Expert consensus on the combined screening of genes and biomarkers for neonatal diseases
Xin-Wen Huang, Ting Zhang, Zhen-Zhen Hu, Zhi-Guo Wang, Xiao-Ping Luo, Yan-Ling Yang, Lian-Shu Han, Xue-Fan Gu, Guang-Ren Xiao, Bao-Sheng Zhu, Ru-Lai Yang, Wei-Peng Wang, Yong-Lan Huang, Jian-Hui Jiang, Hua Wang, Guo-Li Tian, Qiao-Ling Sun, Xin-Mei Mao, Bin Yu, Wen-Bin Zhu

TL;DR
Experts in China propose a standardized framework for combining genetic and biomarker screening in newborns to improve early diagnosis and treatment of inherited metabolic disorders.
Contribution
A multidisciplinary consensus framework for combined genetic and biomarker newborn screening in China, including 154 disease-causing genes and validated methodologies.
Findings
154 disease-causing genes covering 67 inherited metabolic disorders were prioritized based on treatability and early onset.
A combined screening approach using dried blood spots and next-generation sequencing was validated for diagnostic accuracy.
Standardized workflows for sample collection, result interpretation, and dual reporting were established for clinical implementation.
Abstract
Newborn screening (NBS) through disease biomarkers has significantly reduced severe outcomes of congenital disorders. Moreover, exploratory newborn genetic screening programs are increasingly being implemented. This consensus, developed by multidisciplinary experts, aims to standardize the combined screening of genes and biomarkers for neonatal diseases in China, balancing ethical, technical, and clinical considerations. This consensus synthesizes evidence from peer-reviewed literature (PubMed, CNKI, etc.) up to 2024 and integrates clinical experiences from multidisciplinary experts in neonatology, genetics, and laboratory medicine, focusing on disease biomarker-based NBS, newborn genetic screening, and the clinical utility of combined screening. The consensus defines principles for combined screening: (1) disease/gene selection: 154 disease-causing genes covering 67 inherited…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
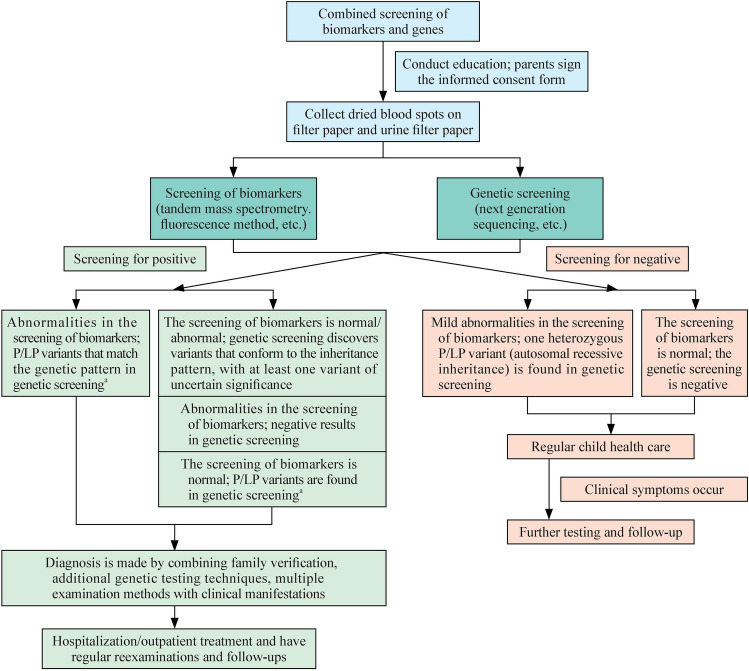 Figure 1
Figure 1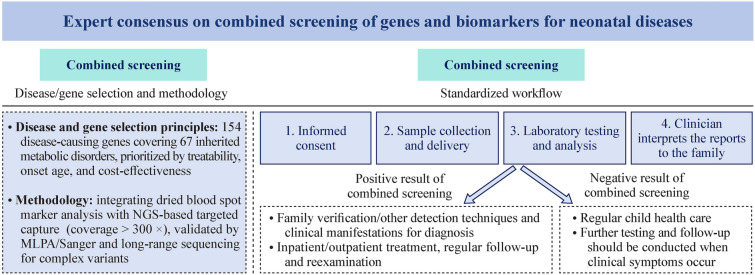 Figure 2
Figure 2- —http://dx.doi.org/10.13039/100022963Key Research and Development Program of Zhejiang Province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Rare Diseases · Metabolism and Genetic Disorders · Folate and B Vitamins Research
Introduction
Newborn screening (NBS) through biomarkers enables early diagnosis and treatment, effectively preventing severe disease outcomes. This study also provides guidance for family reproductive planning, reducing the incidence of birth defects and improving population health. This measure has been recognized internationally as one of the "top ten public health achievements" in the first decade of the twenty-first century [1]. Owing to high entry barriers, many low- and middle-income countries (such as Indonesia, Laos, and Albania) still struggle to provide some, if not all, universal NBS [2]. In China, the coverage rate of NBS has exceeded 98%, with biomarker-based screening remaining the dominant method due to its targeted advantage [3]. Moreover, exploratory newborn genetic screening programs are increasingly being implemented [4–8]. To maximize the protection of the rights and interests of screened newborns and their families, the NBS Working Group, in collaboration with multidisciplinary experts from relevant fields of several societies, has jointly formulated the "Expert consensus on combined screening of genes and biomarkers for neonatal diseases". This consensus adheres to the principles of ethical priority, scientific rigor, forward-thinking, and practicality, aiming to provide actionable guidelines for the standardized application and management of combined biomarkers and genetic screening in disease detection.
