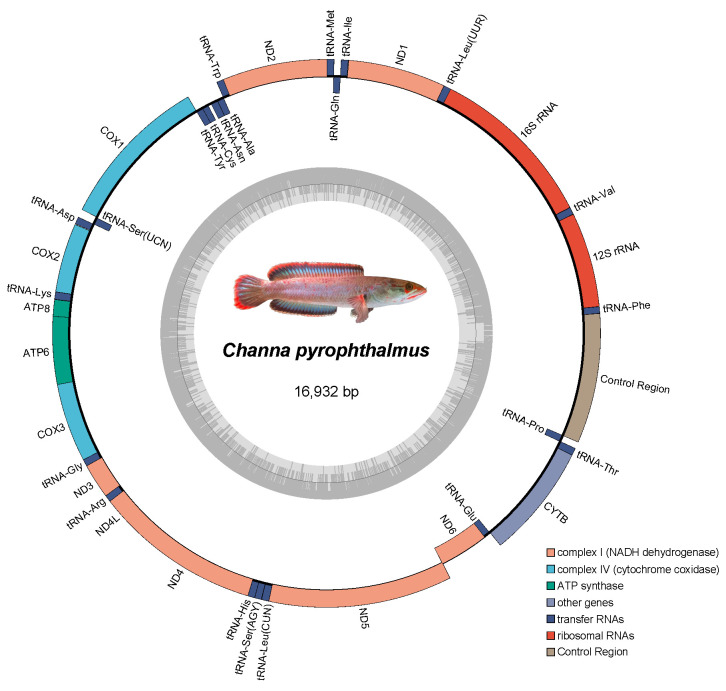 Figure 1
Figure 1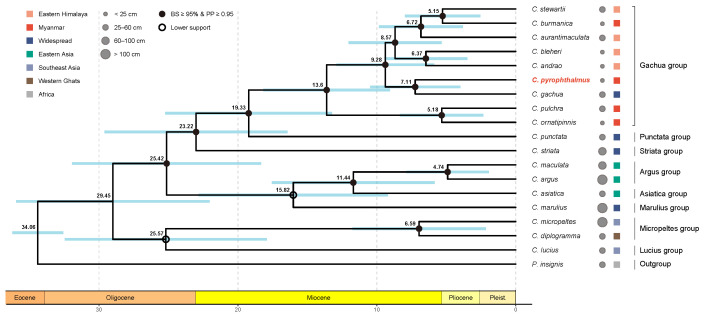 Figure 2
Figure 2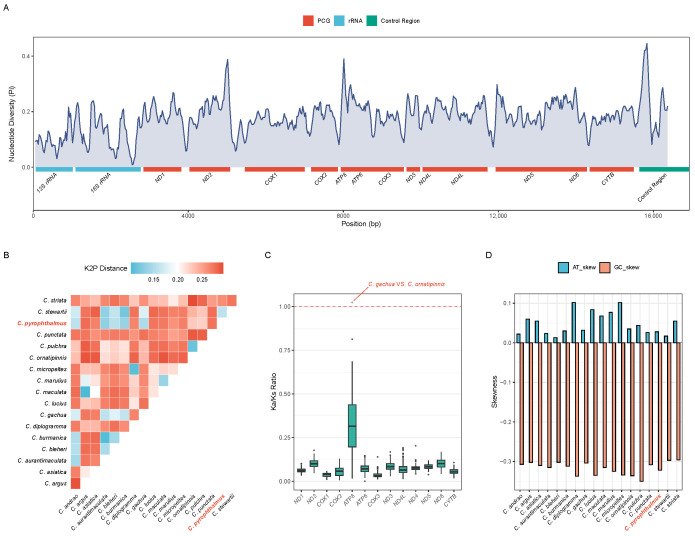 Figure 3
Figure 3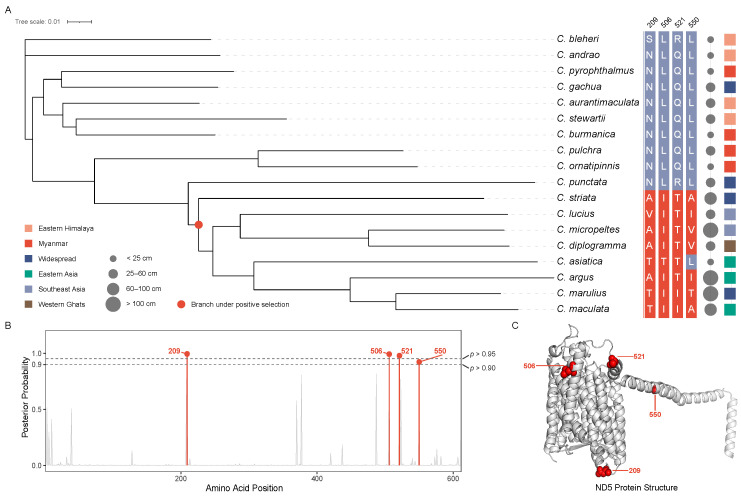 Figure 4
Figure 4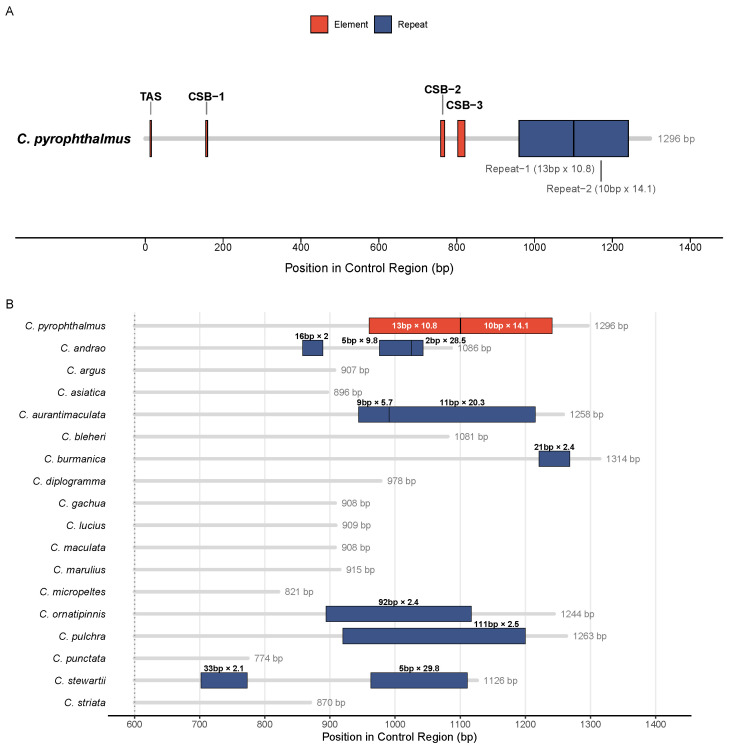 Figure 5
Figure 5- —China Agriculture Research System of MOF and MARA
- —Guangdong Special Support Program
- —National Natural Science Foundation of China
- —Basic and Applied Basic Research Foundation of Guangdong Province
- —Guangdong Provincial Special Fund for Modern Agriculture Industry Technology Innovation Teams
- —Science and Technology Program of Guangzhou
- —Guangdong Province Rural Revitalization Strategy Special Fund
- —Central Public-interest Scientific Institution Basal Research Fund, CAFS
- —China-ASEAN Maritime Cooperation Fund
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Ichthyology and Marine Biology · Fish Biology and Ecology Studies
1. Introduction
Snakeheads, belonging to the family Channidae, are freshwater teleosts easily distinguished by their elongated, cylindrical bodies, formidable dentition, and the possession of a suprabranchial organ facilitating air-breathing [1,2]. Distributed extensively across Africa and Asia, these apex predators hold substantial economic value, serving as key resources for both aquaculture and the ornamental fish trade [3]. Among the channids, the Asian genus Channa represents the most speciose and morphologically diverse lineage, displaying extraordinary variation in life history traits—ranging from dwarf species (<25 cm) inhabiting hill streams to riverine giants exceeding a meter in length [3].
