The High Price of Interrupted Follow‐Up: Catastrophic Progression of Homozygous Familial Hypercholesterolemia—A Case Report and Literature Review
Parham Dastjerdi, Mahdieh Aghababaei, Reza Nikfar, Maryam Aghakouchakzadeh, Reza Mohseni‐Badalabadi, Kaveh Hosseini

TL;DR
A young woman with homozygous familial hypercholesterolemia faced severe heart failure and death due to interrupted medical care during the pandemic.
Contribution
Highlights the catastrophic consequences of disrupted follow-up in homozygous FH patients and emphasizes the need for continuous monitoring.
Findings
Homozygous FH can lead to fatal heart failure despite maximal lipid-lowering therapies.
Interrupted follow-up during the pandemic resulted in severe cardiovascular deterioration.
Early detection and sustained multidisciplinary care are critical for managing HoFH.
Abstract
Familial hypercholesterolemia (FH) is the most common monogenic lipid disorder, primarily resulting from mutations in LDLR, APOB, and PCSK9 genes. These mutations cause persistently high levels of low‐density lipoprotein cholesterol (LDL‐C), predisposing affected individuals to premature atherosclerotic cardiovascular disease (ASCVD). Homozygous FH (HoFH), a rare but severe form, manifests early in life with cutaneous xanthomas and accelerated coronary and aortic disease. Early diagnosis and aggressive, lifelong management are crucial, yet challenges remain, particularly when follow‐up is interrupted. We report the case of a 20‐year‐old female diagnosed with HoFH at age 13 after presenting with xanthomas. Initial evaluation revealed mild to moderate aortic stenosis and early coronary artery involvement. Genetic testing confirmed a homozygous LDLR mutation. Despite treatment with…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
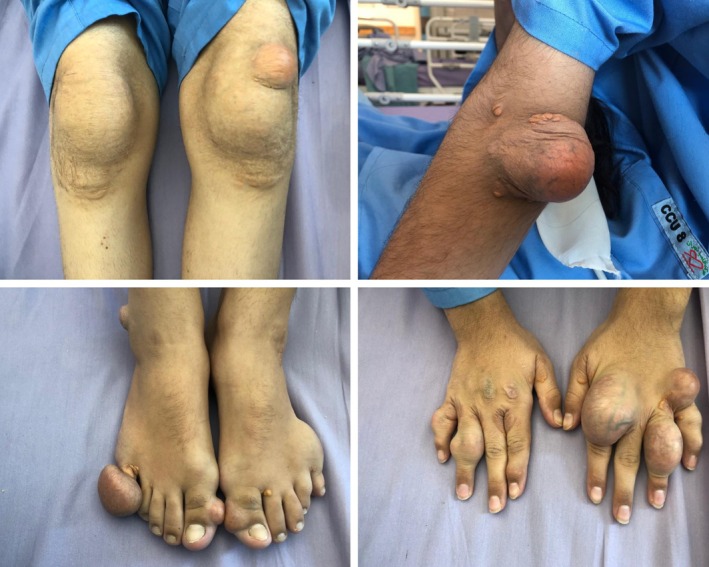 FIGURE 1
FIGURE 1 FIGURE 2
FIGURE 2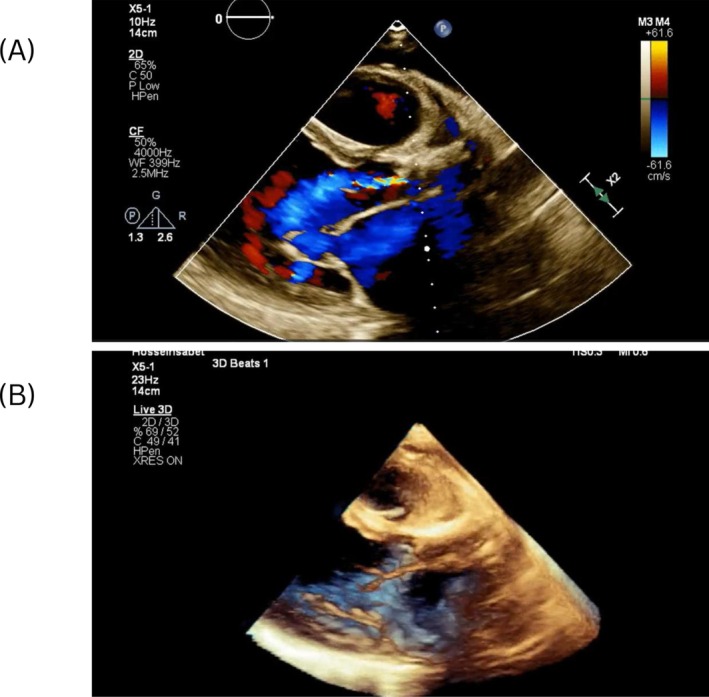 FIGURE 3
FIGURE 3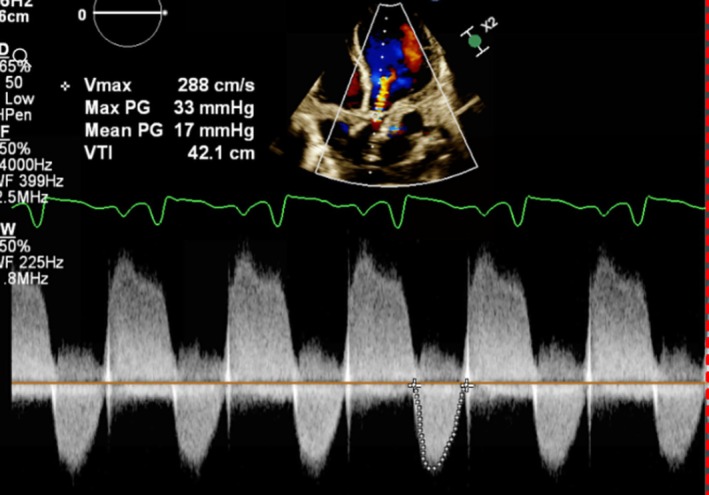 FIGURE 4
FIGURE 4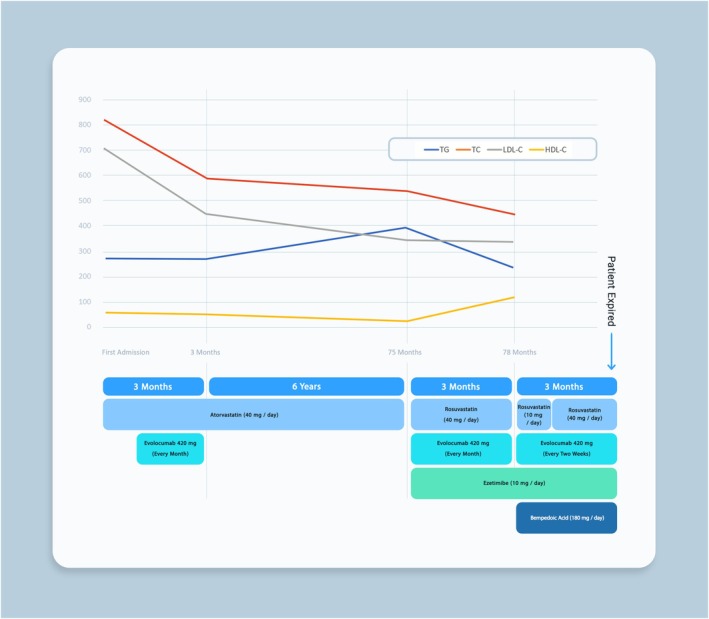 FIGURE 5
FIGURE 5| Category | Differential diagnosis | Mechanism/clinical relevance |
|---|---|---|
| Valvular heart disease | Progression of aortic stenosis (valvular or supravalvular) | Worsening outflow obstruction leading to increased left ventricular pressure and heart failure symptoms |
