Sequence type and strain-level detection of Klebsiella pneumoniae in culture-enriched bacterial metagenomes: comparative performance of mSWEEP and StrainGE bioinformatic tools
Dorota Julia Buczek, Wasifa Kabir, Kenneth Lindstedt, Tommi Mäklin, Harry A. Thorpe, Yutaka Suzuki, Jukka Corander, Ørjan Samuelsen, Arnfinn Sundsfjord

TL;DR
This study compares two bioinformatic tools, mSWEEP and StrainGE, for accurately detecting Klebsiella pneumoniae sequence types in gut samples, showing they outperform traditional methods.
Contribution
The study evaluates and validates the performance of mSWEEP and StrainGE for strain-level detection of K. pneumoniae in metagenomic data.
Findings
Both mSWEEP and StrainGE accurately detect K. pneumoniae sequence types in 93-94% of samples with single STs.
mSWEEP and StrainGE detect within-sample ST diversity in 19 and 15 samples, respectively.
StrainGE reconstructs accurate phylogenetic relationships for strains of the same ST.
Abstract
Klebsiella pneumoniae is a major cause of human infections and is frequently associated with antimicrobial resistance (AMR). Carriage of K. pneumoniae in the gut is a major risk factor for infection and a reservoir for the spread of high-risk clonal lineages and associated AMR determinants. Accurate detection of K. pneumoniae at the subspecies level is therefore essential to better understand K. pneumoniae gut colonization ecology and clonal dissemination. We analysed two recently developed bioinformatic tools, mSWEEP and StrainGE, for sequence type (ST) detection of K. pneumoniae in culture-enriched sweep metagenomes compared to single-colony whole-genome sequencing (WGS). We show that both mSWEEP and StrainGE perform highly accurate ST detection, concordant with culture in 46/49 and 44/49 samples with WGS-detected single STs, respectively, as well as in 2/3 samples with two…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
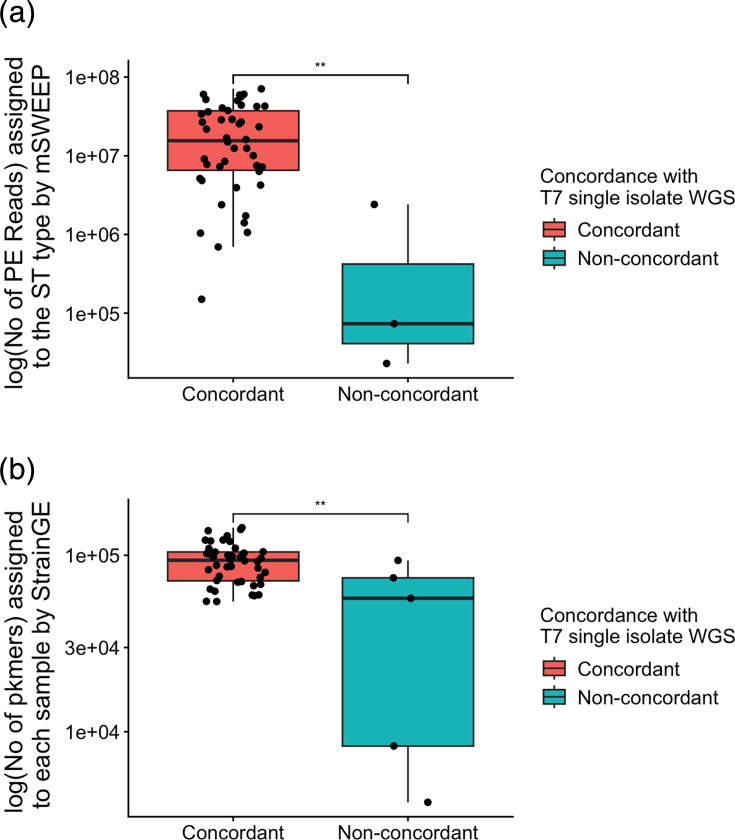 Fig. 1
Fig. 1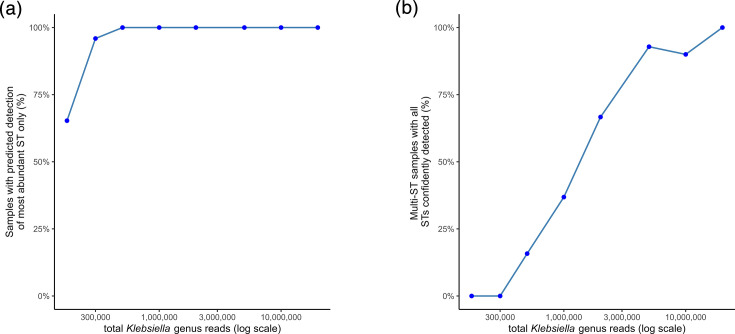 Fig. 2
Fig. 2| Kpn ST20 | Kpn ST25 | Kpn ST26 |
|---|---|---|
| SWE14-WMS* | SWE46-WMS | SWE26-WMS |
| SWE36-WMS | SWE35-WMS* | SWE20-WMS |
| SWE72-WMS | SWE45-WMS | SWE23-WMS |
| – | – | SWE85-WMS* |
| SWE14-WGS reference ST* | SWE35-WGS reference ST* | SWE85-WGS reference ST* |
| ID | Total PE reads per sample post-QC | T7 single colony | mSWEEP | StrainGE | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (%) § | No of PE reads assigned to the ST type | Concordance with T7 single colony | (%) § | pkmers || | score ¶ | Concordance with T7 single colony | ||||||||
