Three-Dimensional Human Liver Micro Organoids and Bone Co-Culture Mimics Alcohol-Induced BMP Dysregulation and Bone Remodeling Defects
Yuxuan Xin, Guanqiao Chen, Mohammad Majd Hammour, Xiang Gao, Fabian Springer, Elke Maurer, Andreas K. Nüssler, Romina H. Aspera-Werz

TL;DR
This study created a 3D human liver-bone model that mimics alcohol-related liver and bone damage, showing how alcohol disrupts bone proteins and weakens bone health.
Contribution
The study introduces a 3D human liver-bone co-culture model that replicates alcohol-induced hepatic osteodystrophy and BMP dysregulation.
Findings
Chronic alcohol exposure activates CYP2E1 and causes liver fibrosis and EMT.
Alcohol reduces BMP2 and increases BMP13, leading to impaired osteoblast function and chondrogenic shifts in bone progenitors.
The model reflects clinical features of chronic liver disease, enabling BMP-targeted therapy screening.
Abstract
What are the main findings? The present study established a long-term 3D human liver–bone co-culture model mimicking alcohol-induced hepatic osteodystrophy (HOD) with fibrogenic liver changes and bone defects.Chronic 50 mM alcohol triggers hepatic CYP2E1 activation, EMT/fibrosis, BMP imbalance (↓BMP2, ↑BMP13), reduced osteoblast mineralization, and chondrogenic shift in bone progenitors. The present study established a long-term 3D human liver–bone co-culture model mimicking alcohol-induced hepatic osteodystrophy (HOD) with fibrogenic liver changes and bone defects. Chronic 50 mM alcohol triggers hepatic CYP2E1 activation, EMT/fibrosis, BMP imbalance (↓BMP2, ↑BMP13), reduced osteoblast mineralization, and chondrogenic shift in bone progenitors. What are the implications of the main findings? The present study provides an organoid-based platform for studying the liver–bone axis and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
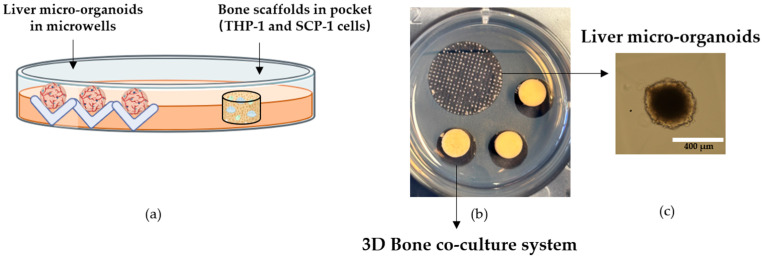 Figure 1
Figure 1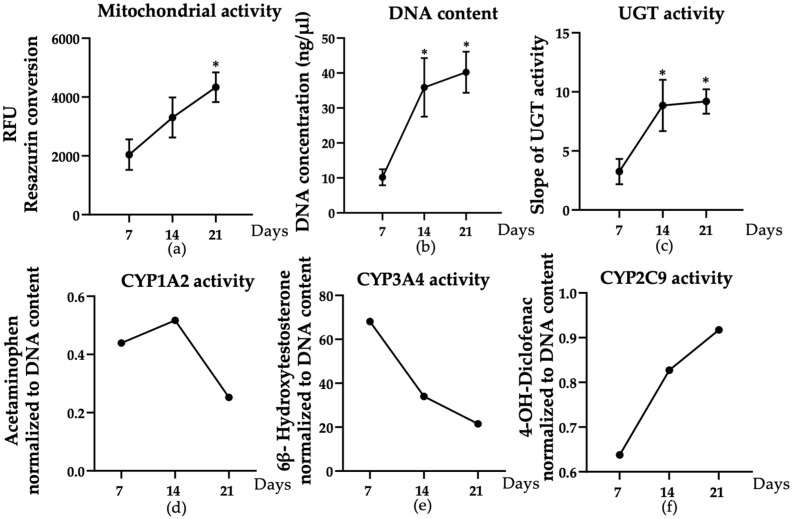 Figure 2
Figure 2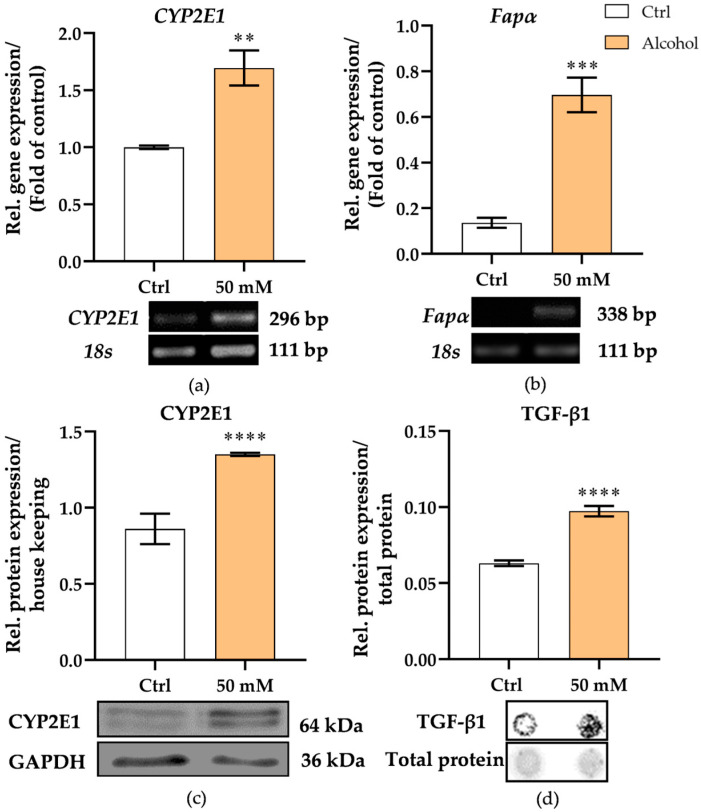 Figure 3
Figure 3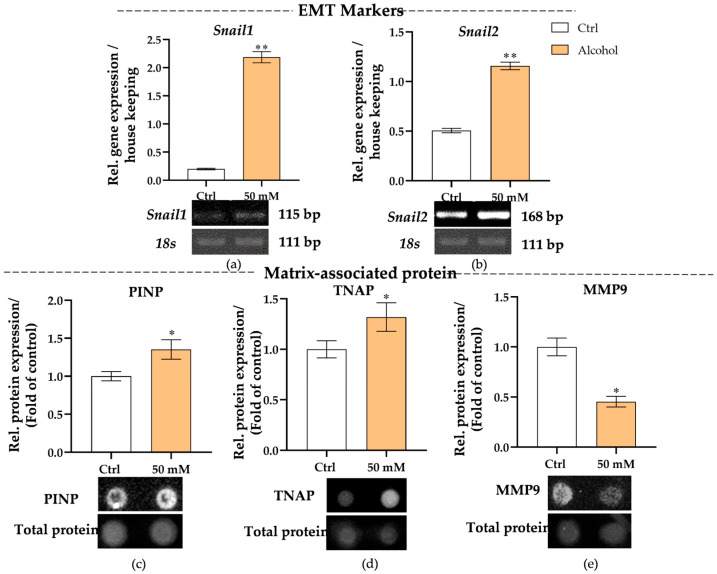 Figure 4
Figure 4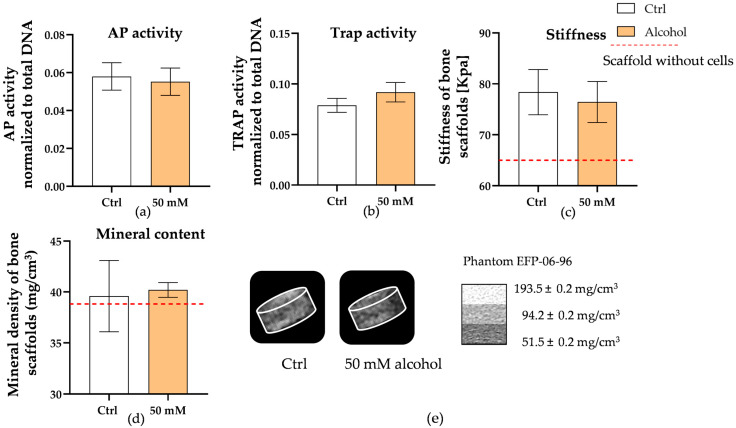 Figure 5
Figure 5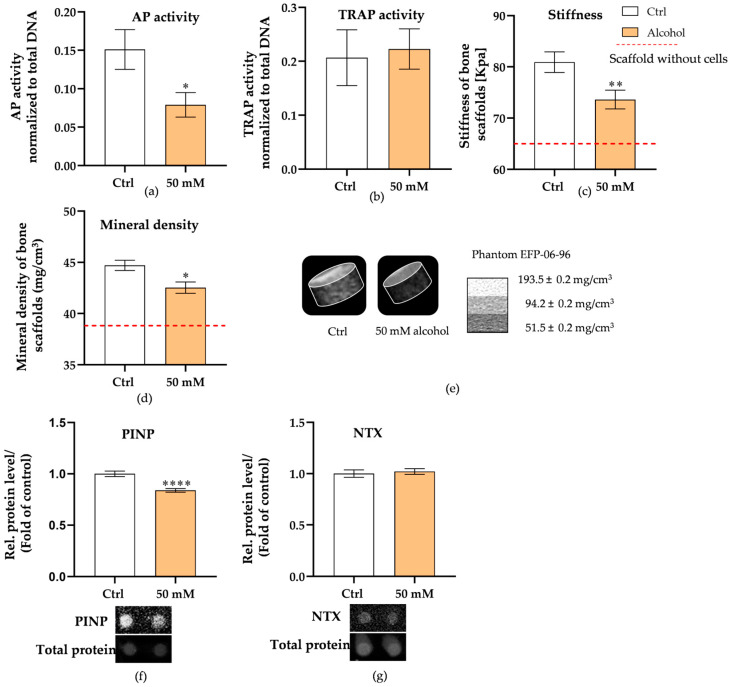 Figure 6
Figure 6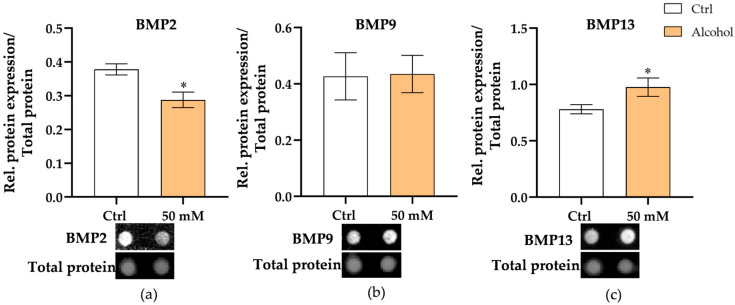 Figure 7
Figure 7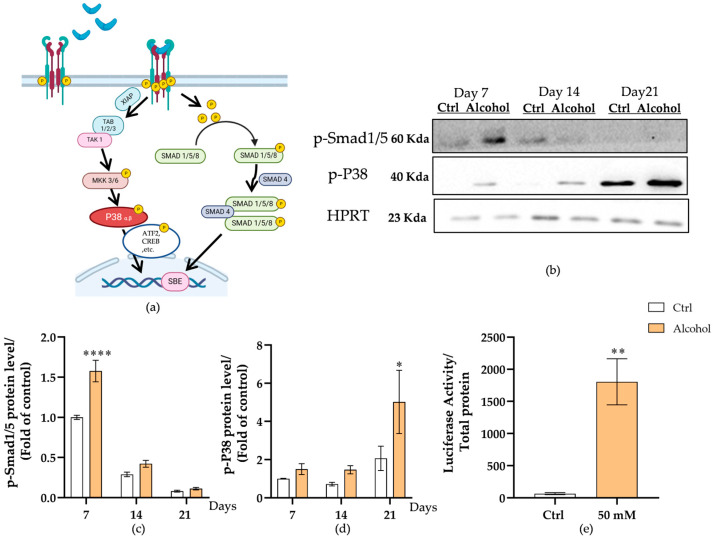 Figure 8
Figure 8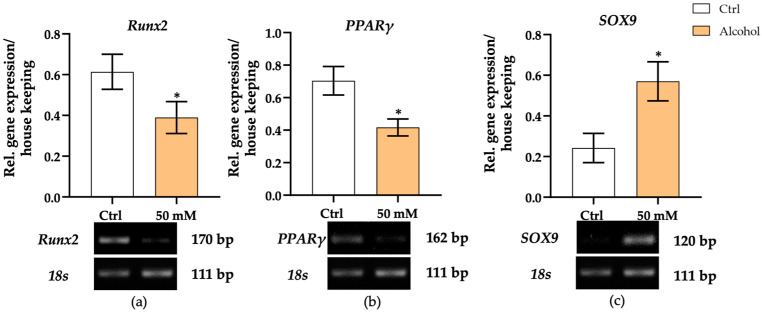 Figure 9
Figure 9Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLiver physiology and pathology · Bone health and osteoporosis research · TGF-β signaling in diseases
1. Introduction
Hepatic osteodystrophy (HOD) is a common complication secondary to chronic liver disease (CLD), mainly characterized by decreased bone mineral density, altered bone microarchitecture, and increased fracture risk [1,2]. This bone metabolic disorder significantly compromises patients’ quality of life and long-term prognosis. Although its underlying pathogenesis remains incompletely understood, growing evidence suggests that liver dysfunction may promote bone loss and disrupt bone