| Ischemic heart disease | Progression of coronary artery disease with ischemic cardiomyopathy | Myocardial ischemia and left ventricular dysfunction causing reduced cardiac output |
| Secondary valvular disease | Severe mitral regurgitation secondary to ventricular remodeling | Left ventricular dilation and dysfunction leading to functional mitral regurgitation and volume overload |
| Non‐cardiac causes | Anemia, pulmonary embolism, or severe pulmonary hypertension | Reduced oxygen delivery or increased pulmonary vascular resistance mimicking or worsening heart failure |
| Summary | Treatment plan | Outcome | Follow‐up | |
|---|---|---|---|---|
| Kolovou‐2006 [ |
|
|
| NA |
| Ozumi‐2005 [ |
|
|
| NA |
| Rasheed‐2024 [ |
|
|
| Follow‐up after 1 month indicated asymptomatic status and regular clinical monitoring. |
| Sahiner‐2016 [ |
|
|
| NA |
| Alenizi‐2020 [ |
|
|
| Regular LDL apheresis and echocardiography. His general condition remained stable with no complication |
| Alkhateeb‐2013 [ |
|
|
|
|
| Chowdhury‐2020 [ |
|
|
| At 12 months, the patient had better exercise tolerance, no daily angina, and a stable mechanical valve with a 10 mmHg mean trans‐prosthetic gradient. |
| Davoodabadi‐2022 [ |
|
|
| NA |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLipoproteins and Cardiovascular Health · Cholesterol and Lipid Metabolism · Dermatological and Skeletal Disorders
Introduction
1
Familial hypercholesterolemia (FH) is the most common monogenic disease, characterized by mutations in genes encoding proteins essential for lipoprotein metabolism—primarily the low‐density lipoprotein receptor (LDLR), apolipoprotein B (APOB), and PCSK9 [1, 2]. These genetic alterations result in lifelong elevated levels of low‐density lipoprotein cholesterol (LDL‐C), establishing the foundation for the early development of atherosclerotic cardiovascular disease (ASCVD) [2]. Homozygous familial hypercholesterolemia (HoFH), which affects approximately one in a million children globally, can lead to coronary heart disease (CHD) within the first decade of life, with many young patients experiencing myocardial infarctions before the age of 20 [3, 4, 5, 6]. In contrast, heterozygous familial hypercholesterolemia (HeFH) impacts about 1 in 250 individuals, potentially affecting an estimated 6.8–8.5 million children and adolescents worldwide [7]. Given the significant burden and early onset of disease, FH should be considered in patients under 20 years of age when LDL levels exceed 160 mg/dL [8].
