A population‐scale multi‐omics blueprint of immune gene regulation
Antonio Benedetto Ventura, Lucia Deligio, Giacomo Volpe

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
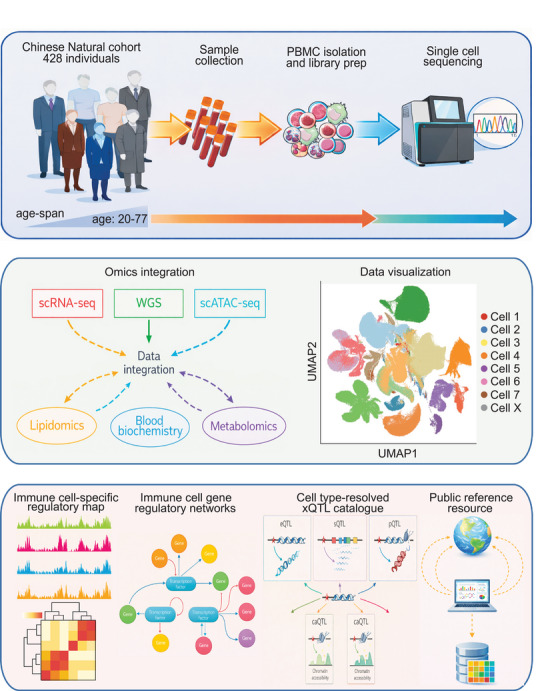 Figure 1
Figure 1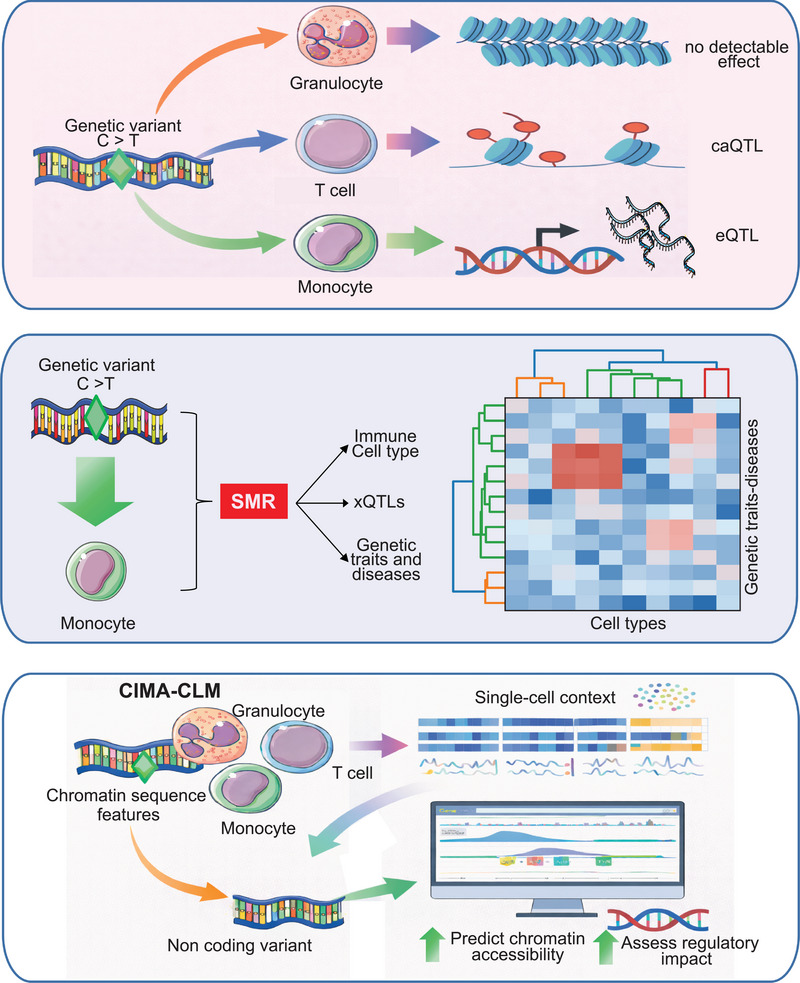 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSingle-cell and spatial transcriptomics · Genetic Associations and Epidemiology · Ferroptosis and cancer prognosis
A central challenge in immunology and human genetics is the understanding of how genetic variation translates into immune diversity and disease susceptibility in a cell type‐specific manner.1 Although genome‐wide association studies (GWAS) have identified thousands of loci associated with immune‐mediated diseases, most variants lie in noncoding regions, obscuring their functional interpretation.2, 3 Bridging this gap requires comprehensive maps of gene regulation that integrate transcriptional output, chromatin accessibility, and genetic variation at single‐cell resolution.4 In this context, the Chinese Immune Multi‐Omics Atlas (CIMA) represents a landmark contribution, providing an unprecedented population‐scale, single‐cell multi‐omics resource for the human immune system.5
The most immediate novelty of CIMA lies in its scale and integrative design. By profiling more than 10 million peripheral blood mononuclear cells from 428 healthy Chinese adults, covering an age span from 20 to 77 years, using both single‐cell RNA sequencing (scRNA‐seq) and single‐cell ATAC sequencing (scATAC‐seq), the authors generate the largest immune single‐cell compendium to date. Importantly, this effort goes beyond descriptive cell atlases6, 7, 8 by integrating whole‐genome sequencing, plasma metabolomics, lipidomics and blood biochemistry, thereby creating a unified framework to study immune variation across molecular layers. Such breadth allows the authors to interrogate how genetic, epigenetic, transcriptional, and physiological variations intersect within the immune system.