homeostasis through systemic metabolic disturbances and dysregulation of liver–bone axis signaling [3,4]. Among the various etiologies of CLD, chronic alcohol abuse represents one of the most prevalent and well-characterized causes [5,6]. According to the World Health Organization, approximately 43% of individuals aged 15 years and older consume alcohol [7], with an average annual pure alcohol intake of 6.2 L per person [8]. In this study, we aim to investigate how liver injury, modeled through chronic alcohol exposure, leads to dysregulation of bone metabolism via impaired liver–bone crosstalk.
Existing studies indicate that chronic alcohol exposure is a major driver of hepatic fibrosis, characterized by excessive extracellular matrix deposition and activation of hepatic stellate cells (HSCs) [9,10]. Emerging evidence suggests that bone morphogenetic proteins (BMPs) play critical roles in this process. In the fibrotic liver, BMP2 expression is often downregulated, impairing hepatocyte regeneration and favoring a profibrotic environment [11]. BMP9, which normally contributes to vascular stability and hepatocellular homeostasis, exhibits dysregulated expression under alcoholic stress, thereby exacerbating endothelial dysfunction and fibrotic remodeling [12]. Conversely, BMP13 is markedly upregulated during hepatic fibrogenesis, where it promotes HSC activation and collagen synthesis [13]. Such fibrosis-associated alterations in hepatic BMP expression may not only accelerate liver injury but also exert systemic effects on distant organs, particularly bone tissue. Indeed, liver injury has been shown to disrupt bone remodeling through both direct and indirect mechanisms. Direct effects may involve the toxicity of alcohol or liver-derived metabolites on osteoblasts and osteoclasts, whereas systemic metabolic disturbances, hormonal imbalances, and chronic inflammation mediate indirect effects. Given that BMPs are key regulators of osteoblast differentiation and bone homeostasis, hepatic BMP dysregulation may represent a crucial molecular link between chronic liver injury and skeletal disorders. In the bone microenvironment, BMP2 is essential for osteoblast differentiation and mineralization [14], while BMP9 regulates vascular integrity and bone metabolism [15]. By contrast, BMP13 is associated with fibrogenic and chondrogenic pathways [16]. Clinical studies have shown that patients with long-term liver disease exhibit decreased circulating BMP2 and increased BMP13 levels, correlating with reduced bone mineral density and cartilage-like changes in the bone marrow [11,13]. Despite these findings, the specific role of liver-derived BMPs in alcohol-induced bone homeostatic imbalance remains unclear, partly due to the lack of physiologically relevant in vitro models capable of recapitulating long-term liver–bone interactions. Therefore, a human in vitro system that accurately reproduces this hepatic BMP imbalance is needed to dissect the underlying mechanisms and identify potential therapeutic targets.