| SWE1* | 46320236 | 27.6 | Kp1 ST4633 | Kp1 | ST4633 | 100 | 12435560 | yes | Kp1 | ST4633 | 96.8 | 70933 | 0.989 | yes |
| SWE2* | 50571824 | 86.2 | Kp1 ST3043 | Kp1 | ST3043 | 95.9 | 42270436 | yes | Kp1 | ST3043 | 96.1 | 102087 | 0.99 | yes |
| SWE3* | 42090900 | 28.1 | Kp1 ST1750 | Kp1 | ST1750 | 92.9 | 10045474 | yes | Kp1 | ST1750 | 82.8 | 139981 | 0.77 | yes |
| Kp4 | ST4096 | 0.7 | 559500 | – | Kp3 | ST4608 | 7.4 | 0.298 | – | |||||
| – | – | – | – | K4 | ST4096 | 6.3 | 0.355 | – | ||||||
| SWE7* | 40182364 | 20.3 | Kp1 ST1862 | Kp1 | ST1862 | 80.4 | 6341806 | Yes | Kp1 | ST1862 | 47.7 | 102166 | 0.833 | yes |
| Kp4 | ST1308 | 12 | 1784194 | – | Kp4 | ST1308 | 48.2 | 0.552 | – | |||||
| Kp4 | ST480-2LV | 7.6 | 1763298 | – | – | – | – | – | ||||||
| SWE8* | 40894246 | 6.1 | Kp1 ST14 | Kp4 | ST4096 | 99.6 | 2408212 | No | Kp4 | ST4096 | 94.3 | 57059 | 0.976 | no |
| SWE11* | 86612976 | 0.9 | Kp1 ST4255 |
| – | 22942 | No |
| – | 8296 | – | no | ||
| SWE12* | 105237936 | 59.5 | Kp1 ST70 | Kp1 | ST70 | 100 | 60290028 | Yes | Kp1 | ST70 | 93.9 | 121728 | 0.992 | yes |
| SWE13* | 99518122 | 34.7 | Kp1 ST45 | Kp1 | ST45 | 100 | 33881356 | Yes | Kp1 | ST45 | 96.8 | 92245 | 0.887 | yes |
| SWE14* | 106158610 | 92.8 | Kp1 ST20 | Kp1 | ST20 | 72.6 | 70926504 | Yes | Kp1 | ST20 | 95.8 | 128854 | 0.992 | yes |
| Kp1 | ST5100 | 27.4 | 26981624 | – | – | – | – | – | ||||||
| SWE18* | 97761156 | 27.1 | Kp1 ST4039 | Kp1 | ST4039 | 99.7 | 25525020 | Yes | Kp1 | ST4039 | 95.7 | 86471 | 0.984 | yes |
| SWE19* | 96593632 | 24.8 | Kp1 ST17 | Kp1 | ST17 | 92 | 21819079 | Yes | Kp1 | ST17 | 83.8 | 94281 | 0.944 | yes |
| Kp1 | ST16 | 7.8 | 1867809 | – | Kp1 | ST1428 | 6 | 0.042 | – | |||||
| SWE20* | 107490984 | 8.2 | Kp1 ST26 | Kp1 | ST26 | 93 | 7465576 | Yes | Kp1 | ST26 | 82.9 | 68390 | 0.91 | yes |
| SWE21* | 101125792 | 47 | Kp1 ST151 | Kp1 | ST151 | 91.9 | 26559926 | Yes | Kp1 | ST151 | 50.6 | 157363 | 0.871 | yes |
| Kp3 ST1209-3LV |
| – | 2808576 | No | Kp3 | ST355 | 9.4 | 0.345 | No | |||||
| – | – | – | – | Kp3 | ST1562 | 6.2 | 0.05 | No | ||||||
| SWE22* | 101959022 | 2.8 | Kp1 ST27 | Kp3 | ST3084 | 63.4 | 1690910 | Yes | Kp3 | ST3084 | 57.2 | 98873 | 0.611 | Yes |
| Kp3 3084 | Kp1 | ST27 | 34.6 | 979378 | yes | Kp1 | ST27 | 32.3 | 0.707 | Yes | ||||