A second major advance is the depth of immune cell annotation. Through iterative clustering and hierarchical classification, the study resolves 73 transcriptionally distinct immune cell types, including rare and transitional populations that are often invisible in bulk or low‐resolution analyses. This refined cellular taxonomy provides the foundation for all downstream analyses and highlights the power of single‐cell approaches to capture immune heterogeneity across sex and age.6, 9, 10 The atlas reveals systematic age‐ and sex‐associated shifts in immune cell composition and molecular programs, reinforcing the notion that immune variation is both structured and context dependent.
One of the most impactful aspects of CIMA is the construction of enhancer‐driven gene regulatory networks for immune cells.11, 12 Using scATAC‐seq data, the authors identify more than 338,000 candidate cis‐regulatory elements, most of which are distal and highly cell‐type specific. By integrating chromatin accessibility with gene expression, they link regulatory elements to target genes and transcription factors, resulting in GRNs that encode cell identities and functional states13 (Figure 1). These networks recover known lineage‐defining regulators while also identifying transcription factors active in rare or poorly characterised cell types. Importantly, the GRNs are not static: age‐associated and sex‐associated regulatory programs are resolved, revealing how immune regulation is remodelled across the human lifespan.13
The integration of genetic variation elevates CIMA from an atlas to a mechanistic resource. By performing cell‐type‐resolved quantitative trait locus (xQTL) mapping, the authors identify 9600 expression quantitative trait loci and 52,361 chromatin accessibility QTLs. Strikingly, a large fraction of these associations is cell type specific, even when the regulated gene or regulatory element is active in multiple cell types (Figure 2). This observation underscores a key principle of immune genetics: the same genetic variant can have distinct regulatory consequences depending on the cellular context.14, 15, 16
Beyond static associations, the study captures dynamic genetic effects along differentiation trajectories in monocytes and B cells, demonstrating that genetic regulation itself can change during cellular maturation. This insight is particularly important for immune‐mediated diseases, many of which involve dysregulated differentiation or activation states rather than altered baseline cell identity.
A highlight of the work is the systematic integration of xQTL results with GWAS data using summary data‐based Mendelian randomisation.17, 18 This analysis uncovers more than 1000 pleiotropic associations linking genetic variants to chromatin accessibility, gene expression, circulating inflammatory proteins, and disease risk, often in a single immune cell type. The example of a variant influencing asthma susceptibility through IKZF4 regulation specifically in regulatory T cells illustrates how CIMA enables precise mapping from genotype to disease‐relevant cellular mechanisms. Such cell‐type‐resolved interpretations are essential for translating genetic discoveries into therapeutic hypotheses.
Another innovative component of the study is the development of CIMA‐CLM, a cell‐type‐specific language model that integrates chromatin sequence features with single‐cell transcriptomic data to predict chromatin accessibility.5 This approach demonstrates strong concordance with experimental data and enables in silico mutagenesis to assess the effects of noncoding variants. By coupling experimental and computational frameworks, CIMA points toward a future in which regulatory variant function can be predicted and prioritised at scale.