Advantages and challenges of disease biomarker-based newborn screening
The international NBS has a history of more than 60 years. With advancements in technologies, such as enzyme-linked immunosorbent assays, time-resolved fluorescence analysis, and tandem mass spectrometry (MS/MS), the range of screened conditions has expanded, and screening efficiency has significantly improved [9, 10]. As a robust approach, traditional disease biomarker-based NBS offers notable strengths: it is cost effective for large-scale population screening, features fast detection speed to meet timely screening needs, and supports high throughput to accommodate the volume of newborns in public health programs. Given that NBS serves as a core public health project, cost effectiveness and operational efficiency are crucial factors that must be prioritized in its nationwide implementation. However, NBS typically relies on dried blood spot (DBS) samples collected on filter paper. Although DBSs can be stored and transported at room temperature, high-temperature or humid conditions may lead to a reduction in or inactivation of enzyme activities [such as glucose-6-phosphate dehydrogenase (G6PD) enzymes] and the degradation of biomarkers (such as Met, Arg, and C2) in filter paper DBSs. Moreover, the accuracy of screening results can be affected by blood spot quality (size, double-spotting, contamination, incomplete saturation, etc.), punch location (center vs. edge of the spot), and hematocrit level [11–14]. Moreover, biomarkers are susceptible to interference from multiple factors, including geographic/ethnic variations [15, 16], seasonal fluctuations [17], neonatal diseases (prematurity, immature development of liver enzymes, liver diseases, immature development of kidneys, jaundice, infections, hypoglycemia, etc.) [18–22], nutrition, therapeutic interventions (extracorporeal life support, blood transfusion, parenteral nutrition, antibiotics, antiepileptic drugs, etc.) [19, 23–25], maternal factors [vitamin B12/carnitine deficiency, maternal inherited metabolic disorders (IMDs)/liver disease, steroid use] [26–28], environmental influences [29], technical limitations of detection methods (MS/MS cannot distinguish isomers, fluorescence assays are affected by anticoagulants, etc.) [30, 31], and cutoff value settings. These variables generate many false negative/false positive results, compromising screening efficiency [32]. More importantly, discrepancies between the results of different biochemical tests have often been observed, enzymatic assays are time-consuming and labor-intensive, and their results are typically semi-quantitative, which further complicates the interpretation of findings [33, 34]. For screen-positive cases, further confirmation requires specialized biochemical testing and genetic testing, leading to a prolonged diagnostic process. This delay may, to some extent, miss the optimal window for early intervention, resulting in irreversible harm to infants.
Advantages and challenges of newborn genetic screening
Newborn genetic screening is being increasingly implemented worldwide, primarily through three models: (1) two-tier newborn genetic screening: for samples that test positive in initial biomarker screening, direct genetic diagnostic techniques such as quantitative polymerase chain reaction (qPCR) or MassArray nucleic acid mass spectrometry are employed, effectively reducing false-positive rates in primary screening [35–37]; (2) disease-specific newborn genetic screening: NBS for conditions such as deafness [38], sickle cell anemia, and spinal muscular atrophy [39, 40] enables early diagnosis of diseases that cannot be detected through biomarker screening; and (3) next generation sequencing (NGS)-based newborn genetic screening: in September 2013, the United States launched the Newborn Sequencing Initiative, utilizing whole-exome sequencing (WES) and whole-genome sequencing (WGS), followed by four major projects. Multiple studies in other countries and institutions in China have also investigated newborn genetic screening programs one after another [8, 41–49]. The First International Conference on Neonatal Sequencing (ICoNS, 2022, Boston, USA) and the International Conference on Newborn Genomic Screening (2023, London, UK) presented various national newborn genetic screening initiatives from the United States, Europe, and Australia, significantly advancing the application of genetic sequencing in NBS.