While animal models have provided valuable insights into alcohol-induced liver and bone pathology, their translational relevance to HOD remains limited due to interspecies differences and ethical constraints [3,17]. Although BMPs share central functions between humans and animal models, there are important differences in the magnitude of response, receptor expression, and specific functional outcomes [18]. Although human in vitro liver–bone co-culture systems have been developed previously, they often lack essential fibrogenic components, such as HSCs, and therefore fail to capture the key pathological features of liver injury [19]. Consequently, the establishment of long-term three-dimensional (3D) co-culture systems that sustain physiological liver–bone interactions and mimic fibrotic progression is urgently required.
In Europe and North America, alcohol-related liver disease represents a leading cause of chronic liver disease and cirrhosis, accounting for nearly half of all cases [20,21,22]. Although the hepatotoxic mechanisms of alcohol have been extensively characterized, the consequences of liver dysfunction on skeletal homeostasis via the liver–bone axis remain poorly understood. Critically, long-term human in vitro systems modeling hepatic BMP dysregulation and its skeletal sequelae are lacking—a pivotal knowledge gap that our study addresses through innovative 3D co-culture models. Our findings elucidate these mechanisms, offering novel insights into hepatic osteodystrophy pathogenesis.
2. Materials and Methods
2.1. Cell Culture
Details of the culture media for all cells are provided in Supplementary Table S1.
HepaRG cells (Biopredic International, Saint Grégoire, France), which are liver progenitor cells able to differentiate into hepatocytes and biliary cells, were maintained in the undifferentiated state in HepaRG Culture medium at 37 °C and 5% CO_2_. During the proliferation phase, HepaRG cells were passaged every 2 weeks. Cell differentiation was induced using 1.7% dimethyl sulfoxide (DMSO; 4720.2, Roth) for another 2 weeks [23]. During HepaRG proliferation and differentiation, medium changes were performed every other day.
The human hepatic stellate cell line, LX-2 (Merck Millipore, Darmstadt, Germany), was cultured in LX-2 culture medium. Cells were maintained in a humidified incubator at 37 °C with 5% CO_2_ [24]. All experiments were conducted using LX-2 cells at passages 5 to 14, with the culture medium refreshed twice weekly.
HUVECs [25] (kindly provided by Prof. Alexander-Friedrich) belonging to the umbilical vein endothelial cell line were cultured in HUVEC culture medium on the 0.1% gelatine-coated flask. All the experiments with HUVECs were performed at passages 7 to 12. The medium was changed twice weekly.
For the human leukemia monocytic cell line, THP-1 (ACC16, DSMZ) cells, as the osteoclastic precursor cells, were cultured as a suspension cell culture in THP-1 culture medium [19]. The cells were maintained in a humidified atmosphere with 5% CO_2_ at 37 °C, and the growing medium was renewed twice weekly.
SCP-1, a mesenchymal stem cell line [26], (kindly provided by Prof. Dr. Matthias Schieker) was used as osteoprogenitor cells and cultured in SCP-1 culture medium at 37 °C in a humidified atmosphere with 5% CO_2_. The growing medium was refreshed twice weekly.
2.2. Chemical and Reagents
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) or Carl Roth (Karlsruhe, Germany).
2.3. Preparation and Sterilization of Agarose Plates
Non-adherent agarose microwell plates were fabricated using mold replication technology, as mentioned previously [27]. Briefly, molten agarose solution (2%, 3.2 mL; Lonza, Basel, Switzerland, 50004) was dispensed into individual wells of a 6-well plate. A polydimethylsiloxane insert (generously provided by Prof. Massoud Vosough), featuring 300 pyramid-shaped microwells with a diameter of 800 µm, was then placed onto the surface. Once the agarose solidified, the insert was removed, creating the desired microwell structure [28]. The plates could be stored at 4 °C for up to 2 weeks if sealed to prevent drying. Before use, the plates were sterilized under ultraviolet light for 1 h.
2.4. Preparation of Human Platelet-Rich Plasma (hPRP) Scaffolds
PRP scaffolds were generated as reported previously [29]. The thoroughly mixed solution was kept on ice and contained 16.0% 2-Hydroxyethyl methacrylate (128635-500G, Sigma), 0.3% Bis-Acrylamide (3039.1, Carl Roth), 0.25 g/L PRP, and ddH_2_O. After 30 min of incubation, di-sodium hydrogen phosphate buffer (T876.1, Carl Roth) was added to the mixture to reach a final concentration of 0.3 M. Subsequently, 0.1% glutaraldehyde (3778.1, Carl Roth), 0.2% Ammonium Persulfate (A3678-25G, Sigma), and 0.2% TEMED (2367.3, Carl Roth) were incorporated into the reaction mixture. The final mixture was transferred to the 6 mm diameter, 3 mm height polystyrene molds (2 mL per mold), then frozen at −20 °C overnight. To facilitate cutting, the molds were transferred at −80 °C for at least half an hour before operation. The resulting scaffolds exhibited uniformity in size, with a height of 4 mm and a diameter of 6 mm. The HEMA-based scaffolds were then transferred to a 1 M CaCl_2_ (CN93.2, Carl Roth) solution and placed on a rotating shaker overnight. Then the scaffolds were transferred into 70% alcohol and incubated overnight on a rotating shaker for at least 12 h. Finally, the scaffolds were washed with sterile Phosphate-Buffered Saline (PBS) 4 times and stored at 4 °C with sterile PBS. Before use, the fabricated scaffolds were placed in a 48-well plate and incubated in THP-1 culture medium for 48 h to verify sterility and ensure proper preconditioning.
2.5. Cell Seeding
2.5.1. Bone Co-Culture System
For the 3D bone co-culture system, THP-1 and SCP-1 cells were seeded onto the PRP scaffold. Following the method previously described [30], THP-1 cells (8 × 10^4^ cells per scaffold, suspended in 15 µL) were applied to the scaffold surface in THP-1 culture medium supplemented with 200 nM phorbol 12-myristate 13-acetate (PMA; Cay10008041, Biomol, Hamburg, Germany). Following a 4 h incubation at 37 °C under humidified conditions with 5% CO_2_, an additional 500 µL of THP-1 culture medium containing 200 nM PMA was added to each scaffold. The following day, the cell culture medium was removed, and SCP-1 cells (1 × 10^4^ cells/15 µL per scaffold) were seeded in bone co-culture differentiation medium. SCP-1 cells were also incubated for 4 h to allow them to adhere, after which, 500 µL of bone co-culture differentiation medium was added to each scaffold [31]. The bone co-culture scaffolds are maintained in a humidified 37 °C environment with 5% CO_2_, with the medium changed twice weekly.
2.5.2. Liver Micro-Organoids
For the 3D liver micro-organoid model, differentiated HepaRG cells, LX-2 cells, and HUVECs were seeded at a ratio of 4:2:1 in pre-prepared agarose microwells within a 6-well plate [28]. To begin, each well was filled with 2 mL of liver micro-organoid culture medium. Then, 1 mL of cell suspension, prepared at a density of 3.0 × 10^5^ cells/mL (1000 cells per microwell) in the same medium, was added to each well. To ensure an even distribution of cells within the microwells, the plate was immediately centrifuged for 3 min at 1200 rpm at room temperature. The spheroids were cultured in a humidified incubator at 37 °C with 5% CO_2_. The liver micro-organoid culture medium was refreshed three times per week.
2.5.3. Development of 3D Human In Vitro Liver–Bone Co-Culture System
When liver micro-organoids had formed, the side of the agarose without cell distribution was hollowed out to create a pocket shape for placing the scaffold. The ratio of liver micro-organoids to bone scaffolds was 300:3 (Figure 1). After combining to form the liver–bone system, the co-culture system was maintained using liver–bone co-culture medium for 28 days [19]. The medium is changed three times per week. The liver–bone co-culture system is cultured in a humidified incubator, maintaining a temperature of 37 °C and 5% CO_2_.
2.5.4. Stimulation of the Liver–Bone System with Alcohol
Absolute alcohol (20821.330, VWR, Radnor, PA, USA) was stored at 4 °C. To prepare alcohol-containing medium, 3, 6, or 12 μL of alcohol was added to 1 mL of complete medium to achieve final concentrations of 50, 100, and 200 mM, respectively. The co-cultured system was stimulated with alcohol daily, and the complete medium was refreshed twice a week. To minimize alcohol evaporation, the culture plates were covered with sealing membranes (391-1262, VWR, Radnor, USA) during incubation.