| SWE23* | 94301508 | 9.5 | Kp1 ST26 | Kp3 | ST681 | 47.8 | 3761724 | Yes | Kp1 | ST26 | 50 | 109651 | 0.621 | Yes |
| Kp3 ST681 | Kp1 | ST26 | 42.6 | 3463628 | Yes | Kp3 | ST681 | 44.2 | 0.776 | Yes | ||||
| SWE34† | 43039094 | 42.2 | Kp1 ST157 | Kp1 | ST157 | 100 | 16855580 | Yes | Kp1 | ST157 | 93.6 | 84573 | 0.988 | Yes |
| SWE35† | 44126160 | 44.6 | Kp1 ST25 | Kp1 | ST10 | 84.3 | 15888556 | – | Kp1 | ST10 | 78.7 | 104240 | 0.932 | – |
| Kp1 | ST25 | 13.2 | 7214704 | Yes | Kp1 | ST25 | 12 | 0.525 | Yes | |||||
| SWE36† | 57118608 | 68.1 | Kp1 ST20 | Kp1 | ST20 | 100 | 37630002 | Yes | Kp1 | ST20 | 94.4 | 109208 | 0.989 | Yes |
| SWE40† | 42285706 | 6.4 | Kp1 ST4649 | Kp1 | ST4649 | 96.5 | 2387544 | Yes | Kp1 | ST4649 | 88.1 | 67195 | 0.951 | Yes |
| Kp1 | ST3164 | 5.4 | 10618180 | – | Kp1 | ST1565 | 29.3 | 0.139 | – | |||||
| SWE41† | 32664222 | 88.4 | Kp1 ST405 | Kp1 | ST405 | 100 | 26725162 | Yes | Kp1 | ST405 | 95 | 97582 | 0.992 | Yes |
| SWE42 | 42938740 | 26.2 | Kp1 ST4248 | Kp1 | ST4248 | 72.3 | 7280928 | Yes | Kp1 | ST4248 | 69 | 85974 | 0.906 | Yes |
| SWE43† | 43039094 | 18.5 | Kp1 ST4643 | Kp1 | ST4643 | 100 | 7481886 | Yes | Kp1 | ST4643 | 94.4 | 70385 | 0.96 | Yes |
| SWE44† | 55848022 | 74.7 | Kp1 ST23 | Kp1 | ST23 | 100 | 40507736 | Yes | Kp1 | ST23 | 48.6 | 109471 | 0.928 | Yes |
| – | – | – | – | Kp1 | ST1660 | 47 | 0.153 | – | ||||||
| SWE45† | 51827146 | 59.7 | Kp1 ST25 | Kp1 | ST25 | 86.2 | 23336270 | Yes | Kp1 | ST25 | 57.5 | 121952 | 0.938 | Yes |
| Kp1 | ST3164 | 5.4 | 10618180 | – | Kp1 | ST1565 | 29.3 | 0.139 | – | |||||
| SWE46† | 37854950 | 87.9 | Kp1 ST25 | Kp1 | ST461 | 60.2 | 21880254 | – | Kp1 | ST461 | 27.9 | 119817 | 0.832 | – |
| Kp1 | ST25 | 34.6 | 16104768 | Yes | Kp1 | ST25 | 18.5 | 0.413 | Yes | |||||
| SWE47† | 77698532 | 9.9 | Kp1 ST11 | Kp1 | ST11 | 95.6 | 4850910 | Yes | Kp1 | ST11 | 65.9 | 75996 | 0.91 | Yes |
| SWE70† | 69417118 | 4.3 | Kp1 ST37 | Kp1 | ST37 | 63.7 | 1722882 | Yes | Kp1 | ST37 | 90.2 | 62521 | 0.971 | Yes |
| Kp1 | ST256 | 21.3 | 2247810 | – | – | – | – | – | ||||||
| Kp1 | ST2370 | 11.3 | 2232222 | – | – | – | – | – | ||||||
| SWE71† | 50416960 | 0.6 | Kp1 ST2248 | Kp1 | ST2248 | 70.3 | 150136 | Yes | Kp1 | ST2248 | 52.9 | 54645 | 0.796 | Yes |
| SWE72† | 57398656 | 28.8 | Kp1 ST20 | Kp1 | ST20 | 97.8 | 12432384 | Yes | Kp1 | ST20 | 71.9 | 93006 | 0.947 | Yes |
| SWE73† | 53382614 | 55.9 | Kp1 ST10 | Kp1 | ST10 | 100 | 28641772 | Yes | Kp1 | ST10 | 91.9 | 99970 | 0.908 | Yes |