On the basis of the current research status of newborn genetic screening, WES, WGS, and targeted gene panels demonstrate relatively high diagnostic yields and clinical utility for critically ill neonates in neonatal intensive care units (NICUs) and hospitalized newborns. In particular, rapid WGS or WES, which reduces sequencing and interpretation time, has proven to be an effective diagnostic tool for critically ill newborns and infants suspected of having a genetic disorder, with reported diagnostic yields ranging from 30% to 57% [50]. However, owing to certain limitations in genetic sequencing technologies (e.g., detection of structural variants and pseudogenes) and variant interpretation [e.g., interpretation of variants of uncertain significance (VUS)], the sensitivity and specificity of screening may be compromised, resulting in a certain degree of false-negative and false-positive findings. In addition, there remains controversy regarding how to report carrier status identified through genetic screening. Consequently, newborn genetic screening cannot fully replace traditional NBS and should serve only as a complementary approach. Furthermore, owing to variations in research objectives, target populations (including NICU patients and hospitalized newborns) [51, 52], diverse technologies, methods and solutions selected by combined NBS (such as WES, WGS, gene panels, hotspot mutation detection, and qPCR), and highly heterogeneous disease selection criteria [53, 54], a standardized system for newborn genetic screening has yet to be established.
Advantages of combining newborn disease biomarkers with genetic screening
The integration of genetic sequencing into NBS is increasingly recognized as a valuable addition, with pilot programs underway in some regions. However, large-scale implementation depends on local economic conditions and healthcare capacity, making the key questions “where, when, and how” to implement it effectively. In NBS, screening for more diseases is not necessarily better, as expanding the scope of screening introduces greater complexity in terms of technical, psychological, social, economic, and ethical considerations. Only when implemented in conjunction with measures to improve child healthcare and when the screened diseases meet criteria such as severe harm, absence of early specific symptoms, being preventable and treatable, and having established therapeutic protocols and medications can it genuinely reduce infant mortality and promote health equity.
With advancements in genomic sequencing technologies, the cost of genetic testing continues to decline. The results of disease biomarker testing serve as corroborative evidence for genetic findings, facilitating more straightforward report interpretation and shortening the turnaround time. Moreover, the results of genetic screening contribute to reducing both false-positive and false-negative rates in NBS and enable rapid disease subtyping and differential diagnosis [55, 56]. The combination of both approaches allows for faster and more accurate disease diagnosis, achieving the goal of early treatment. This is particularly critical for IMD with acute onset in the early neonatal stage, as it enables targeted, rapid, and effective interventions, thereby saving lives and reducing the occurrence of severe complications such as neurological disorders. Most importantly, large-scale population screening can be effectively implemented only through integration with existing, well-established NBS systems.
Methods of combined screening
Principles for the combined screening of disease and gene selection, and associated diseases and genes
The general principles for disease selection in combined screening include the following: (1) diseases must have biomarkers suitable for screening; (2) there should be a clear gene–biomarker–disease association; (3) pathogenic variants can be reliably detected on available genomic sequencing platforms and bioinformatics workflows; (4) the age of onset or treatable age should be less than 5 years; (5) the disease should have a high incidence or mortality rate; (6) there should be effective treatments or interventions that significantly alter disease progression; (7) consideration of the sensitivity and specificity of biomarkers and genetic testing methods; and (8) a favorable cost‒benefit ratio for the combined screening program.
General principles for gene selection in combined screening [57, 58] include the following: (1) genes associated with routine NBS conditions, such as G6PD, PAH, and PTS; (2) genes linked to diseases screened by MS/MS or other methods in newborns, including MMACHC, MMUT, SLC25A13, SLC22A5, and GCDH, which have been confirmed as common pathogenic variants in the Chinese population; (3) clear gene‒disease relationships, as defined by the Clinical Genome Resource (ClinGen) Center of the National Institutes of Health in the United States; (4) the inheritance pattern of genes is definite; and (5) maximizing coverage of clinically common monogenic disorders while balancing technical sensitivity and fully considering the cost-effectiveness of the screening program.