Historically, the taxonomy of Channa has proven challenging. Morphological conservatism and high phenotypic plasticity have obscured species boundaries, resulting in several unresolved species complexes, notably surrounding Channa striata, Channa marulius, and Channa gachua [4,5]. The C. gachua group is particularly problematic, representing a widespread and taxonomically intricate lineage centered on the Eastern Himalaya and the broader Indo-Burma Biodiversity Hotspot [3]. However, recent integrative taxonomic efforts have begun to disentangle this diversity, revealing numerous endemic species previously subsumed under broad nominal taxa [6,7]. One such species is Channa pyrophthalmus, a recently described dwarf species endemic to the Tanintharyi Region of Myanmar—a critical center of endemism within the hotspot. Notably, due to its striking bright orange suborbital patch and steel-blue lips, this species has recently gained economic importance in the international ornamental fish trade, where it is marketed under the name “Fire and Ice Snakehead” [7]. Geographically isolated west of the Tenasserim barrier, C. pyrophthalmus occupies a restricted range distinct from its widespread congeners, making it an excellent model for investigating allopatric speciation and local adaptation [7].
Mitochondrial genomes (mitogenomes) offer robust, high-resolution markers for reconstructing phylogenies and exploring molecular evolution, given their maternal inheritance, lack of recombination, and rapid evolutionary rates [8,9,10]. While single-locus markers like COX1 and CYTB are standard for barcoding [5,11], they often provide insufficient resolution for resolving deep nodes or detecting complex signals such as adaptive selection. Recent efforts have successfully characterized complete mitogenomes for various Channa species, including Channa argus [12,13,14], Channa maculata [13,14,15], C. striata [16], and the Gachua group members like Channa stewartii and Channa pulchra [17]. Despite this progress, genomic data for C. pyrophthalmus remains absent, leaving gaps in our understanding of how phenotypic traits and biogeographical events in the Indo-Burman Ranges (IBR) have shaped this distinct lineage.
Here, we report the complete mitochondrial genome of C. pyrophthalmus obtained via next-generation sequencing. Through a comprehensive analysis against 17 other Channa mitogenomes, we aim to elucidate genomic architecture and structural variations, with specific attention to the control region. Concurrently, we reconstruct a robust time-calibrated phylogeny to evaluate biogeographical hypotheses concerning the uplift of the Indo-Burman Ranges. Finally, by investigating selective pressures on protein-coding genes, we seek to uncover the molecular basis of adaptation, offering new insights into the mitochondrial bioenergetic constraints underlying body size evolution in freshwater teleosts.
2. Materials and Methods
2.1. Sample Collection and Molecular Identification
A specimen of C. pyrophthalmus was purchased from a licensed commercial ornamental fish vendor in the Baiyi Ornamental Fish Market (Guangzhou, China). This species is known to be imported from Myanmar for the aquarium trade. Species identification was confirmed based on diagnostic morphological characters following the original description [7] and further validated by COX1 sequence comparison with reference sequences from type material (Figure S1). The specimen has been deposited in the China-ASEAN Fisheries Resources Database (Voucher No. Cpy250302003). Genomic DNA extraction was carried out using the E.Z.N.A. Tissue DNA Kit (Omega Bio-tek, Norcross, GA, USA) according to the manufacturer’s instructions, with quality and concentration quantified via the TBS 380 fluorometer (Invitrogen, Carlsbad, CA, USA) and Pico Green Assay (Life Technologies, Carlsbad, CA, USA). We constructed the sequencing library using the Illumina TruSeq^TM^ Nano DNA Sample Prep Kit (Illumina, San Diego, CA, USA). The experimental protocol was approved by the Animal Ethics Committee of the Pearl River Fisheries Research Institute, Chinese Academy of Fishery Sciences (LAEC-PRFRI-2024-12-04).
2.2. Mitogenome Sequencing and Assembly
Genomic sequencing was conducted on the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA). We processed raw reads with Trimmomatic v0.39 to eliminate adaptors and low-quality bases, followed by de novo assembly using GetOrganelle v1.7.5 [18].