Clinically, FH is manifested by elevated plasma LDL‐C concentrations, the presence of tendon xanthomas or corneal arcus, and a family history of premature CHD [9]. Diagnosis can be achieved through clinical assessment, detailed evaluation of personal and family history, or genetic testing [9]. Due to the high risk of premature ASCVD, particularly involving the coronary arteries, patients with FH require aggressive treatment [10, 11, 12]. Statins are the first‐line therapy, with ezetimibe as a second‐line option; PCSK9 inhibitors are employed less frequently [13]. In cases of HoFH, aortic supravalvular and/or valvular stenosis may also be observed and can be a primary cause of mortality [14, 15, 16].
In this case report and literature review, we present a detailed report of a young female with familial hypercholesterolemia whose clinical course exemplifies the challenges of managing advanced HoFH. The case highlights the interplay between a strong genetic predisposition and the progressive development of cardiovascular complications. By synthesizing the clinical evolution observed in our patient with current literature on the genetic basis, diagnostic challenges, and evolving treatment strategies in FH, we emphasize the critical need for early detection, continuous follow‐up, and aggressive, individualized management. This report aims to provide insights into the importance of proactive care and the potential consequences of disrupted monitoring, thereby informing clinicians about strategies to mitigate severe aortic and coronary complications in these patients. This case report was prepared in compliance with the CARE (CAse REport) guidelines to ensure completeness and transparency in reporting [17].
Case History / Examination
2
A 20‐year‐old female with a known history of FH diagnosed at age 13 was referred to our center for progressive worsening of exertional dyspnea, paroxysmal nocturnal dyspnea, and orthopnea over several months.