2.6. Measurement of Mitochondrial Activity
The Resazurin conversion assay was used to measure the metabolic activity of the cells [19]. For the liver micro-organoids, the spheroids were collected from agarose and washed with PBS prior to analysis. Then, 300 µL of 0.0025% resazurin working solution was used for a total of 300 liver micro-organoids, as the background 100 µL resazurin working solution without incubation with cells was used. From this, three 100 µL aliquots of liver micro-organoids in the resazurin working solution were transferred to a 96-well plate. After incubation for 30, 60, 90, and 120 min at 37 °C, the plate reader FLUOstar Omega Plate Reader V. 5.70 (BMG Labtech, Ortenberg, Germany) was used to measure the fluorescence produced by the resorufin at ex/em 544 nm/590–10 nm. For the bone portion, scaffolds were first washed with PBS, followed by the immediate addition of 500 µL of a 0.0025% resazurin solution. Scaffolds without cells served as background controls. After a 2 h incubation at 37 °C, a 96-well plate was filled with 4 × 100 µL of each scaffold sample to allow for the evaluation of the fluorescence by a similar detection method as liver micro-organoids.
2.7. Measurement of UDP-Glucuronosyltransferase (UGT) Activity (Phase II Enzymes)
To assess UGT activity, a fluorescence-based assay using 4-methylumbelliferone (4-MU) was performed. A total of 200 μL of 6.25 μM 4-MU, prepared in HepaRG plain medium, was applied to 300 liver micro-organoids. From this mixture, 100 μL was transferred into the wells of a 96-well plate containing the liver micro-organoids to generate duplicate samples. Additionally, 100 μL of the same working solution was added to a well without cells to serve as a background control [32]. The plate was incubated, and fluorescence was measured at excitation/emission wavelengths of 355 nm/460 nm using an Omega FLUOstar Plate Reader V. 5.70 (BMG Labtech, Ortenberg, Germany) at the following time points: 30, 60, 90, and 120 min. DNA concentration from the liver micro-organoids was quantified and used as a normalization factor.
2.8. Measurement of Alkaline Phosphatase (AP) Activity
AP activity detection was used to evaluate osteogenic differentiation and bone-forming cell function [33]. Scaffolds, washed with PBS, were incubated in 500 µL of AP reaction buffer (1 mM MgCl_2_, 50 mM glycine, 3.5 mM pNPP, and 100 mM Tris buffer; pH 10.5) for 2 h at 37 °C. The conversion of 4-nitrophenyl-phosphate to 4-nitrophenol was quantified photometrically (λ = 405 nm; Omega FLUOstar Plate Reader V. 5.70, BMG Labtech, Ortenberg, Germany) in quadruplicate from 100 µL samples of the AP reaction buffer. The experimental values obtained were adjusted using a background control (consisting of cell-free scaffolds in AP reaction buffer). The data were normalized using the data from the total DNA content of the bone co-culture system [34].
2.9. Measurement of Tartrate-Resistant Acid Phosphatase (TRAP) Activity
To evaluate osteoclast function, the TRAP level (a late-stage marker of osteoclast activity) was quantified [35]. In each assay, 30 µL of supernatant was combined with 90 µL of TRAP activity assay solution (containing 5 mM pNPP, 50 mM disodium tartrate at pH 5.5, and 100 mM sodium acetate) and incubated at 37 °C for 6 h. Subsequently, 90 µL of 1 M NaOH per well was added to inhibit further enzymatic activity. The reaction product, 4-nitrophenol, was quantified at 405 nm using a photometer. The experimental values were adjusted by subtracting the background control, which consisted of scaffolds without cells in the TRAP activity assay solution, and the data were normalized to the DNA content of the bone co-culture system [34].
2.10. DNA Isolation and Quantification
DNA measurements were performed to assess cell number and to normalize enzymatic activity values. For the liver micro-organoids, samples were first collected from the agarose microplate, washed with PBS, and then treated with 100 µL of 98 °C 50 mM NaOH. After vortexing, samples were stored at −20 °C for at least 24 h. The next day, they were thawed for 30 min (98 °C) and mixed with 110 μL of 0.1 M Tris (pH 8.0), and 100 µL of the mixture was transferred into a 96-well plate (V-type) and centrifuged at 20,000× g for 10 min. For the bone portion, bone scaffolds were washed with PBS, and 250 µL of 98 °C 50 mM NaOH was added to each scaffold. After 5 min of incubation, the scaffolds were frozen at −20 °C for at least 24 h. The next day, 275 μL of 0.1 M Tris (pH 8.0) was added to the thawed samples. The mixture was mixed thoroughly, transferred to a 96-well plate (V-type), and centrifuged at 20,000× g for 10 min. To determine the DNA concentration, absorption was measured at λ = 260 nm for all samples using a BMG Labtech LVIS, and the data were processed with Omega analysis software MARS software 3.42 [19].
2.11. Intracellular Lactate Dehydrogenase (LDH) Activity
LDH release into the culture medium is a widely used indicator of cell membrane damage [36]. However, in our in vitro liver–bone co-culture system, it is not feasible to distinguish the cellular origin of LDH detected in the supernatant, since both liver micro-organoids and bone-derived cells contribute to the extracellular LDH pool. To specifically assess intracellular LDH activity, assessment via CytoTox-ONE (Promega, Madison WI, USA) was performed in each tissue type. Liver micro-organoids and the bone system were collected separately and subjected individually to lysis with 1% Triton X-100 to fully release intracellular LDH. Subsequently, 50 μL of each lysate was mixed with 50 μL of LDH reaction working solution, and absorbance was measured at 490 nm using a FLUOstar Omega plate reader V. 5.70 (BMG Labtech, Ortenberg, Germany). The background absorbance, measured from wells without cells containing the LDH reaction working solution and Triton X-100, was subtracted from all sample values.
2.12. Luciferase Reporter Assay
To evaluate the nuclear activation of the BMP signaling pathway, a luciferase reporter assay was conducted using an adenoviral vector carrying a luciferase reporter gene under the control of a BMP-responsive element (Ad-BRE). SCP-1 cells were seeded into 96-well plates and infected with Ad-BRE for 24 h. After infection, cells were washed with PBS, then treated with the supernatant from liver micro-organoids stimulated by 50 mM alcohol. Following 48 h of stimulation, lysis buffer was added to each well to lyse the cells. The plates were subsequently stored at −80 °C for at least 1 h. Then, 30 μL of cell lysate was collected from each well and mixed with an equal volume (30 μL) of luciferase assay reagent. Luciferase activity was performed according to the manufacturer’s instructions, using the Steady-Glo Luciferase Assay System (Promega, Madison, WI, USA), and normalized to total protein content, measured with Lowry [37].
2.13. Semi-Quantitative Reverse-Transcription Polymerase Chain Reaction (RT-PCR) Analysis
Gene expression levels were determined by RT-PCR [28]. Total RNA was isolated from liver micro-organoids and bone system samples using TriFast reagent (Peqlab, Erlangen, Germany), and RNA concentration was determined with an Omega FLUOstar Plate Reader V. 5.70 (BMG Labtech, Ortenberg, Germany). Complementary DNA (cDNA) was generated using a commercial reverse transcription kit (Thermo Fisher Scientific, Waltham, MA, USA). Then, PCR amplification was performed according to the manufacturer’s instructions for Biozym Red HS Taq Master Mix (Vienna, Austria). Primer sequences and PCR conditions for target genes are shown in Supplementary Table S2; 18S rRNA served as a housekeeping gene. PCR products were resolved on 1.8% agarose gels containing ethidium bromide and separated by electrophoresis at 90 V for 60 min. Band intensities were subsequently quantified using ImageJ software V. 1.54G (NIH, Bethesda, MD, USA).