Preliminary annotation was performed via the MITOS2 web server, utilizing the vertebrate mitochondrial code. To ensure high accuracy, we imported the sequences into Geneious Prime v2025.1.3 (Biomatters Ltd., Auckland, New Zealand) for manual curation, refining start and stop codons through comparative alignment with orthologous sequences of other Channa species. The final circular map was visualized using OGDRAW v1.3.1 [19]. Additionally, tRNA secondary structures were predicted using tRNAscan-SE v2.0 [20] and rendered via the Forna web server (http://rna.tbi.univie.ac.at/forna/, accessed on 23 November 2025).
2.3. Comparative Genomic Analysis
For comparative purposes, we compiled a dataset comprising 17 Channa mitogenomes and one outgroup Parachanna insignis from GenBank (details in Table S1) [16,17,21]. We assessed nucleotide diversity ( ) via sliding window analysis in DnaSP v6 [22], setting a window size of 100 bp with a 25 bp step. Synonymous (Ks) and non-synonymous (Ka) substitution rates were also estimated using DnaSP v6, with Ka/Ks ratios calculated to infer selection pressures on each protein-coding gene (PCG). Evolutionary metrics, including pairwise genetic distances (Kimura-2-parameter, K2P) and AT/GC-skewness, were computed using PhyloSuite v2 [23]. We also calculated Relative Synonymous Codon Usage (RSCU) to evaluate codon usage bias across the dataset. The structural organization of the control region was analyzed to identify conserved motifs (TAS, CSB) and tandem repeats. Tandem repeats were detected using the Tandem Repeats Finder (TRF) server (https://tandem.bu.edu/trf/trf.html, accessed on 10 December 2025) and manually verified.
2.4. Phylogenetic Reconstruction and Divergence Time Estimation
Phylogenetic inferences were based on a concatenated dataset of 13 protein-coding genes (PCGs). Sequences were aligned using MAFFT v7 [24] and subsequently trimmed with Gblocks. We assessed substitution saturation using DAMBE7 [25]. Phylogenetic tree reconstruction employed both Maximum Likelihood (ML) and Bayesian Inference (BI) frameworks. ML analysis was conducted in IQ-TREE v3 [26] using the best-fit model (GTR+F+R4) determined by ModelFinder [27], with robustness assessed via 5000 bootstrap replicates. BI analysis was performed using MrBayes v3.2 [28], running for 2 million generations (sampling every 1000 generations) and discarding the initial 25% as burn-in. The resulting topologies were visualized and refined in iTOL (https://itol.embl.de/, accessed on 22 December 2025) [29].
We estimated divergence times using BEAST v2.7 [30]. The dataset was analyzed using the SRD06 partitioning strategy [31], dividing the nucleotide alignment into two partitions: combined 1st and 2nd codon positions, and the 3rd codon position. Substitution models (GTR+G) and rate heterogeneity parameters were unlinked across partitions, while the clock model and tree topology were linked. The analysis applied a Yule speciation prior and an uncorrelated relaxed clock (log-normal). To calibrate the tree, we used a secondary calibration point at the Channidae crown node based on the time estimates from Calibration Scheme 1 of Rüber et al. [3], which incorporates the fossil Parachanna fayumensis. Accordingly, we set a Log-Normal prior (offset = 33.0 Ma, log mean = 0.1, log SD = 0.8). We constrained the monophyly of the genus Channa. The MCMC chain ran for 50 million generations, with convergence confirmed in Tracer v1.7 [32] (ESS > 200). A maximum clade credibility (MCC) tree was generated using TreeAnnotator after a 10% burn-in.
2.5. Selection Pressure and Structural Analysis
To screen for signals of adaptive evolution, we employed the adaptive Branch-Site Random Effects Likelihood (aBSREL) method within HyPhy (https://www.hyphy.org/, accessed on 10 December 2025) [33]. Statistical significance was determined using the Likelihood Ratio Test (LRT) at p < 0.05. Where significant positive selection was detected, we mapped specific selected sites onto the 3D protein structure predicted by AlphaFold3 (https://alphafoldserver.com/, accessed on 10 December 2025) [34] and visualized them using PyMOL v3.1.0.