Her hypercholesterolemia had first manifested at 2 years of age with cutaneous xanthomas over the hands, elbows, and legs. At age 13, hospitalization for xanthoma progression led to diagnosis. At that time, transthoracic echocardiography (TTE) revealed mild to moderate aortic stenosis (pressure gradient [PG] 46 mmHg, mean gradient [MG] 26 mmHg), left ventricular ejection fraction (LVEF) 60%, and mild mitral regurgitation (MR). Coronary computed tomography angiography (CTA) showed mild coronary artery stenosis. Genetic testing confirmed a homozygous pathogenic LDLR mutation (c.389C>G; rs879254509). Her lipid profile was markedly abnormal: total cholesterol 818 mg/dL, LDL‐C 710 mg/dL, HDL‐C 56 mg/dL, triglycerides 272 mg/dL.
Initial treatment included atorvastatin 40 mg daily and two courses of evolocumab (three subcutaneous injections of 140 mg each), which reduced total cholesterol to 586 mg/dL and LDL‐C to 457 mg/dL, with minimal change in triglycerides. She was discharged on atorvastatin, propranolol, captopril, and aspirin. Follow‐up was interrupted during the COVID‐19 pandemic.
On her current presentation at age 20, she appeared dyspneic at rest. Physical examination revealed extensive cutaneous xanthomas (Figure 1) and bilateral corneal arcus (Figure 2).
Extensive cutaneous xanthomas on the patient's extremities, including the knees, elbows, feet, and hands.
Bilateral corneal arcus in both eyes of the patient.
Differential Diagnosis, Investigations, and Treatment
3
Although the patient carried a confirmed diagnosis of HoFH, the acute presentation with worsening dyspnea, orthopnea, and paroxysmal nocturnal dyspnea warranted consideration of other potential causes of decompensation. In the context of severe dyslipidemia and prior aortic valve disease, differentials are included in Table 1.
2D and 3D echocardiography with continuous wave (CW) Doppler revealed severe left ventricular systolic dysfunction (EF 20%), marked ventricular dilation, severe MR, and calcified aortic stenosis (PG 44 mmHg, MG 26 mmHg) with an annulus diameter of 18–19 mm. Figure 3 presents the 2D and 3D color Doppler echocardiography cuts of the parasternal view of the patient, showing the supravalvular aortic stenosis (AS). Figure 4 demonstrates the severe AS and aortic insufficiency (AI) using CW color Doppler in the 4‐chamber view.
(A) 2D color Doppler echocardiography of the parasternal view showing supravalvular AS. (B) 3D echocardiography of the parasternal view demonstrating the same supravalvular AS.
CW Doppler showing severe AS in the 4‐chamber view.
Coronary angiography demonstrated critical ostio‐proximal left main (LM) stenosis, mild proximal left anterior descending (LAD) stenosis, moderate proximal left circumflex (LCX) stenosis, and significant ostio‐proximal and mid‐right coronary artery (RCA) stenosis. Gastroenterology consultation and liver biopsy confirmed stage F2 hepatic fibrosis. Admission lipid profile showed: total cholesterol 539 mg/dL, LDL‐C 351 mg/dL, HDL‐C 27 mg/dL, triglycerides 392 mg/dL.
These findings prompted the initiation of further lipid‐lowering therapy. Following a consultation with a clinical pharmacist, her lipid‐lowering regimen was adjusted. Rosuvastatin 40 mg daily was substituted for atorvastatin 40 mg daily, ezetimibe 10 mg daily was added, and the patient was prescribed evolocumab injection 420 mg per month. Heart failure management was also optimized, including dapagliflozin, metoprolol succinate, furosemide, and spironolactone.