2.14. Dot Blot Analysis
Secreted proteins in culture supernatant were detected by dot blot [19]. Using a 96-well dot blotter (Carl Roth, Karlsruhe, Germany), 100 µL of supernatant was transferred onto a wet nitrocellulose membrane with a vacuum pump. Ponceau S staining was used for visualization and quantification of total proteins. After being blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline/Tween 20 (TBS-T, 9127.1, Carl Roth, Karlsruhe, Germany) for 1 h, membranes were incubated with primary antibodies at 4 °C overnight. The antibodies used are summarized in Supplementary Table S3. After washing with TBS-T, membranes were exposed to the appropriate secondary antibodies for 2 h. Protein signals were visualized using a chemiluminescence detection system consisting of 100 mM TRIS, 250 mM luminol (Carl Roth, Karlsruhe, Germany), 90 mM p-coumaric acid (Carl Roth, Karlsruhe, Germany), and 30% (v/v) H_2_O_2_ (Carl Roth, Karlsruhe, Germany). Signal intensities were quantified using ImageJ software.
2.15. Western Blot Analysis
The samples were lysed in a freshly prepared RIPA buffer supplemented with protease and phosphatase inhibitors. After quantification with Lowry, proteins (50 µg per sample) were separated by performing sodium dodecyl sulfate–polyacrylamide gel (SDS–PAGE), according to their molecular weight, and transferred to nitrocellulose membranes. Ponceau S staining was used for the visualization of total proteins. After blocking in 5% BSA in TBS-T for 1 h, the membranes were incubated at 4 °C overnight with the primary antibody; the antibodies used are summarized in Table S3. The following day, the membranes were washed by TBS-T, then secondary antibodies were applied for 1 h at room temperature and washed with TBS-T again. For signal development, they were detected by chemiluminescence and quantified by ImageJ [28].
2.16. Mineral Content of Bone Scaffold
The mineral content of the bone scaffold was measured using quantitative CT scans on a clinical CT scanner with 128-slice capability. Scanning was conducted with the following settings: 80 kV tube voltage, effective tube current of 500 mA, 16 × 0.3 mm acquisition with a slice thickness of 0.4 mm, and a pitch of 0.4. The images were then reconstructed using a high-resolution V80u kernel and iterative image reconstruction technology at grade 5, and displayed in a bone window. The rectangular axial field-of-view was approximately 10 cm × 10 cm with a matrix resolution of 512 × 512 pixels. The resulting digital imaging and communications in medicine (DICOM) images were imported into ImageJ software via the “DICOM sort” plugin, and the stack was cropped to focus on the region of interest. The mean integrated density for each scaffold was calculated and normalized against the reference block (Phantom EFP-06-96) [38].
2.17. Stiffness of Bone Scaffold
Scaffold stiffness was calculated using the Young’s modulus method [19]. The 4 mm scaffolds were compressed four times uniaxially by 10% (speed = 5 mm/min) of the original height by using a ZwickiLine Z 2.5TN material testing machine (ZwickRoell GmbH & Co. KG, Ulm, Germany). An X-force HP 5N sensor was used to measure the applied load in real-time. The load-deformation data were then converted into a stress–strain curve using the height and area of the uncompressed scaffold. Young’s modulus for the region of linear elastic deformation was determined through the following calculation: Young’s modulus [MPa] = (applied force [N] × initial scaffold height [mm])/(area of the scaffold [mm^2^] × change in height [mm]).
2.18. Statistical Analysis
The data are presented as means ± the standard error of the mean (SEM). All the experiments were repeated at least three times (N) with two or three technical replicates (n). Statistical analyses were performed using GraphPad Prism software (GraphPad Software 8.0, La Jolla, CA, USA). The data of the two groups were compared with the Mann–Whitney test. The data of multiple groups were compared with the non-parametric Kruskal–Wallis test, followed by Dunn’s multiple comparison test. A two-way ANOVA test followed by Tukey’s multiple comparisons was used when two independent variables were compared among groups. A p < 0.05 was considered statistically significant.
3. Results
3.1. Long-Term Viability and Functional Maintenance of In Vitro Liver Micro-Organoids for at Least 21 Days
To evaluate the long-term stability of the liver micro-organoids, functional and viability parameters were monitored over a 21-day culture period. Mitochondrial activity, as measured by resazurin reduction, increased continuously throughout the culture (p = 0.0454) (Figure 2a), reflecting sustained cellular viability. DNA content also increased significantly by day 14 (p = 0.0263) and remained stable thereafter (p = 0.0105), indicating cell proliferation and maintenance (Figure 2b). For functional assays, phase II enzyme activity, as assessed by UGT function, increased between days 7 and 14 (p = 0.0484) and remained stable until day 21 (p = 0.0366) (Figure 2c). Simultaneous measurement of cytochrome P450 basal enzyme activity revealed dynamic changes in enzyme specificity. CYP1A2 activity peaked on day 14 (Figure 2d), while CYP3A4 activity gradually decreased after day 7 but remained at a high level overall compared to levels detected in micro-organoids containing only HepaRG cells [19] (Figure 2e). In contrast, CYP2C9 activity increased steadily throughout the culture period (Figure 2f). Overall, these results indicate that the liver micro-organoids model maintains basic viability and hepatic function over 21 days, supporting its use in long-term in vitro studies.
3.2. Induction of Alcohol-Metabolizing Enzyme and Fibrogenic Markers Following Alcohol Exposure in In Vitro Liver Micro-Organoids
With the long-term functionality of the liver micro-organoids model established, we then proceeded to assess the alcohol metabolization capacity of liver micro-organoids and the expression of fibrotic markers after daily exposure to alcohol. An alcohol concentration of 50 mM was chosen to stimulate the micro-organoids daily, as this is equivalent to the alcohol intake of four bottles (330 mL) of beer [39], which is considered excessive alcohol consumption. Additionally, daily 50 mM alcohol exposure showed no cytotoxicity on liver micro-organoids (Supplementary Figure S1). After daily 50 mM alcohol treatment, the expression of CYP2E1, which is a key alcohol-metabolizing enzyme in the liver, was significantly increased at both mRNA (p = 0.0022) and protein levels (p < 0.0001) (Figure 3a,c). Concurrently, the mRNA level of fibroblast activation protein α (Fapα), a well-established marker of hepatic stellate cell activation, was significantly upregulated (p = 0.0002) (Figure 3b), indicating that LX-2 cells were activated under alcohol stimulation. In addition, alcohol treatment resulted in a significant increase in Transforming Growth Factor Beta 1 (TGF-β1) protein levels (p < 0.0001) (Figure 3d), further supporting the initiation of a profibrotic response. These findings suggest that alcohol exposure activates metabolic and profibrotic signaling pathways in the liver micro-organoids.
3.3. Alcohol Promotes EMT-Regulating Gene Expression, Matrix-Associated Protein Release, and Liver Damage Markers in Liver Micro-Organoids
Previous results have shown that alcohol can be metabolized in the liver micro-organoids and induce the activation of stellate-like cells. To further investigate whether alcohol metabolism in the liver micro-organoids led to pathological remodeling, we examined the expression of epithelial–mesenchymal transition (EMT) markers and extracellular matrix (ECM) related proteins. Gene expression analysis showed that both Snail1 and Snail2 were significantly upregulated (p = 0.0022) after 21 days of 50 mM daily alcohol stimulation (Figure 4a,b), indicating that EMT-related transcriptional programs were activated. Meanwhile, ECM remodeling was assessed by measuring key matrix-associated proteins in the culture supernatant. The levels of type I procollagen N-terminal propeptide (PINP) in the alcohol-treated group were significantly increased at 21 days (p = 0.0379) (Figure 4c). In contrast, the matrix-degrading enzyme matrix metalloproteinase 9 (MMP9) was significantly decreased after alcohol exposure (p = 0.0286) (Figure 4e), indicating a decrease in matrix turnover and a shift towards fibrotic matrix accumulation. Additionally, daily alcohol exposure significantly increases the levels of tissue-nonspecific alkaline phosphatase (TNAP) (p = 0.0411) (Figure 4d) on liver micro-organoids, indicating injury. These findings, therefore, demonstrate that alcohol exposure in the liver model induces early fibrotic remodeling characterized by EMT activation, altered ECM homeostasis, and liver damage.