3. Results
3.1. Genome Organization and Characterization
The newly sequenced mitochondrial genome of C. pyrophthalmus forms a closed circular molecule of 16,932 bp in length (GenBank Accession No. PX764270). It exhibits the typical vertebrate mitochondrial gene arrangement, comprising 13 protein-coding genes (PCGs), 22 tRNAs, 2 rRNAs, and a control region (Table 1, Figure 1). Nucleotide composition is biased toward A+T (55.4%). Regarding start codons, most PCGs utilize standard ATG, with the exception of COX1, which initiates with GTG. Incomplete stop codons (T––) were observed in four genes. These are likely completed via post-transcriptional polyadenylation.
RSCU analysis highlighted Leucine as the most frequently utilized amino acid, contrasting with Cysteine, which was the least frequent (Figure S2). Consistent with the genome-wide A+T bias, codons ending in A or T were preferentially selected. Structural analysis of the 22 tRNAs showed that they range from 65 to 75 bp in length and fold into typical cloverleaf structures, with the notable exception of tRNA-Ser(AGY), which lacks the dihydrouridine arm (Figure S3).
3.2. Phylogenetic Relationships and Divergence Times
Both ML and BI analyses yielded congruent topologies with robust nodal support across all major clades (Figure 2 and Figure S5). C. pyrophthalmus was unambiguously resolved as the sister taxon to C. gachua, a placement supported by maximal values (BS = 99, PP = 1.00), firmly embedding it within the Gachua group. The designation of major species groups within the genus follows the comprehensive phylogenetic framework established by Rüber et al. [3].
Our time-calibrated phylogeny (Figure 2) dates the origin of the family Channidae (the Parachanna–Channa split) to the Oligocene, approximately 29.45 Ma (95% HPD: 21.1–35.0 Ma). The radiation of the Gachua group appears to have initiated in the Middle Miocene (∼19.3 Ma). More specifically, the speciation event separating C. pyrophthalmus from its sister species C. gachua is estimated at 7.1 Ma (95% HPD: 4.0–10.5 Ma). This Late Miocene divergence coincides with the intensification or accelerated deformation phase of the Indo-Burman Ranges.
3.3. Comparative Genomic Landscape
A comparative assessment of 18 Channa mitogenomes revealed distinct evolutionary heterogeneity (Figure 3). Sliding window analysis of nucleotide diversity ( ) identified the control region, ATP8, and ND2 as mutational hotspots, whereas 12S rRNA exhibited high conservation (Figure 3A). In terms of genetic distance (K2P), C. pyrophthalmus is most divergent from C. argus and shows the closest affinity to other members of the Gachua group (Figure 3B).
Selection pressure analysis generally indicated pervasive purifying selection across the 13 PCGs, with median Ka/Ks ratios consistently falling below 1.0 (Figure 3C). COX1 displayed the lowest ratio, underscoring its functional rigidity. However, an exception to this trend was observed in the ATP8 gene: a specific pairwise comparison between C. gachua and C. ornatipinnis yielded a Ka/Ks ratio of 1.02. Given the short length of the ATP8 gene, this elevated Ka/Ks ratio should be interpreted cautiously, as estimates for small genes are more sensitive to stochastic variation and lineage-specific effects. This represents the only instance across the entire mitogenome where the neutrality threshold was exceeded, suggesting accelerated evolution in this lineage. Additionally, strand-specific mutational bias was evident across all species, characterized by positive AT-skew and negative GC-skew in the majority of PCGs (Figure 3D).
4. Signatures of Adaptive Evolution
The aBSREL analysis uncovered a dichotomy in selective regimes between different lineages (Figure 4A, Table S2). The terminal branch leading to C. pyrophthalmus showed no evidence of positive selection (Mean ω ≈ 0.10), indicating a history of strong purifying selection that aligns with its specialized ecological niche. Conversely, we detected significant episodic positive selection (LRT, p = 0.022) at Node 5, which represents the ancestral lineage of the large-bodied clade (comprising C. striata, C. marulius, C. argus, etc.). On this ancestral branch, a small subset of sites (∼2.0%) in the ND5 gene evolved under intense positive selection ( = 116.2), driving the weighted mean to 2.40. This signal reflects episodic positive selection acting on a very small proportion of sites along this ancestral branch, while the majority of codons remained under strong purifying selection. Structural mapping of the four positively selected sites (209, 506, 521 and 550) onto the ND5 protein (Figure 4B) reveals that they are situated within the transmembrane domain and the lateral helix, pointing to potential functional adaptations in proton translocation or membrane interaction.