| SWE74† | 40635788 | 92.4 | Kp1 ST27 | Kp1 | ST27 | 100 | 36190262 | Yes | Kp1 | ST27 | 94.5 | 98679 | 0.989 | Yes |
| SWE75† | 62898100 | 96 | Kp1 ST485 | Kp1 | ST485 | 93.8 | 50181476 | Yes | Kp1 | ST485 | 81.3 | 143314 | 0.909 | Yes |
| Kp1 | ST35 | 6.2 | 20176438 | – | Kp1 | ST35 | 13.1 | 0.444 | – | |||||
| SWE84* | 62548172 | 1.4 | Kp1 ST200 | Kp1 | ST200 | 90.1 | 694590 | yes | Kp1 | ST200 | 76.2 | 54800 | 0.973 | Yes |
| SWE85* | 62401154 | 8.4 | Kp1 ST26 | Kp1 | ST26 | 74.9 | 3934072 | Yes | Kp1 | ST26 | 44.7 | 82744 | 0.897 | Yes |
| Kp1 | ST2791 | 15.9 | 1912382 | – | Kp1 | ST27 | 40.2 | 0.384 | – | |||||
| Kp1 | ST27 | 8.6 | 1625020 | – | Kp1 | ST2791 | 9.6 | 0.059 | – | |||||
| SWE86* | 64269478 | 95.7 | Kp1 ST515 | Kp1 | ST515 | 100 | 60569230 | Yes | Kp1 | ST515 | 97 | 100861 | 0.989 | Yes |
| SWE87* | 66225340 | 1.8 | Kp1 ST4658 | Kp1 | ST4658 | 89.2 | 1039800 | Yes | Kp1 | ST4658 | 85.2 | 59624 | 0.922 | Yes |
| SWE88* | 49928990 | 7.4 | Kp1 ST37 | Kp1 | ST37 | 71.1 | 1407258 | Yes | Kp1 | ST37 | 86.3 | 59010 | 0.964 | Yes |
| Kp1 | ST2370 | 16.1 | 2612108 | – | – | – | – | – | ||||||
| Kp1 | ST256 | 12.8 | 2607288 | – | – | – | – | – | ||||||
| SWE89* | 58437944 | 76.5 | Kp1 ST14 | Kp1 | ST14 | 100 | 44014250 | Yes | Kp1 | ST14 | 97.6 | 96020 | 0.987 | Yes |
| SWE90* | 56103864 | 44.3 | Kp1 ST1106 | Kp1 | ST1106 | 31.3 | 7787674 | Yes | Kp2 | ST1681 | 66.2 | 137705 | 0.53 | – |
| Kp2 | ST1681 | 6.3 | 6433926 | – | Kp1 | ST1106 | 25.1 | 0.417 | Yes | |||||
| Kp1 | ST449 | 5.4 | 46277106 | – | – | – | – | – | ||||||
| SWE91* | 63984542 | 93.9 | Kp1 ST1496 | Kp1 | ST1496 | 100 | 59207556 | Yes | Kp1 | ST1496 | 97.8 | 104627 | 0.986 | Yes |
| SWE92* | 54636568 | 91 | Kp1 ST35 | Kp1 | ST35 | 94.6 | 42732572 | Yes | Kp1 | ST35 | 97.8 | 97993 | 0.983 | Yes |
| Kp1 | ST449 | 5.4 | 46277106 | – | – | – | – | – | ||||||
| SWE93* | 58366906 | 0.3 | Kp1 ST20 | Kp1 | ST159 | 49.5 | 73494 | No |
| – | 3981 | – | No | |
| SWE94* | 61225402 | 8.8 | Kp1 ST704 | Kp1 | ST704 | 96.7 | 5146204 | Yes | Kp1 | ST704 | 96.3 | 64242 | 0.949 | Yes |
| SWE95* | 61898468 | 20.1 | Kp1 ST4632 | Kp1 | ST4632 | 67 | 8478160 | Yes | Kp1 | ST4632 | 57.2 | 87724 | 0.881 | Yes |
| SWE96* | 58234202 | 47.1 | Kp1 ST1145 | Kp1 | ST1145 | 100 | 26859338 | Yes | Kp1 | ST1145 | 97 | 79918 | 0.991 | Yes |
| SWE97* | 67057622 | 80.6 | Kp1 ST704 | Kp1 | ST704 | 96.5 | 52001674 | yes | Kp1 | ST704 | 77.3 | 120447 | 0.985 | Yes |