3.4. Alcohol Does Not Significantly Affect Bone Co-Culture System Function
Before combining the liver and bone system together, we first need to clarify the direct effects of alcohol on the bone co-culture system. We continuously exposed the bone co-culture system to 50 mM alcohol for up to 28 days. Direct exposure to 50 mM alcohol does not affect bone metabolic activity (Supplementary Figure S2). Regarding function, we examined osteoblast activity, which was assessed by AP activity and showed no difference between the alcohol-treated group and the control group on day 21 (Figure 5a). Similarly, osteoclast activity, measured by TRAP assay, did not differ significantly between the control and alcohol-treated groups at the same time point (Figure 5b). This suggests that direct alcohol exposure does not affect the osteogenic and osteoclast functions of the bone co-culture system. To further evaluate the functional outcome of these cellular changes, we measured stiffness and mineral density of the bone scaffolds after 28 days of direct alcohol exposure. Both parameters showed comparable values between the control and alcohol-treated groups (Figure 5c,d). In summary, these findings indicate that direct exposure of the bone co-culture system to 50 mM alcohol did not significantly alter osteoblast and osteoclast activity and function, nor did it significantly affect the overall mechanical properties of the bone scaffold.
3.5. Alcohol Exposure Weakens Bone Quality by Inhibiting Mineralization in Liver–Bone Co-Culture System
Following the confirmation of liver and bone system metabolic activity under alcohol exposure (Supplementary Figure S3), we then assessed the effects of an alcohol-induced pathological liver on bone metabolism. We analyzed key osteoblast and osteoclast markers in the liver–bone co-culture system at day 21. The AP activity was significantly reduced in the alcohol group (p = 0.0249) (Figure 6a), indicating impaired bone formation. TRAP activity (Figure 6b) did not differ significantly between the two groups, indicating that osteoclast activity remained stable. Then, we measured scaffold stiffness and bone mineral density. Compared with the control group, 50 mM alcohol treatment significantly reduced the stiffness (p = 0.0082) (Figure 6c) and the mineral density (p = 0.0117) of the bone scaffold (Figure 6d) after continuous 28-day exposure. To further investigate the effects of alcohol on the bone co-culture system, the PINP protein level in supernatant, a marker of bone collagen synthesis (Figure 6f), was also measured and demonstrated a reduction in the alcohol group (p < 0.0001), possibly reflecting the disruption of bone remodeling. The N-terminal telopeptide (NTX) (Figure 6g), a product of collagen degradation that plays a crucial role in bone remodeling and can be used as an indicator of osteoclast activity, showed no significant difference between the two groups. These findings suggest that alcohol-induced liver disease may negatively affect bone metabolism by inhibiting osteoblast function and maintaining osteoclast activity, ultimately leading to impaired bone homeostasis, as demonstrated by a decrease in bone mineral density and bone scaffold stiffness.
3.6. Alcohol Exposure Downregulates BMP2 and Upregulates BMP13 in Liver Micro-Organoids
Based on previous results demonstrating that the deleterious effects of alcohol on bone are predominantly associated with impaired liver function—affecting primarily bone-forming cells—we then investigated how alcohol influences the hepatic secretion profile of BMPs. BMPs were selected for analysis because of the importance of BMP signaling in bone remodeling and osteogenic differentiation, and the existing evidence indicates that their expression patterns are altered under fibrotic conditions [40], suggesting a potential mechanistic link between liver dysfunction and changes in bone homeostasis mediated by these signaling molecules. Since the liver has been reported to secrete BMP2, BMP9, and BMP13 [11,13,41], we measured the BMP protein levels in the culture supernatants. BMP2 levels were significantly decreased in the alcohol group compared to the control (p = 0.026) (Figure 7a), while BMP13 levels were significantly increased (p = 0.026) (Figure 7c). No significant difference was observed in BMP9 levels between the two groups (Figure 7b). These results suggest that alcohol exposure induces selective alterations in BMP ligand secretion, characterized by reduced BMP2 (promoter of osteogenesis) and increased BMP13 (inhibit osteogenesis) levels, which could shift the balance of progenitor differentiation, impacting bone homeostasis.
3.7. Hepatic BMPs Trigger Bone BMP Pathway Activation with Downstream Nuclear Signaling
To investigate whether BMP signaling can be properly activated in bone-forming progenitor cells, we examined Smad-dependent and Smad-independent pathways (Figure 8a). Over time, the level of p-Smad1/5 exhibited an overall decreasing trend, while p-P38 showed a progressive increase. When comparing between groups, both p-Smad1/5 and p-P38 levels were consistently higher in the alcohol group than in the control group at all time points (Figure 8b). Quantitative analysis (Figure 8c) further confirmed that p-Smad1/5 levels were elevated in the alcohol group across all time points, especially on day 7 (p < 0.0001), whereas p-P38 levels (Figure 8d) demonstrated a time-dependent increase, becoming significantly higher than control at day 21 (p = 0.0166). To assess whether alcohol-induced liver-derived factors activated BMP signaling at the nuclear level, we performed a luciferase reporter assay using an adenoviral BMP-responsive element reporter construct (Ad-BRE-Luciferase) in SCP-1 cells. As shown in Figure 8e, luciferase activity was significantly increased in cells treated with the conditioned medium collected from liver micro-organoids exposed to 50 mM alcohol for 24 h compared with cells treated with culture medium from the control group (p = 0.0022). This suggests that alcohol-exposed liver micro-organoids secrete BMPs or BMP-inducing factors, which subsequently activate the BMP signaling pathway in SCP-1 cells. The downstream effectors of this pathway translocate to the nucleus of target cells, regulating the differentiation lineage of bone-forming progenitor cells.
3.8. Alcohol Impairs Osteogenic Differentiation by Downregulating RUNX2 and Modulating Lineage-Associated Transcription Factors
Since altered secretion levels of BMP2 and BMP13 promoters of osteogenesis and chondrogenesis [16,42], respectively, were detected in the supernatant of liver micro-organoids treated with alcohol, and BMP signaling was shown to occur appropriately in bone-forming progenitor cells, we sought to elucidate the differentiation potential of these progenitors. To achieve this, we analyzed the expression levels of master transcriptional factors involved in osteogenic, adipogenic, and chondrogenic lineages. After 7 days of co-culture under alcohol treatment, RNA was extracted from SCP-1 cells to assess early lineage commitment. Gene expression analysis revealed a significant downregulation of RUNX2, a master regulator of osteogenesis, in the alcohol group compared to control (p = 0.0315) (Figure 9a). Similarly, PPARγ, a transcription factor associated with adipogenic differentiation, was also markedly reduced (p = 0.0188) (Figure 9b). In contrast, SOX9, a key transcription factor for chondrogenic lineage, showed a significant increase in the alcohol-treated group (p = 0.0244) (Figure 9c), suggesting a possible shift in differentiation potential. These results suggest that signals derived from alcohol-exposed liver micro-organoids may disrupt osteogenic and adipogenic programming in SCP-1 cells, while favoring chondrogenic tendencies.
4. Discussion
In this study, we successfully developed a long-term in vitro model that mimics alcohol-induced HOD using a 3D co-culture system that integrates liver micro-organoids and bone scaffolds [19]. Now, numerous in vitro studies have investigated the direct effects of alcohol on bone cells, primarily focusing on the inhibitory impact of alcohol on osteoblast proliferation, differentiation, and activity, which ultimately contributes to reduced bone formation and an increased risk of osteoporosis. These studies consistently demonstrate alcohol’s deleterious effects on bone-forming cells, including the induction of oxidative stress, cellular senescence, and impaired mineralization [43,44,45]. However, despite these insights, current in vitro models remain limited as they neglect the potential systemic influence of the liver—a key organ in both alcohol metabolism and the regulation of bone homeostasis. The liver produces and secretes multiple endocrine and paracrine factors, such as BMPs, fibroblast growth factor 21, and TGFβ, all of which critically modulate bone remodeling and cellular function [46,47]. The consideration of liver-derived factors is essential for a comprehensive understanding of alcohol-related skeletal disorders. Integrating hepatic components into bone models enables the investigation of systemic liver–bone interactions and provides mechanistic insight into how hepatic injury contributes to bone deterioration. Our findings highlight the central role of the BMP signaling pathway in mediating alcohol-induced liver–bone crosstalk and suggest that its dysregulation may represent a core mechanism underlying HOD pathogenesis.