5. Structural Variation in the Control Region
The control region of C. pyrophthalmus is characterized by standard conserved elements, including the termination-associated sequence (TAS) and three conserved sequence blocks (CSB-1, CSB-2, CSB-3) (Figure 5A). However, a broader comparison across the genus revealed significant structural plasticity in tandem repeats (Figure 5B). Specifically, C. pyrophthalmus contains two distinct sets of microsatellite-like tandem repeats within the variable 3′ domain. This architecture contrasts sharply with the extensive macro-repeats found in the C. pulchra and C. ornatipinnis lineages, highlighting the rapid evolutionary dynamics of this non-coding region.
6. Discussion
6.1. Phylogenetic Placement and Mitogenomic Conservatism in Channa
Resolving phylogenetic relationships within the genus Channa has historically been hindered by extensive morphological conservatism and phenotypic plasticity, particularly within species complexes such as the Gachua group and the Striata group [4,5]. Although single-locus mitochondrial markers like COX1 and CYTB are widely used for rapid species identification, they often lack the resolution required to disentangle recent radiations and cryptic lineages [3,5,35]. By leveraging complete mitochondrial genomes, our study provides a robust phylogenetic framework that definitively positions C. pyrophthalmus as the sister lineage to C. gachua sensu stricto, with maximal nodal support across both maximum likelihood and Bayesian inference analyses. This result independently corroborates the recent morphological delimitation of C. pyrophthalmus and firmly embeds this taxon within the Gachua group [7,36].
Beyond resolving phylogenetic placement, our mitogenomic data clarify conflicting reports regarding structural rearrangements in Channa mitochondrial genomes. Previous studies proposed lineage-specific insertions between tRNA-Met and ND2 as potential synapomorphies within the Gachua group [37]. However, the absence of such rearrangements in C. pyrophthalmus and in multiple reference mitogenomes examined here indicates that these features are likely homoplastic or population-specific events rather than clade-defining characters. Collectively, these findings highlight a high degree of structural conservatism in mitochondrial gene order across Channa, despite the remarkable ecological and morphological diversity of the genus [17,38].
6.2. Late Miocene Diversification and Indo-Burman Vicariance
Our time-calibrated phylogeny dates the divergence between C. pyrophthalmus and its sister lineage C. gachua to approximately 7.1 Ma. Geologially, the orogenesis of the Indo-Burman Ranges (IBR) is a protracted process; recent magnetostratigraphic studies indicate that the IBR had already emerged as a significant topographic barrier by the Early Miocene (∼23 Ma) [39,40].
The temporal lag between this initial uplift and the later biological divergence (∼7.1 Ma) suggests that ephemeral hydrological connectivity likely persisted across the IBR long after its initial rise. We propose that dynamic river capture events in the headwaters of the paleo-Brahmaputra and paleo-Irrawaddy systems maintained gene flow during the Mid-Miocene. Consequently, the intensification of crustal shortening and tectonic deformation in the Late Miocene—potentially coupled with the distinct geological dynamics of the southern IBR—constituted the decisive vicariant event. This tectonic reorganization finally severed these residual riverine connections, driving allopatric divergence [39,40,41].
Biogeographically, C. pyrophthalmus is confined to the Tanintharyi Region west of the Bilauktaung Range. Situated at the crossroads of the Indochinese and Sundaic faunas, this region likely functioned as a “coastal refugium” during regional tectonic upheaval [42]. The combined effects of mountain uplift and drainage isolation effectively encircled ancestral populations, fostering long-term genetic isolation and ultimately leading to the narrow endemism observed today. This pattern mirrors diversification processes documented in other freshwater taxa from the Indo-Burma region, underscoring the pivotal role of geological vicariance in shaping ichthyofaunal diversity [43].