The broader importance of CIMA extends beyond immunology. From a population genetics perspective, the atlas addresses a long‐standing bias toward European ancestry in functional genomics resources.1 The focus on a large Chinese cohort reveals regulatory variants that are rare or absent in other populations, emphasising the need for ancestry‐diverse datasets to fully understand human biology and disease. For the wider scientific community, CIMA provides a template for how population‐scale single‐cell atlases can be constructed and analysed at a comparable scale and resolution.
CIMA will also be invaluable as a reference resource. Its publicly accessible portal enables researchers to explore immune cell–specific regulatory landscapes, query xQTLs, and intersect their own data with a richly annotated atlas. Future studies of immune‐mediated diseases, vaccine responses, ageing and inflammation can leverage CIMA to interpret transcriptional or genetic signals in a precise cellular context. Moreover, the GRNs and regulatory annotations generated here offer a foundation for experimental validation and functional perturbation studies.
In summary, the CIMA represents a decisive step toward a mechanistic understanding of immune diversity. By uniting population‐scale genetics with single‐cell multi‐omics and advanced computational modelling, CIMA transforms how regulatory variation in the immune system can be studied. Its impact will be felt not only in immunology and human genetics but also across systems biology, precision medicine and computational genomics, serving as both a discovery engine and a lasting community resource.
Looking ahead, CIMA provides a foundation for extending immune regulatory maps to additional ancestries, disease states, longitudinal sampling and environmental perturbations. Integrating this framework with functional perturbation experiments, spatial profiling and clinical cohorts will enable causal dissection of immune regulation and accelerate the translation of genetic discoveries into precision immunology.
AUTHOR CONTRIBUTIONS
Antonio Benedetto Ventura and Lucia Deligio wrote the manuscript and prepared the figures. Giacomo Volpe supervised the work and contributed to writing and editing the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors affiliated with the IRCCS Istituto Tumori “Giovanni Paolo II”, Bari, are responsible for the views expressed in this article, which do not necessarily represent the Institute. The authors declare no conflict of interest.
ETHICS STATEMENT
This article is an editorial/commentary and does not report original research involving human participants or animals. Therefore, no ethical approval or informed consent was required.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sirugo G , Williams SM , Tishkoff SA . The missing diversity in human genetic studies. Cell. 2019;177:26‐31. doi:10.1016/j.cell.2019.02.048 30901543 10.1016/j.cell.2019.02.048PMC 7380073 · doi ↗ · pubmed ↗
- 2Farh KK , Marson A , Zhu J , et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337‐343. doi:10.1038/nature 13835 25363779 10.1038/nature 13835 PMC 4336207 · doi ↗ · pubmed ↗
- 3Westra H , Peters MJ , Esko T , et al. Systematic identification of trans e QT Ls as putative drivers of known disease associations. Nat Genet. 2013;45:1238‐1243. doi:10.1038/ng.2756 24013639 10.1038/ng.2756 PMC 3991562 · doi ↗ · pubmed ↗
- 4Papalexi E , Satija R . Single‐cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol. 2018;18:35‐45. doi:10.1038/nri.2017.76 28787399 10.1038/nri.2017.76 · doi ↗ · pubmed ↗
- 5Yin J , Zheng Y , Huang Z , et al. Chinese Immune Multi‐Omics Atlas. Science. 2026;391:eadt 3130. doi:10.1126/science.adt 3130 41505528 10.1126/science.adt 3130 · doi ↗ · pubmed ↗
- 6Zheng GXY , Terry JM , Belgrader P , et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. doi:10.1038/ncomms 14049 28091601 10.1038/ncomms 14049 PMC 5241818 · doi ↗ · pubmed ↗
- 7Hao Y , Hao S , Andersen‐Nissen E , et al. Integrated analysis of multimodal single‐cell data. Cell. 2021;184:3573‐3587 e 3529. doi:10.1016/j.cell.2021.04.048 34062119 10.1016/j.cell.2021.04.048PMC 8238499 · doi ↗ · pubmed ↗
- 8Han L , Wei X , Liu C , et al. Cell transcriptomic atlas of the non‐human primate Macaca fascicularis. Nature. 2022;604:723‐731. doi:10.1038/s 41586‐022‐04587‐3 35418686 10.1038/s 41586-022-04587-3 · doi ↗ · pubmed ↗