On the basis of these principles and considering diseases already included in national or regional NBS programs (via DBS or urine filter paper tests), through expert consensus, the recommended target diseases for combined screening include 154 disease-causing genes covering 67 IMDs, specifically organic acid metabolism disorders, amino acid metabolism disorders, urea cycle disorders, fatty acid β-oxidation disorders, carnitine transport disorders, creatine synthesis and transport disorders, peroxisomal diseases, and lysosomal storage diseases. The detailed diseases and traditional screening methods used are shown in Tables 1 and 2. Individual laboratories may adjust corresponding combined screening methods according to their technical capabilities and local disease prevalence.Table 1. Diseases recommended for combined biomarker and genetic screeningNumberDiseaseClassificationGeneInheritance patternSpecific biomarkers—bloodSpecific biomarkers—urineOrganic acid metabolism disorders 1Methylmalonic acidemiaType MutMMUTARC3, C3/C2, methylmalonic acid, methylcitric acidMethylmalonic acid, methylcitric acidType AMMAAARMethylmalonic acid, methylcitric acidType BMMABARMethylmalonic acid, methylcitric acidType CblCMMACHCARC3, C3/C2, homocysteineMethylmalonic acid, methylcitric acid, homocysteineType CblDMMADHCARMethylmalonic acid, methylcitric acid, homocysteineType CblFLMBRD1ARMethylmalonic acid, methylcitric acid, homocysteineType CblJABCD4AR–Type CblXHCFC1XLR 2Propionic acidemiaPCCAARC3, C3/C2, methylcitric acidMethylcitric acid, 3-hydroxypropionic acid, propionylglycinePCCBAR 3Combined malonic acid and methylmalonic aciduriaACSF3AR–Methylmalonic acid, methylcitric acid, malonic acid 4Malonyl-CoA decarboxylase deficiencyMLYCDARC3DCMalonic acid* 5IsobutyrylglycinuriaACAD8ARC4– 6Ethylmalonic encephalopathyETHE1ARC4, C5– 7β-Ketothiolase deficiencyACAT1ARC4OH, C5:1, C5OH– 8Isovaleric acidemiaIVDARC5Isovalerylglycine, 3-hydroxyisovaleric acid 92-MethylbutyrylglycinuriaACADSBARC5– 102-Methyl-3-hydroxybutyric aciduriaHSD17B10ARC5:1, C5OH–HADH2AR 11Glutaric acidemiaType IGCDHARC5DCGlutaric acid, 3-hydroxyglutaric acidType IIISUGCTAR– 12Multiple carboxylase deficiencyHolocarboxylase synthetase deficiencyHLCSARC5OH, C3, C3/C2–Biotinidase deficiencyBTDARBiotinidase activity (L)– 133-Hydroxy-3-methylglutaracidemiaHMGCLARC5OH–HMGCS2AR 143-Methylcrotonyl-CoA carboxylase deficiencyMCCC1ARC5OH3-Methylcrotonylglycine, 3-hydroxyisovaleric acidMCCC2AR 153-Methylglutaconic acidemiaType IAUHARC5OH–Type IITAFAZZINXLRTAZXLRType IIIOPA3ARType VDNAJC19ARType VISERAC1ARType VII A/BCLPBARType VIIIHTRA2ARType IXTIMM50AR 16AlcaptonuriaHGDAR–Homogentisic acidAmino acid metabolism disorders 17Maple syrup urine diseaseBCKDHAARLeu, Ile, Val–BCKDHBARDBTARDLDAR 18TyrosinemiaType IFAHARTyr, SA–Type IITATARTyr–Type IIIHPDARTyr– 19HyperphenylalaninemiaPhenylalanine hydroxylase deficiencyPAHARPhe, Phe/Tyr–Pterin-4a-carbinolamine dehydratase deficiencyPCBD1AR6-Pyruvoyl-tetrahydrobiopterin synthase deficiencyPTSARDihydropteridine reductase deficiencyQDPRARGuanosine triphosphate cyclohydrolase deficiencyGCH1ARSepiapterin reductase deficiencySPRAR 20Methionine cycle disorderHomocysteinemiaCBSARMet, homocysteineHomocysteineMTHFRARMet (L), homocysteineMTRRARHomocysteineMTRARHomocysteineMethionine adenosyltransferase I/III deficiencyMAT1AAR/ADMet–Glycine N-methyltransferase deficiencyGNMTARS-adenosylhomocysteine hydrolase deficiencyAHCYARAdenosine kinase deficiencyADKAR 21HyperprolinemiaType IPRODHARPro–Type IIALDH4A1AR 22Non-ketotic hyperglycinemiaGLDCARGly–AMTARGCSHAR 23HyperlysinemiaAASSARLys– 24CystinuriaSLC3A1AR–Cys, Orn, Lys, ArgSLC7A9AR 25Glutathione synthetase deficiencyGSSAR5-Oxoproline– 26Aromatic L-amino acid decarboxylase deficiencyDDCAR3-O-methyldopa–Urea cycle disorders 27ArgininemiaARG1ARArgArg 28Argininosuccinic acidemiaASLARCit, ASAASA 29Carbamoyl phosphate synthetase I deficiencyCPS1ARCit (L), Gln– 30N-acetylglutamate synthase deficiencyNAGSARCit (L), Gln– 31Ornithine transcarbamylase deficiencyOTCXLRCit (L), Gln– 32Carbonic anhydrase VA deficiencyCA5AARCit (L), Gln– 33Argininosuccinate synthetase deficiencyASS1ARCitCit 34Citrin deficiencySLC25A13ARCit, Met, Arg, PheCit 35Hyperornithinemia–hyperammonemia–homocitrullinemia syndromeSLC25A15AROrnOrotic acid, uracil 36Ornithine aminotransferase deficiencyOATAROrn–Fatty acid β-oxidation disorders 372, 4-Dienoyl-CoA reductase deficiencyNADK2ARC10:2– 38Very-long-chain acyl-CoA dehydrogenase deficiencyACADVLARC14:1, C14:2, C14, C12:1, C12– 39Isolated and trifunctional protein deficiencyHADHAARC14OH, C14:1OH, C16OH, C16:1OH, C18OH, C18:1OH–HADHBAR 40Medium chain acyl-CoA dehydrogenase deficiencyACADMARC6, C8, C10, C8/C10– 41Short-chain acyl-CoA dehydrogenase deficiencyACADSARC4– 42Short chain 3-hydroxyacyl-CoA dehydrogenase deficiencyHADHARC4OH– 43Multiple acyl-CoA dehydrogenase deficiencyETFAARC4–C18–ETFBARETFDHAR 44Medium chain ketoyl-CoA thiolase deficiencyC6OH, C8, C10OH–Primary carnitine deficiency 45Primary carnitine