6.3. Metabolic Evolution Underlying the Giant–Dwarf Dichotomy
A striking contrast in selective regimes emerges when comparing dwarf, range-restricted snakeheads with their large-bodied, widely distributed congeners. The terminal lineage leading to C. pyrophthalmus exhibits pervasive purifying selection across mitochondrial protein-coding genes, consistent with an evolutionary trajectory characterized by niche specialization and relative physiological stability. In contrast, the ancestral branch of the giant-bodied Channa clade shows evidence of episodic positive selection acting on the ND5 gene, with a small subset of sites experiencing markedly elevated values.
ND5 encodes a core subunit of mitochondrial Complex I and plays a central role in proton translocation and oxidative phosphorylation efficiency [44,45,46]. Structural mapping of positively selected sites indicates that positively selected substitutions are concentrated within transmembrane regions and lateral helices, where they may influence proton pumping mechanics or membrane interactions [47,48,49]. Similar patterns of adaptive evolution in ND5 have been documented in teleost lineages facing high energetic demands, such as migratory salmonids and high-altitude fishes [50,51,52]. Taken together, these observations suggest that adaptive modifications of mitochondrial bioenergetics may have provided the physiological infrastructure for the evolution of large body size and active predatory lifestyles in giant snakeheads.
6.4. Lineage-Specific Acceleration of ATP8 and Cryptic Diversity
Among mitochondrial genes, ATP8 displays the highest evolutionary rate across Channa, a pattern commonly attributed to relaxed functional constraints and short gene length. Notably, however, a lineage-specific signal of accelerated evolution was detected between C. gachua and C. ornatipinnis. Although estimates for short genes are inherently sensitive to stochastic effects [53], the restricted phylogenetic distribution of this signal suggests that it may reflect localized adaptive pressures rather than genome-wide relaxation.
The lineages exhibiting accelerated ATP8 evolution predominantly inhabit fast-flowing hill streams, environments that impose sustained swimming demands and elevated metabolic turnover. Comparable signatures of ATP8 adaptation have been reported in rheophilic and high-altitude teleosts, where modifications to ATP synthase are hypothesized to enhance energy production efficiency under challenging conditions [54,55,56,57]. Crucially, this finding resolves a discrepancy with a previous study [17], who reported a Ka/Ks ratio < 1.0 for ATP8 in the same species comparison. We attribute this conflicting signal to the distinct phylogeographic origins of the reference sequences employed: Wang et al. utilized a lowland or introduced C. gachua from Guangzhou [16], whereas our analysis employed a native highland lineage from Yunnan [58]. This finding underscores the necessity of resolving the taxonomy of the C. gachua complex, as pooling distinct lineages obscures critical signals of adaptive evolution.
6.5. Structural Plasticity of the Mitochondrial Control Region
In contrast to the conserved architecture of coding regions, the mitochondrial control region emerges as a focal point of evolutionary dynamism within Channa. Comparative analyses reveal pronounced heterogeneity in control region length and repeat architecture, driven primarily by lineage-specific expansions and contractions of tandem repeat elements. C. pyrophthalmus possesses two distinct sets of microsatellite-like repeats within the variable 3′ domain, a configuration that differs markedly from the extensive macro-repeat arrays observed in species such as C. pulchra and C. ornatipinnis.
This structural plasticity is consistent with replication slippage as a major mechanism shaping control region evolution in teleost mitogenomes [59]. Importantly, the species-specific nature of these repeat motifs suggests that non-coding regions of the mitochondrial genome evolve under relaxed selective constraints, accumulating variation at a pace far exceeding that of protein-coding genes. As such, the control region represents a promising molecular marker for resolving shallow phylogeographic structure and population differentiation within Channa, particularly in species complexes where coding regions provide limited resolution [60,61].
6.6. Methodological Considerations and Data Limitations
While mitochondrial genomes provide powerful insights into phylogenetic relationships and lineage-specific evolutionary processes, several methodological considerations warrant attention. This study is based on a single mitochondrial genome of C. pyrophthalmus, which limits the assessment of intraspecific variation. As a maternally inherited locus, mitochondrial DNA captures only a subset of the species’ evolutionary history, and potential mitonuclear discordance cannot be ruled out without nuclear markers [62]. Furthermore, given the narrow endemic status of C. pyrophthalmus, future studies integrating nuclear genomics would be valuable not only for corroborating these evolutionary patterns but also for assessing genetic diversity to inform conservation strategies. Given that C. pyrophthalmus is a narrow-range endemic targeted by the international aquarium trade, establishing such genetic baselines is urgent for monitoring potential overexploitation [63].