After 3 months, the patient's lipid profile was as follows: triglycerides at 241 mg/dL, total cholesterol at 456 mg/dL, LDL‐C at 341 mg/dL, and HDL‐C at 115 mg/dL. Due to elevated liver function tests, the rosuvastatin dosage was reduced from 40 mg daily to 10 mg daily. Additionally, given the inadequate lipid control, the evolocumab dosage was increased from 420 mg monthly to 420 mg every 2 weeks. Once liver function tests normalized, the rosuvastatin dosage was gradually restored to 40 mg daily, and the patient was initiated on bempedoic acid 180 mg daily. The sequential changes in lipid‐lowering therapy and corresponding lipid profile fluctuations are illustrated in Figure 5, highlighting the patient's response over time.
Timeline of lipid‐lowering therapy adjustments and corresponding lipid profile changes.
Given the severity of her disease, the medical team initially recommended aortic valve replacement (AVR) and coronary artery bypass grafting (CABG) as the primary treatment options. However, due to the high surgical risk, the decision was made to postpone surgery and assess the patient's response to the final adjustments in lipid‐lowering therapy, including the initiation of bempedoic acid.
Outcome and Follow‐Up
4
Before re‐evaluation and planned follow‐up laboratory testing, the patient suffered sudden death on the day of her scheduled appointment. No further interventions, such as percutaneous coronary intervention (PCI), transcatheter aortic valve implantation (TAVI), or consideration of combined heart–liver transplantation, could be undertaken.
Discussion
5
FH is an autosomal dominant genetic disorder marked by significantly elevated LDL‐C levels and an increased risk of premature ASCVD. It results primarily from mutations in the LDLR gene, which impairs LDL‐C clearance. Additional mutations in APOB and PCSK9 contribute to FH by reducing LDL receptor binding affinity and enhancing receptor degradation, respectively [18]. In heterozygous FH (HeFH), a single mutated allele leads to a twofold rise in LDL‐C, whereas homozygous FH (HoFH)—caused by mutations in both alleles—can increase LDL‐C levels to more than five times normal values [19, 20]. Over 1200 LDLR variants have been identified, with specific mutations in APOB (e.g., R3500Q) and gain‐of‐function mutations in PCSK9 further worsening receptor function [18, 21, 22].
HoFH presents with an aggressive phenotype. When LDL receptor activity is < 2%, prognosis is poor, with many patients not surviving beyond their twenties without intervention. Even partial receptor activity (2%–25%) can lead to severe coronary and aortic valve disease by age 30 [19]. Early manifestations include cutaneous and interdigital xanthomas in childhood, and LDL‐C levels often exceed 500 mg/dL in untreated individuals [20, 23]. The progression of the disease is intimately linked to both the magnitude and duration of LDL‐C elevation, with myocardial infarctions and sudden cardiac death reported in children as young as 4 years old [4, 24, 25]. In addition to coronary complications, many HoFH patients suffer from supravalvular and aortic valve stenosis, often necessitating surgical intervention later in life [26, 27, 28]. Lipid accumulation in the cornea, which manifests as corneal arcus, further underscores the severity of the condition [29]. Our patient demonstrated several of these classic features. From an early age, she developed cutaneous xanthomas on her extremities—a hallmark of the disease. By her early twenties, bilateral corneal arcus was evident, reflecting profound lipid accumulation. Imaging studies confirmed advanced cardiovascular involvement, revealing severe aortic stenosis, significant coronary artery disease with critical ostio‐proximal stenoses, and supravalvular aortic stenosis.