deficiencySLC22A5ARC0 (L), C2 (L), C3 (L)– 46Carnitine palmitoyl transferase II deficiencyCPT2ARC0 (L), C16, C18, C0/(C16 + 18) (L)- 47Carnitine palmitoyl transferase I deficiencyCPT1AARC0, C16 (L), C18 (L), C0/(C16 + 18)– 48Carnitine-acylcarnitine translocase deficiencySLC25A20ARC0 (L), C16, C18, C0/(C16 + 18)–Peroxisomal disorders 49AdrenoleukodystrophyABCD1XLRC26:0 Lysophosphatidylcholine– 50Zellweger syndromePEX1ARC26:0 Lysophosphatidylcholine–PEX10ARPEX11ARPEX12ARPEX13ARPEX14ARPEX16ARPEX19ARPEX2ARPEX26ARPEX3ARPEX5ARPEX6ARCreatine synthesis and transport disorders 51Guanidinoacetate methyltransferase deficiencyGAMTARGuanidinoacetic acid, creatinine (L), guanidinoacetic acid/creatinineCreatinine (L), guanidinoacetic acid 52Arginine:glycine transaminase deficiencyGATMAR–Creatinine (L), guanidinoacetic acid 53Creatine transporter deficiencySLC6A8XLR–CreatinineLysosomal storage disorders 54MucopolysaccharidosisType IIDUAARα-L-iduronidase (L), mucopolysaccharide–Type IIIDSXLRIduronate-2-sulfatase (L), mucopolysaccharideType IIIBNAGLUARα-N-acetylglucosaminidase (L)Type IVAGALNSARN-acetyl-galactosamine-6-sulfatase (L), mucopolysaccharideType VIARSBARN-acetyl-galactosamine-4-sulfatase (L) 55Krabbe diseaseGALCARβ-Galactocerebrosidase (L)– 56Gaucher diseaseGBAARAcid-β-glucocerebrosidase (L)– 57Fabry diseaseGLAXLDα-Galactosidase A (L)– 58Niemann–Pick disease type A/BSMPD1ARAcid sphingomyelinase (L)– 59Pompe diseaseGAAARAcid α-glucosidase (L)–Others 60Pyruvate carboxylase deficiencyPCARCit– 61Congenital adrenal hyperplasiaCYP21A2AR17-Hydroxyprogesterone–CYP11B1AR 62GalactosemiaGalactokinase deficiencyGALK1ARTotal galactose–Galactose isomerase deficiencyGALEAR–Galactose mutarotase deficiencyGALMAR–Classical galactosemiaGALTARTotal galactose, galactose-1-phosphate uridyltransferase (L)– 63Glucose-6-phosphate dehydrogenase deficiencyG6PDXLDG6PD (L)– 64Glutamate formylaminotransferase deficiencyFTCDARFormiminoglutamic acid, C4– 65Congenital hypothyroidismPAX8ARThyroid stimulating hormone, thyroxine (L)THRAARTHRBARTSHBARTSHRARTGARTPOARDUOXA2ARDUOX2AR 66Severe combined immunodeficiency caused by adenosine deaminase deficiencyADAARAdenosine, 2’-deoxyadenosine 67Duchenne muscular dystrophyDMDXLDCreatine kinaseAR* autosomal recessive, AD autosomal dominant, XLR X-linked recessive, XLD X-linked dominant, C0 carnitine, C2 acetylcarnitine, C3 propionylcarnitine, C3DC malonylcarnitine, C4 butyrylcarnitine, C4OH 3-hydroxy-butyryl carnitine, C5 isovaleryl carnitine, C5:1 tiglyl carnitine, C5OH 3-hydroxy-isovaleryl carnitine, C5DC glutaryl carnitine, C6 hexanoyl carnitine,* C6OH* 3-hydroxy-hexanoyl carnitine, C8 octanoyl carnitine, C10 decanoyl carnitine, C10OH 3-hydroxy-decanoyl carnitine, C12 dodecanoyl carnitine, C14 tetradecanoyl carnitine, C14:2 tetradecadienoyl carnitine, C16 palmitoyl carnitine, C18 stearoyl carnitine, C18:1 oleyl carnitine, C26:0 hexacosanoyl carnitine, C14OH 3-hydroxy myristoyl carnitine, C16OH 3-hydroxy palmitoyl carnitine, C16:1OH 3-hydroxy palmitoleyl carnitine, C18:1OH 3-hydroxy olely carnitine, SA succinylacetone, ASA argininosuccinic acid, G6PD glucose-6-phosphate dehydrogenaseTable 2Summary of the traditional methods used currentlyDiseasesScreening methodsOrganic acid metabolism disordersMS/MS, colorimetric assay, fluorometric assayAmino acid metabolism disordersMS/MS, fluorometric assayUrea cycle disordersMS/MSFatty acid β-oxidation disordersMS/MSPrimary carnitine deficiencyMS/MSPeroxisomal disordersMS/MSCreatine synthesis and transport disordersMS/MSLysosomal storage disordersMS/MS, fluorometric assayOthers Pyruvate carboxylase deficiencyMS/MS Congenital adrenal hyperplasiaFluorometric assay GalactosemiaPaigen test, colorimetric assay, fluorometric assay Glucose-6-phosphate dehydrogenase deficiencyFluorometric assay Glutamate formylaminotransferase deficiencyMS/MS Congenital hypothyroidismFluorometric assay, ELISA Severe combined immunodeficiency caused by adenosine deaminase deficiencyMS/MS Duchenne muscular dystrophyMS/MS, ELISA, fluorometric assayMS/MS includes flow-injection tandem mass spectrometry, liquid chromatography tandem mass spectrometry, ELISA enzyme linked immunosorbent assay
Informed consent, sample collection, detection and reporting for combined screening
Informed consent
Nurses should provide comprehensive education on relevant knowledge to the guardians of neonates and truthfully inform the guardians of all key information regarding the combined screening, including target diseases covered in the screening, screening techniques and their limitations, costs, general procedures, sample collection methods, the resulting inquiry process, potential risks, etc. Based on full disclosure and adherence to the principle of informed choice, a written informed consent form should be signed with the guardian to clarify the rights and obligations of both parties during the screening process to ensure the smooth implementation of screening and safeguard the legitimate rights and interests of both parties.