In addition, divergence time estimates rely on a secondary calibration point due to the paucity of confidently assignable internal fossils within Channidae. Although this approach provides a reasonable temporal framework for comparative inference, absolute age estimates should be treated with caution [64]. Importantly, the evolutionary interpretations advanced in this study are grounded primarily in relative divergence patterns and their congruence with regional geological events. Future research integrating broader geographic sampling and genome-wide nuclear data will be essential for testing the robustness of these hypotheses and for advancing a more comprehensive understanding of snakehead evolution [65].
7. Conclusions
By generating the first complete mitochondrial genome of C. pyrophthalmus, this study resolves the phylogenetic placement of a recently described, geographically restricted snakehead and firmly embeds it within the Gachua group as the sister lineage to C. gachua sensu stricto. Comparative mitogenomic analyses reveal a conserved coding architecture contrasted by pronounced structural plasticity in the mitochondrial control region, underscoring its potential as a marker for resolving shallow evolutionary patterns.
Integrating time-calibrated phylogenetic inference with selection analyses, our results suggest that Late Miocene geological vicariance in the Indo-Burman region and lineage-specific shifts in mitochondrial bioenergetics jointly contributed to diversification within the genus. The contrasting selective regimes between dwarf and giant snakeheads highlight a potential link between mitochondrial metabolic evolution, body size divergence, and ecological specialization. Together, these findings provide a comprehensive framework for understanding diversification of freshwater snakeheads and establish a foundation for future population-level and nuclear genomic investigations.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Britz R. Dahanukar N. Anoop V.K. Philip S. Clark B. Raghavan R. Rüber L. Aenigmachannidae, a new family of snakehead fishes (teleostei: channoidei) from subterranean waters of south India Sci. Rep.2020101608110.1038/s 41598-020-73129-632999397 PMC 7527459 · doi ↗ · pubmed ↗
- 2Courtenay W.R.Jr. Williams J.D. Snakeheads (Pisces, Channidae): A Biological Synopsis and Risk Assessment Technical Report 1251 U.S. Geological Survey Reston, VA, USA 200410.3133/cir 1251 · doi ↗
- 3Rüber L. Tan H.H. Britz R. Snakehead (teleostei: Channidae) diversity and the eastern himalaya biodiversity hotspot J. Zool. Syst. Evol. Res.20205835638610.1111/jzs.12324 · doi ↗
- 4Conte-Grand C. Britz R. Dahanukar N. Raghavan R. Pethiyagoda R. Tan H.H. Hadiaty R.K. Yaakob N.S. Rüber L. Barcoding snakeheads (teleostei, channidae) revisited: Discovering greater species diversity and resolving perpetuated taxonomic confusions P Lo S ONE 201712 e 018401710.1371/journal.pone.018401728931084 PMC 5606936 · doi ↗ · pubmed ↗
- 5Modeel S. Chaurasia M. Siwach S. Dolkar P. Negi R.K. Negi R.K. Mitochondrial perspective on species complexes and evolutionary dynamics within genus Channa Biochem. Genet.202510.1007/s 10528-025-11157-540531408 · doi ↗ · pubmed ↗
- 6Praveenraj J. Uma A. Moulitharan N. Singh S.G. A new species of dwarf Channa (teleostei: Channidae) Meghalaya, Northeast India Copeia 2019107617010.1643/CI-18-079 · doi ↗
- 7Britz R. Tan H.H. Rüber L. Four new species of Channa Myanmar (teleostei, Labyrinthici, Channidae)Raffles Bull. Zool.2024721
- 8Boore J.L. Animal mitochondrial genomes Nucleic Acids Res.1999271767178010.1093/nar/27.8.176710101183 PMC 148383 · doi ↗ · pubmed ↗