Timely diagnosis of HoFH is essential to initiate aggressive treatment strategies and improve long‐term outcomes. Clinically, HoFH is suspected in individuals presenting with markedly elevated LDL‐C levels—often exceeding 500 mg/dL (13 mmol/L)—in conjunction with a family history of premature cardiovascular events or the presence of physical stigmata such as xanthomas emerging in early childhood [30]. Genetic testing plays a pivotal role in confirming the diagnosis, as it can identify pathogenic variants in genes like LDLR, APOB, or PCSK9, thereby differentiating HoFH from severe forms of HeFH [30]. In clinical practice, several validated diagnostic frameworks are available, including the MedPed criteria [31], the Dutch Lipid Clinic Network scoring system [32], the Simon Broome Register criteria [33], and population‐specific guidelines such as the Japanese FH criteria [34], with genetic testing serving as a confirmatory tool when available. In resource‐limited settings or when genetic testing is not accessible, diagnosis is primarily based on these clinical tools together: untreated LDL‐C levels above 500 mg/dL or treated levels above 300 mg/dL, particularly when coupled with early‐onset xanthomas or a known family history of hypercholesterolemia [30]. Supportive imaging studies—such as carotid intima‐media thickness (CIMT), coronary artery calcium (CAC) scoring, and echocardiography—often reveal significant atherosclerosis and aortic valve disease, further aiding in the diagnostic process [19]. However, the overlap in phenotypic expression and the variability in genetic mutations necessitate an interdisciplinary approach, involving genetic counseling, lipidology, and cardiology, to confirm the diagnosis and design an appropriate treatment plan.
The management of HoFH is complex and requires an individualized, stepwise approach depending on disease severity and treatment response. High‐intensity statins (simvastatin, atorvastatin, or rosuvastatin) form the cornerstone of therapy, though they are often insufficient due to limited LDL receptor function. Therefore, combination therapy with ezetimibe, bile acid sequestrants, and fibrates is frequently required [20]. In our case, despite maximally tolerated statins and ezetimibe, LDL‐C levels remained markedly elevated, prompting the initiation of evolocumab. PCSK9 inhibitors like evolocumab and alirocumab have been shown to lower LDL‐C by approximately 30% in patients with receptor‐defective mutations, as evidenced by the TESLA Part B trial [13, 35].
More recently, novel agents such as lomitapide, a microsomal triglyceride transfer protein (MTP) inhibitor, and evinacumab, an angiopoietin‐like protein 3 (ANGPTL3) inhibitor, have demonstrated LDL‐C reductions of up to 50% and have become valuable options for refractory cases [36, 37, 38]. The ELIPSE HoFH trial and subsequent pediatric studies have confirmed evinacumab's efficacy, with LDL‐C reductions approaching 48%–49% even in patients already on maximum lipid‐lowering therapy [38, 39]. Unfortunately, these therapies were not available in our country, limiting the options for our patient. More recently, bempedoic acid, an oral adenosine triphosphate citrate lyase inhibitor, has shown promise by providing an additional 15%–20% reduction in LDL‐C in patients with ASCVD or HeFH on maximal statin therapy, although its efficacy in HoFH remains to be fully established [40, 41, 42]. In our patient, bempedoic acid was introduced as an adjunct after prior therapies failed to achieve optimal control; however, she passed away before the clinical impact of this addition could be fully evaluated. When pharmacotherapy fails to achieve target LDL‐C levels, lipoprotein apheresis becomes a crucial option, typically achieving 50%–70% LDL‐C reduction per session [43, 44]. In our patient's case, we were considering lipoprotein apheresis as the next step if bempedoic acid proved insufficient. Unfortunately, due to her untimely death, this intervention could not be pursued. In advanced cases with progressive, treatment‐resistant atherosclerosis, liver transplantation may be considered as a last resort, providing functional LDL receptors but introducing the burden of lifelong immunosuppression and procedural risks [14].
The management of HoFH, particularly when complicated by CAD and aortic stenosis, remains exceptionally challenging. Patients with HoFH often experience aggressive cardiovascular disease that necessitates high‐risk, complex surgical interventions. For instance, a reported case of two cousins with HoFH required intricate procedures including the Bentall operation, mitral valve replacement, and CABG due to extensive atherosclerotic involvement [45]. Long‐term management also demands continuous monitoring and specialized care. A longitudinal study of 39 HoFH patients revealed that, despite early and aggressive lipid‐lowering therapy, many progressed to severe CAD and aortic valve disease, ultimately necessitating surgical intervention [46]. Furthermore, delayed diagnosis can have catastrophic outcomes. A case report from Egypt detailed two young women, aged 21 and 27, who were diagnosed with HoFH only after the onset of severe vascular complications, including aortic stenosis and multi‐vessel CAD—with one patient dying at the age of 21 [47]. Table 2 provides details on eight cases of complicated HoFH patients.