Sample collection, detection and reporting
Preparation of the sample collection card
It is necessary to fill in the blood collection cards carefully, ensuring clear handwriting and complete registration. The newborn common genetic and metabolic disease screening blood collection cards with legible handwriting and complete information, including basic information, such as name, sex, referring hospital, date of birth, and blood collection date of the neonate, were accurately collected. Moreover, relevant information, such as the name, contact number, contact address of the guardian, and blood collector, was collected. The barcode at the designated position on the sample should be affixed properly to avoid sample confusion effectively and ensure that each sample can be accurately identified and tracked.
Sample collection
The collection of heel blood should be strictly controlled within 2–7 days after the birth of the newborn, no later than 20 day postpartum. For low-birthweight infants (< 1800 g) and preterm infants (< 37 gestational weeks), blood sample collection is repeated in the first month of life at 15 and 30 days [59]. Two additional 8-mm diameter blood spots are needed beyond routine screening to meet combined testing needs. After the blood samples are collected, they should be allowed to dry naturally at room temperature for more than 4 hours to make DBSs on filter paper. During the drying process, placing the samples under an electric hair dryer near a heating radiator, on an electric stove, in sunlight or under a strong light source for baking, or stacking them together, is strictly prohibited. Moreover, contact between the samples and other surfaces should be avoided to prevent the samples from being contaminated or damaged. The qualified DBSs on filter paper should be packaged independently in a moisture proof manner, sealed, and then temporarily stored in a refrigerator at 2–8 ℃. They should be promptly delivered to the designated laboratory in an environment of less than 25 ℃ for testing to ensure the quality and stability of the samples.
Sample detection and reporting
The detection laboratory should be equipped with advanced detection equipment, including time-resolved fluorescence analysis, MS/MS analysis, liquid phase capture sequencing, and NGS, along with robust bioinformatics analysis capabilities. Both biomarker detection reports and genetic testing reports should be issued to relevant personnel simultaneously within 15 working days after sample collection to provide a basis for subsequent diagnosis and treatment in a timely manner.
Additional information in the report should include interpretation and annotations of results, limitations of the testing methodology and the clinical significance of the findings.
Handling special cases
For IMDs with significantly abnormal biomarkers and early onset manifestations in the neonatal period, treatment should be initiated immediately to avoid missing the critical therapeutic window. During the treatment process, upon receiving genetic detection results, a more precise and comprehensive treatment plan should be further formulated according to the detailed genetic findings.
Genetic screening testing protocol
For the genetic testing component of combined screening, it is recommended to employ NGS-based targeted capture techniques (including liquid‒phase hybridization capture or multiplex PCR enrichment) to analyze the exon4ic regions and flanking sequences of the target genes, with coverage extended to known pathogenic or likely pathogenic (P/LP) variants in non-coding regions. Quality control of the sequencing data should adhere to standard NGS guidelines, ensuring that the coverage depth of the target area is greater than 300 × , that the depth of more than 99.5% of the target sequence area reaches more than 20 × and that the depth of more than 99% of the target sequence area reaches more than 100 × . In the secondary analysis pipeline, the use of GATK or other industry-validated analytical tools for sequencing data analysis is recommended, with the reference genome options being GRCh37/hg19 or GRCh38/hg38. For tertiary analysis, annotation should be performed via software, such as variant effect predictor or ANNOVAR, followed by filtering of known pathogenic variants on the basis of major databases, including ClinVar and the Human Gene Mutation Database, which curates known or suspected disease-causing variants. As supplementary evidence for the assessment of variant pathogenicity, bioinformatics software can be employed to predict the impact of variants: REVEL or CADD for missense variant prediction and SpliceAI or dbscSNV for evaluating potential splicing effects. All sequence variants should be named according to the Human Genome Variation Society nomenclature standards, and each variant should be classified for pathogenicity in accordance with the variant interpretation guidelines of the American College of Medical Genetics and Genomics and the ClinGen of the National Institutes of Health.