In terms of structure, incorporating LX-2 cells and HUVECs into the liver model substantially improves its physiological and pathological relevance compared to HepaRG-only organoids. HepaRG cells supply essential metabolic activity for xenobiotic processing [48] but lack the fibrogenic and vascular components needed to mimic chronic liver disease. Adding LX-2 cells introduces a fibrotic element through extracellular matrix production and pro-fibrogenic signaling [28,49], while the presence of HUVECs enhances vascularization, nutrient exchange, and overall tissue organization [28,50]. Unexpectedly, the liver organoid showed reductions in CYP1A2 and CYP3A4 protein levels, coupled with elevated CYP2C9 activity, which could be explained by the direct protein–protein interactions among these enzymes. Specifically, CYP3A4 engages in heteromeric complex formation with CYP2C9 through hydrophobic interactions at their N-terminal transmembrane domains, potently suppressing CYP2C9 catalytic activity. Dose evidence from CYP3A4 knockdown in human hepatocytes demonstrates that lowering CYP3A4 abundance (~60% reduction) relieves this inhibition, enhancing CYP2C9 activity (~74% increase) independently of changes in CYP2C9 mRNA expression; this effect persists even at saturated cytochrome P450 reductase (CPR) levels, implicating physical docking rather than mere CPR competition. CYP1A2 downregulation parallels CYP3A4 trends and may arise from analogous P450-P450 heteromerization or study-specific factors like oxidative stress, inflammation, or xenobiotic induction patterns, which disproportionately suppress CYP1A2 relative to other isoforms [51,52].
Building upon this framework, selecting an appropriate pathological stimulus is essential for effectively modeling disease-relevant interactions within the liver–bone axis. Alcohol serves as a particularly valuable model for inducing liver fibrosis in the context of HOD research, as it closely replicates the multifactorial, progressive, and systemic nature of human alcoholic liver disease (ALD) [53]. Unlike acute or artificially induced fibrosis models—such as those generated by carbon tetrachloride (CCl_4_) or TGF-β1 administration [28,54]—chronic alcohol exposure-induced hepatic injury not only triggers stellate cell activation and extracellular matrix accumulation but also impairs hepatic endocrine and metabolic function, leading to secondary systemic effects, such as malnutrition, hormonal dysregulation, and low-grade inflammation [55,56]. These mechanisms more accurately reflect the chronic pathological processes observed in patients with ALD and their systemic consequences, including alterations in bone metabolism. By contrast, the CCl_4_ model primarily induces direct hepatocyte toxicity and acute fibrotic scarring, lacking the chronic metabolic and immunological components integral to ALD [54,57]. Similarly, TGF-β-driven fibrosis effectively stimulates fibrogenic signaling but fails to reproduce the nutritional, endocrine, and inflammatory perturbations that profoundly influence bone homeostasis in alcohol-related disease [28]. Moreover, chronic alcohol consumption impairs bone matrix quality through both direct and indirect mechanisms. Directly, ethanol suppresses osteoblast differentiation by inhibiting Runx2/Wnt signaling, thereby reducing the synthesis of essential matrix proteins, such as type I collagen, osteocalcin, and osteopontin, while promoting mesenchymal stem cell differentiation into adipocytes, which causes fatty marrow infiltration and defective mineralization. Ethanol also enhances osteoclastogenesis via RANKL upregulation, increasing bone resorption. Indirectly, alcohol-induced liver dysfunction disrupts vitamin D activation and IGF-1 production—critical regulators of osteoblast function and matrix deposition—leading to reduced growth factor availability in bone and greater matrix fragility. Furthermore, alcohol promotes hypocalcemia through intestinal malabsorption and vitamin D deficiency, compounded by hypercortisolemia and hypogonadism, which lower estrogen/testosterone levels necessary for bone maintenance, ultimately shifting remodeling toward net resorption and accelerating osteopenia.
Accordingly, we combined the liver and bone components to generate a liver–bone co-culture system, examining how alcohol-induced liver fibrosis perturbs bone homeostasis. Previous studies have reported divergent mechanisms underlying alcohol-induced bone loss. Some suggest that alcohol promotes osteoclastogenesis via RANKL-mediated pathways, often as a result of elevated inflammatory signaling [58,59]. By contrast, other studies emphasize that the dominant effect of alcohol is the suppression of osteoblast differentiation and function, rather than a direct enhancement of osteoclastic activity [60,61]. Our findings are consistent with the latter mechanism: despite prolonged alcohol exposure, osteoclast function remained unchanged, while osteoblast activity, mineral deposition, and RUNX2 expression were significantly reduced. These results underscore the importance of targeting osteoblast dysfunction in future therapeutic strategies for alcohol-related bone disease. However, several factors may contribute to the lack of observable osteoclast activation in our system. As shown in previous studies, inflammatory factors are the main cause of osteoclast functional alterations. However, our liver micro-organoids lack Kupffer cells and therefore cannot produce large amounts of inflammatory cytokines in response to external stimuli as a normal liver does. The absence of Kupffer cells underestimates inflammatory contributions to the liver–bone axis; their omission limits recapitulation of macrophage-driven fibrogenesis observed clinically. Future studies need to incorporate Kupffer cells or iPSC-derived macrophage co-cultures to comprehensively model innate immunity’s role in HOD. Furthermore, alcohol-induced changes in osteoclast function may be a secondary consequence of impaired osteoblast activity rather than a direct effect, and such changes might require prolonged exposure, higher alcohol concentrations, or the presence of inflammatory mediators to manifest [62].
This selective suppression of osteogenesis, in the absence of overt osteoclast activation, prompted us to prioritize pathways governing mesenchymal lineage allocation—most notably the BMP axis [63]. BMPs exert multiple effects in promoting the differentiation of bone mesenchymal stem cells (BMCs) toward osteogenic, adipogenic, or chondrogenic lineages [64,65]. The cellular and therapeutic actions of BMPs are mediated through downstream signaling cascades that are initiated when BMPs bind to transmembrane serine/threonine kinase receptors. This interaction activates specific intracellular signaling pathways—primarily the SMAD-dependent and SMAD-independent routes—that regulate transcription programs involved in cell differentiation and tissue development [66]. Previous studies have demonstrated that during pathological liver remodeling, the expression and secretion of several BMP family members, including BMP2, BMP9, and BMP13, are markedly altered [13,41,67]. Notably, BMP2 expression tends to decrease following hepatic fibrosis. This reduction may be attributed to the loss of hepatocyte function and the replacement of parenchymal tissue with extracellular matrix components, which limits the capacity of the liver to produce osteogenic cytokines [67]. In contrast, BMP9, which is mainly produced by hepatic sinusoidal endothelial cells, often exhibits a more complex regulatory pattern—its expression may transiently increase during early injury to support vascular remodeling but subsequently decline as fibrosis progresses [12]. BMP13, another member associated with fibrogenic processes, has been reported to be upregulated in activated hepatic stellate cells, suggesting its potential involvement in extracellular matrix deposition and tissue stiffness regulation [13]. These differential changes in BMP expression imply that liver pathology may not only alter local hepatic signaling but also influence systemic BMP availability. Since BMP2, BMP9, and BMP13 are key regulators of BMCs differentiation, such hepatic alterations could indirectly affect bone remodeling and regeneration. Our findings, therefore, highlight a possible link between liver injury-induced BMP dysregulation and bone homeostasis under pathological conditions. Our results suggest that alcohol disrupts bone homeostasis by differentially regulating BMP secretion (↓BMP2 and↑BMP13). Although BMP2 and BMP13 (GDF6) both activate the Smad1/5/8 pathway via type I BMP receptors, they differ in receptor affinity and functional outcomes. BMP2 binds both ALK3 and ALK6 with similar affinity, promoting robust osteogenic differentiation across cell types [68,69]. In contrast, BMP13 preferentially binds ALK6 [69,70], limiting its signaling to specific cells and favoring tenogenic or chondrogenic programs. By competing for type II receptors without efficiently activating ALK3, BMP13 may also antagonize BMP2 activity. These distinctions in receptor selectivity, target activation, and tissue distribution provide a molecular basis for cell fate divergence.