Conclusion
6
Lifelong monitoring of HoFH patients is essential to ensure timely treatment adjustments and make critical decisions as new manifestations arise. In the case presented, the patient's discontinuation of follow‐up visits due to the COVID‐19 pandemic had severe consequences. The absence of regular monitoring meant that necessary adjustments to medications and treatments were not made, and significant changes in EF and other clinical parameters went unnoticed by the care team. Consequently, the patient returned to our center with significantly progressed disease and in critical condition. This case underscores the vital importance of consistent follow‐up and proactive management in preventing the rapid deterioration often seen in HoFH patients.
Author Contributions
Parham Dastjerdi: conceptualization, investigation, methodology, project administration, supervision, visualization, writing – original draft, writing – review and editing. Mahdieh Aghababaei: conceptualization, project administration, supervision. Reza Nikfar: investigation, writing – original draft. Maryam Aghakouchakzadeh: conceptualization, investigation, methodology, validation, writing – review and editing. Reza Mohseni‐Badalabadi: supervision, validation. Kaveh Hosseini: conceptualization, data curation, investigation, methodology, project administration, supervision, validation, writing – review and editing.
Funding
The authors have nothing to report.
Ethics Statement
The authors have nothing to report.
Consent
The patient and their family provided written informed consent for the publication of this case report.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1A. S. Wierzbicki , S. E. Humphries , and R. Minhas , “Familial Hypercholesterolaemia: Summary of NICE Guidance,” BMJ 337 (2008): a 2423.18753174 10.1136/bmj.a 1095 · doi ↗ · pubmed ↗
- 2S. Ziółkowska , N. Kijek , I. Zendran , E. Szuster , and E. Barg , “Familial Hypercholesterolemia–Treatment Update in Children, Systematic Review,” Pediatric Endocrinology, Diabetes, and Metabolism 28, no. 2 (2022): 152–161.35848473 10.5114/pedm.2022.116112 PMC 10214937 · doi ↗ · pubmed ↗
- 3M. Gautschi , M. Pavlovic , and J.‐M. Nuoffer , “Fatal Myocardial Infarction at 4.5 Years in a Case of Homozygous Familial Hypercholesterolaemia,” in JIMD Reports‐Case and Research Reports, 2011/2 (Springer, 2011), 45–50.10.1007/8904_2011_45PMC 350983923430853 · doi ↗ · pubmed ↗
- 4K. Widhalm , C. B. Binder , A. Kreissl , et al., “Sudden Death in a 4‐Year‐Old Boy: A Near‐Complete Occlusion of the Coronary Artery Caused by an Aggressive Low‐Density Lipoprotein Receptor Mutation (W 556R) in Homozygous Familial Hypercholesterolemia,” Journal of Pediatrics 158, no. 1 (2011): 167.20659736 10.1016/j.jpeds.2010.06.027 · doi ↗ · pubmed ↗
- 5D. L. Sprecher , E. J. Schaefer , K. M. Kent , et al., “Cardiovascular Features of Homozygous Familial Hypercholesterolemia: Analysis of 16 Patients,” American Journal of Cardiology 54, no. 1 (1984): 20–30.6331147 10.1016/0002-9149(84)90298-4 · doi ↗ · pubmed ↗
- 6J. L. Goldstein , “Familial Hypercholesterolemia,” (1974).
- 7O. Maliachova and S. Stabouli , “Familial Hypercholesterolemia in Children and Adolescents: Diagnosis and Treatment,” Current Pharmaceutical Design 24, no. 31 (2018): 3672–3677.30317987 10.2174/1381612824666181010145807 · doi ↗ · pubmed ↗
- 8F. Civeira , “Hypercholesterolemia I Po Mo F. Guidelines for the Diagnosis and Management of Heterozygous Familial Hypercholesterolemia,” Atherosclerosis 173, no. 1 (2004): 55–68.15177124 10.1016/j.atherosclerosis.2003.11.010 · doi ↗ · pubmed ↗