Deficiency of 21-hydroxylase (21-OHD), which causes congenital adrenal hyperplasia, is attributed to mutations in the CYP21A2 gene. This gene has a highly homologous pseudogene, CYP21A1P, with 98% and 95% sequence identity in exons and introns, respectively. Among pathogenic mutations in 21-OHD, 95% arise from recombination events between the functional gene and pseudogene: approximately 75% due to mitotic gene conversion, where variants in CYP21A1P are transferred to CYP21A2, and approximately 20%–25% caused by meiotic non-allelic homologous recombination. Owing to the limited read length of conventional NGS, misalignment may occur when highly homologous regions are analyzed. For CYP21A2 gene mutation screening, we recommend the use of either PCR-based NGS or long-read sequencing technologies, followed by the use of multiplex ligation-dependent probe amplification combined with Sanger sequencing for further validation [60].
Citrin deficiency, caused by pathogenic variants in SLC25A13, is frequently associated with complex structural variations, including IVS4ins6kb and IVS16ins3kb. NGS and specific bioinformatics algorithms are employed to screen for mutations in the SLC25A13 gene. When necessary, long-range PCR was used for further detection.
Principles for the determination and handling of combined screening results
A positive combined screening result refers to the discovery of P/LP variants that are highly correlated with clinical (early onset cases) and biomarkers and have a matching genetic pattern: (1) autosomal inheritance: in the dominant inheritance pattern, a heterozygous P/LP variant is present for the disease indicated by the biomarkers in the genetic screening; in the recessive inheritance pattern, a biallelic P/LP variant is present for the disease indicated by the biomarkers in the genetic screening and (2) X-linked inheritance: for diseases indicated by biomarkers in genetic screening, there is a P/LP variation in the gene on the X chromosome. In the case of dominant inheritance patterns, all patients carrying the mutation will be affected; in recessive inheritance patterns, female homozygotes and male hemizygotes are affected, whereas female heterozygotes are usually normal; in incomplete dominant inheritance patterns, female homozygotes and male hemizygotes are affected, and female heterozygotes can have a clinical manifestation spectrum ranging from completely asymptomatic to severely symptomatic [61]. This phenomenon occurs, because skewed X chromosome inactivation (XCI) leads to preferential inactivation of the X chromosome carrying the wild-type allele [62], resulting in a greater proportion of mutant cells than wild-type cells [63, 64], which in turn causes clinical symptoms in female heterozygotes. In some affected females, the proportion of mutant cells is greater than 95%, indicating a severe skew in XCI [65]. In this genetic pattern, XCI is the main factor determining the severity of clinical involvement in female heterozygotes [66]. Fabry disease and ornithine transcarbamylase deficiency both follow this genetic pattern [66, 67].
The following three situations require further confirmation: (1) normal characteristic indicators but genetic testing has identified P/LP variations that conform to genetic patterns (such as neonatal intrahepatic cholestasis caused by citrin deficiency, multiple acyl-CoA dehydrogenase deficiency and other diseases); (2) markedly abnormal characteristic indicators but no P/LP variants identified that align with the clinical phenotype and expected genetic mode in genetic testing; and (3) genetic testing has identified variants that conform to the genetic pattern, among which there is at least one VUS.
For the above situations, family verification and/or other genetic sequencing techniques should be carried out, combined with urine organic acid analysis, enzyme activity determination, very long-chain fatty acid determination, amino acid determination, blood ammonia detection, liver function tests, etc., to rule out maternal genetic metabolic disorders as well as the influences of nutrition, diseases and drugs on the infant and mother. A definite diagnosis should be made through comprehensive analysis. If there are already clinical manifestations and/or metabolic disorders, the infant should be immediately admitted to the hospital for treatment; if there are no clinical manifestations and the metabolism is relatively stable, outpatient treatment can be adopted, with regular reexaminations, follow-ups and management.
A negative combined screening indicates that the newborn shows no clinical symptoms during the neonatal period, has normal disease biomarkers, and that no P/LP variants consistent with the genetic pattern are identified through genetic screening. In such cases, no follow-up is needed.
Special circumstances of heterozygous variants in autosomal recessive inherited metabolic disorders
In autosomal recessive inherited metabolic disorders (ARIMDs), a pathogenic variant at a single locus in certain diseases can lead to partial deficiency of enzyme function, causing mild abnormalities in characteristic markers.