Compared with existing studies, several in vivo studies have reported that alcohol exposure can impair BMP2 function by inhibiting BMP receptor activity [67]. Furthermore, some studies have demonstrated that the overall expression level of BMP2 is significantly decreased during the development of fibrosis [11,71]. Alcohol-induced inhibition of BMP2 may not only hinder liver tissue repair but also disrupt bone remodeling, contributing to decreased bone density through the liver–bone axis. In contrast, BMP13 appears to be upregulated during liver fibrosis, although relevant studies remain limited [41,72]. Consistent with these reports, our model also showed similar BMP secretion patterns, supporting its reliability. Further analysis of protein-level signaling revealed that phosphorylated Smad1/5 (p-Smad1/5) levels increased significantly on day 7, coinciding with the upregulation of SOX9. These results suggest that alcohol may initiate chondrogenic gene programs via the BMP13–Smad1/5–SOX9 signaling axis, rather than promoting osteogenic differentiation through the SMAD-dependent BMP2–RUNX2 pathway. In contrast, phosphorylated p38 (p-p38) expression did not show a significant increase on day 7 but gradually rose by day 21, suggesting it may function as a regulatory factor in maintaining SOX9 expression or promoting chondrocyte maturation at later stages. Although we did not observe a change in total BMP9, prior studies indicate that biological activity depends on the proportion of dimeric, disulfide-stabilized forms [73]. Chronic alcohol exposure can perturb protein folding pathways in hepatocytes and might thereby influence BMP9 bioactivity despite stable abundance.
The temporal dynamics of the BMP-signaling-pathway components revealed distinct regulatory patterns in our system. The SMAD-dependent component (p-Smad1/5) declined over time in both groups, consistent with its early role in lineage commitment and subsequent downregulation during cell maturation [74]. However, its levels are downregulated as cells enter maturation and matrix production phases. In contrast, p-p38 levels increased over time and was further enhanced by alcohol, likely reflecting stress and inflammatory responses in the liver–bone co-culture, since this SMAD-independent BMP branch is known to respond to oxidative stress and inflammatory cytokines [75]. Since p-p38 can promote SOX9 expression, this shift may redirect differentiation toward the chondrogenic lineage [76]. Overall, alcohol appears to suppress osteogenic signaling while activating stress-related BMP pathways, contributing to impaired MSC differentiation and bone loss.
Both osteogenesis and adipogenesis are metabolically demanding processes that rely heavily on mitochondrial integrity and oxidative phosphorylation [77]. Alcohol metabolism, primarily via hepatic CYP2E1 activity, leads to the generation of reactive oxygen species, which impairs mitochondrial membrane potential, ATP production, and redox balance [78,79]. These metabolic disruptions can inhibit master transcription factors such as RUNX2 and PPARγ, thereby suppressing osteoblast and adipocyte lineage commitment. In contrast, chondrogenesis, regulated by SOX9, is comparatively less dependent on mitochondrial function and is often upregulated under hypoxic, low-energy, or inflammatory conditions. Moreover, systemic inflammatory signals from the liver, including elevated IL-6 and TNF-α, may selectively inhibit osteogenic and adipogenic programs while permissively supporting fibrocartilage differentiation [80,81,82]. Additionally, the antagonistic interaction between SOX9 and RUNX2 may further amplify this fate divergence, as the inhibition of osteogenesis can indirectly favor chondrogenic dominance [83]. The enhanced chondrogenesis may reflect a stress-adaptive response, as mesenchymal stem cells under oxidative or inflammatory stress tend to favor the chondrogenic lineage for its lower energy demand and protective matrix production [78].
These mechanistic insights have important implications for the treatment of alcohol-induced bone disorders, particularly HOD, where conventional bone-targeted therapies may not sufficiently address the underlying systemic drivers. While standard approaches to treating bone loss typically involve the use of osteoanabolic agents or antiresorptive drugs [84]—such as parathyroid hormone analogs, vitamin D supplementation, or bisphosphonates—our findings suggest that such interventions may be insufficient in the context of HOD. In this condition, bone dysfunction is primarily driven by chronic liver injury and its systemic consequences, including altered cytokine secretion profile, oxidative stress, and metabolic imbalances. Therefore, achieving long-term improvement in bone health requires addressing the underlying hepatic pathology. Therapeutic strategies aimed at reducing liver inflammation and oxidative stress—such as antioxidants and anti-inflammatory agents—may help limit the production of bone-damaging mediators [85]. Additionally, restoring hepatocyte function and promoting hepatic repair could indirectly support bone remodeling by rebalancing liver–bone signaling axes, including IGF-1, HGF, and FGF pathways.
A key strength of this study is the novel 3D liver–bone co-culture system, which captures inter-organ BMP signaling and pathological crosstalk unattainable in monoculture or single-organoid models. However, the 28-day alcohol exposure, while physiologically relevant (50 mM ≈ 4 beers/day), cannot fully replicate decades-long human consumption patterns. Additionally, the static in vitro setup lacks mechanical loading, vascular perfusion, and systemic endocrine/hormonal influences present in vivo, which should be considered when extrapolating these findings to clinical hepatic osteodystrophy. Future studies incorporating dynamic bioreactors and multi-organ chips will be valuable to address these gaps.
5. Conclusions
In summary, we successfully established a long-term human in vitro liver–bone co-culture model that mimics key features of alcohol-induced HOD. We established a physiologically relevant, long-term human 3D in vitro liver–bone co-culture system that integrates key hepatic cell types (hepatocytes, hepatic stellate cells, and endothelial cells) with bone cell types (osteoblasts and osteoclasts). This model mimics the dynamic and reciprocal interactions between the liver and bone under chronic alcohol exposure. Compared to previous studies, it offers enhanced control over cellular composition, microenvironment, and exposure duration, making it well-suited for investigating the mechanistic basis of HOD. Furthermore, this platform offers a unique opportunity to investigate inter-organ signaling mechanisms, with a particular focus on BMP-mediated communication, which plays a central role in osteoblast differentiation. Our findings highlight the central role of BMP imbalance—specifically, the downregulation of BMP2 and upregulation of BMP13—in mediating the shift from osteogenic to chondrogenic lineage commitment. This mechanistic insight, coupled with the controllability and translational potential of our in vitro platform, provides a valuable tool for studying inter-organ metabolic crosstalk in CLD. In addition, this model aligns with the 3R principles by reducing dependence on animal experiments and offering an ethical, scalable platform for therapeutic screening.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ehnert S. Aspera-Werz R.H. RuoßM. Dooley S. Hengstler J.G. Nadalin S. Relja B. Badke A. Nussler A.K. Hepatic Osteodystrophy-Molecular Mechanisms Proposed to Favor Its Development Int. J. Mol. Sci.201920255510.3390/ijms 2010255531137669 PMC 6566554 · doi ↗ · pubmed ↗
- 2Collier J. Bone disorders in chronic liver disease Hepatology 2007461271127810.1002/hep.2185217886334 · doi ↗ · pubmed ↗
- 3Lu K. Shi T.S. Shen S.Y. Shi Y. Gao H.L. Wu J. Lu X. Gao X. Ju H.X. Wang W. Defects in a liver-bone axis contribute to hepatic osteodystrophy disease progression Cell Metab.202234441457.e 710.1016/j.cmet.2022.02.00635235775 · doi ↗ · pubmed ↗
- 4Gao H. Peng X. Li N. Gou L. Xu T. Wang Y. Qin J. Liang H. Ma P. Li S. Emerging role of liver-bone axis in osteoporosis J. Orthop. Transl.20244821723110.1016/j.jot.2024.07.008PMC 1140791139290849 · doi ↗ · pubmed ↗
- 5Osna N.A. Donohue T.M.Jr. Kharbanda K.K. Alcoholic Liver Disease: Pathogenesis and Current Management Alcohol Res.20173814716110.35946/arcr.v 38.2.0128988570 PMC 5513682 · doi ↗ · pubmed ↗
- 6Nagarjuna D. Karthikeyan E. Alcohol-associated liver disease: A review Gastroenterol. Endosc.20253658510.1016/j.gande.2025.01.003 · doi ↗
- 7World Health Organization. Regional Office for Europe Alcohol Taxation and Pricing Policies and Their Impact on Alcohol Consumption in Ukraine from 2011–2021: Country Report World Health Organization Geneva, Switzerland 2024
- 8World Health Organization Global Information System on Alcohol and Health. Levels of Consumption Available online: https://www.who.int/data/gho/data/themes/topics/topic-details/GHO/levels-of-consumption(accessed on 22 January 2026)