For multimeric proteins, variant proteins can disrupt the formation, function and stability of homomeric or heteromeric multimers through dominant-negative effects or interfere with the interactions between wild-type proteins and other molecules, thereby impairing the partial function of wild-type proteins [68]. For example, citrullinemia type 1 (CTLN1) is recessively inherited and caused by variants in the ASS1 gene, which encodes argininosuccinate synthase (ASS). The crystal structure of human ASS is a functional tetramer consisting of two identical dimers. The most common variant, p.R363W, likely affects the oligomerization of the protein via a dominant‐negative effect, which affects the other normal counter partner. This caused an increase in citrulline levels in carriers [69–71]. The heteromeric multimeric enzyme methylcrotonyl-CoA carboxylase is a heteropolymer composed of MCCCα and MCCCβ subunits (encoded by MCCC1 and MCCC2, respectively). Mutations at specific loci in either MCCC1 or MCCC2 can exert a dominant-negative effect: the mutant protein product interferes with the normal function of the wild-type MCC complex, resulting in suppressed MCC enzyme activity. This functional impairment further leads to mildly elevated levels of metabolites associated with 3-methylcrotonyl-CoA carboxylase deficiency in heterozygotes [72].
The impact of the dominant negative effect varies, and sometimes, it can even lead to significant functional impairment of the protein. The c.791G > A (R264H) mutation site of the MAT1A gene in hypermethioninemia alters the residue position at the dimer interface, preventing substrate binding and resulting in loss of activity and causing disease. This alteration at the site is dominant inheritance. c.769G > A (p.G257R) is located in a critical structural domain of the protein. This variant may affect the dimeric interaction of the protein. It has been reported in patients with hypermethioninemia in homozygous, compound heterozygous and single heterozygous forms, suggesting that it may exist in both dominant and recessive modes [73]. These two variants generally do not significantly increase the methionine content and often do not require special treatment. However, they may be detected via biomarker-based screening or newborn genetic screening, necessitating regular monitoring of methionine levels.
In general, patients with heterozygous variations in ARIMD typically do not exhibit clinical symptoms. However, whether regular monitoring and management are necessary for these individuals remains to be further studied.
Establishment of a combined screening system
Only when the combined screening system is closely integrated with measures to improve children’s healthcare and becomes a public health measure for obtaining high-quality medical services can it tangibly improve children’s health. Accordingly, genetic screening should be incorporated into existing biomarker-based screening systems.
Within the existing management system, provincial/municipal NBS centers should establish integrated platforms for biomarker and genetic testing. Within 48 hours after the birth of an infant, a series of tasks, including obtaining informed consent, sample collection, delivery, testing, diagnosis, treatment, follow-up assessment, and comprehensive management, should be carried out simultaneously with the screening of biomarkers. The personnel structure of combined screening should cover individuals with professional backgrounds in laboratory science, molecular genetics, bioinformatics, clinical medicine, etc. All personnel must strictly follow the duties and technical procedures for diagnosis and treatment as stipulated in relevant regulations. The quality control center conducts quality control to determine the concentration of biomarkers, DNA extraction, library preparation, amplification, etc., to ensure the accuracy and reliability of the test results. A complete data analysis and information management system is established to provide a standardized communication mechanism for traditional NBS laboratories and newborn genetic screening laboratories, ensuring that results and data can be transmitted accurately and promptly. At the same time, a combined screening team composed of testing personnel, genetic counselors, and clinical physicians should be formed to establish diagnosis and preliminary treatment plans.
In conclusion, this expert consensus defines a comprehensive framework for neonatal combined genetic and biomarker screening, outlining clear disease/gene selection criteria, standardized operational procedures, and specific strategies for managing special cases (Fig. 1). While this approach holds great promise for improving infant health outcomes, its widespread implementation faces technical, ethical, and validation challenges. Future efforts must focus on optimizing cost-effectiveness, improving data interpretation, and establishing a standardized national system. Integrating this screening into established newborn screening programs will ultimately reduce the burden of treatable genetic diseases and advance health equity.Fig. 1. Diagnosis and treatment pathway for combined screening of biomarkers and genes. P/LP pathogenic/likely pathogenic. ^a^AD inheritance: one heterozygous P/LP variant; AR inheritance: biallelic P/LP variants; X-linked recessive inheritance: female biallelic or male hemizygous P/LP variants; X-linked dominant or incomplete dominant inheritance: female heterozygous or male hemizygous P/LP variants; the dominant mutation locus of the AR genetic disease MATD: MAT1A Arg264His (R264H)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Neonatal Inherited Metabolic Disease Screening Group, Birth Defects and Control Professional Committee, Chinese Preventive Medicine Association; Neonatology Group, Pediatrics Branch, Chinese Medical Association. Expert consensus on neonatal genetic screening in China: application of high-throughput sequencing in single-gene disease screening. Chin J Pract Pediatr. 2019;38:31–6 (in Chinese).