| – | – | – | – | Kp1 | ST253 | 19 | 0.038 | – | ||||||
| SWE98* | 57525522 | 8.3 | Kp1 ST4657 | Kp1 | ST4657 | 73.7 | 4241620 | Yes |
| 84.7 | 74444 | 0.902 | No | |
| Kp1 | ST584 | 11.9 | 3802654 | – | Kp1 | ST584 | 7 | 0.232 | – | |||||
| Kp1 | ST1832 | 7.8 | 1589170 | – | Kp1 | ST1832 | 4.7 | 0.102 | – | |||||
| Kp1 | ST2004 | 6.6 | 3346678 | – | Kp1 | ST2004 | – | – | – | |||||
| SWE99* | 61137200 | 2.3 | Kp1 ST461 | Kp1 | ST461 | 74.7 | 1060320 | Yes | Kp1 | ST461 | 93.2 | 59455 | 0.937 | Yes |
| Kp1 | ST3660 | 22.3 | 1179836 | – | – | – | – | – | ||||||
| SWE100* | 55978856 | 27.9 | Kp1 ST375 | Kp1 | ST375 | 99.7 | 14979132 | Yes | Kp1 | ST375 | 95.5 | 74785 | 0.972 | Yes |
| SWE101* | 59994.99 | 15.6 | Kp1 ST2042 | Kp1 | ST2042 | 77.9 | 9101702 | Yes | Kp1 | ST2042 | 97 | 71886 | 0.965 | Yes |
| Kp1 | ST4660 | 22.1 | 9022308 | – | – | – | – | – | ||||||
| SWE102* | 66525008 | 86.3 | Kp1 ST2039 | Kp1 | ST2039 | 100 | 56193170 | Yes | Kp1 | ST2039 | 96.5 | 102765 | 0.992 | Yes |
| SWE103* | 71616674 | 41.7 | Kp1 ST4660 | Kp1 | ST4660 | 74.2 | 29058728 | Yes | Kp1 | ST2042 | 94.4 | 93507 | 0.976 | No |
| Kp1 | ST2042 | 25.8 | 28862122 | – | – | – | – | – | ||||||
| WMS samples with concordance (+) and discrepancies (-) between mSWEEP and StrainGST ST results, respectively, compared to the single-colony WGS ST | ||||
|---|---|---|---|---|
|
|
|
|
|
|
| 44 | 0 | 2 | 3 | |
|
| 2 | 0 | 0 | 1 |
- —http://dx.doi.org/10.13039/100016190 Trond Mohn stiftelse
- —http://dx.doi.org/10.13039/501100007137 Helse Nord RHF
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntibiotic Resistance in Bacteria · Genomics and Phylogenetic Studies · Bacterial Identification and Susceptibility Testing
Data Summary
All data related to this work are reported in the article. Metagenomic data (raw MGI reads with human residues removed) are publicly available in ENA under BioProject PRJEB87654.
Introduction
Klebsiella pneumoniae (Kpn) is a gram-negative bacterium occupying a wide range of environmental niches. In humans, Kpn acts as a commensal and an opportunistic pathogen colonizing the gastrointestinal tract, causing infections of the urinary tract, respiratory tract and bloodstream in immunosuppressed patients, neonates and elderly [16]. Kpn is considered an important reservoir and vector in the dissemination of antimicrobial resistance (AMR), and gut carriage is the most important risk factor for clinical infections [68].
Kpn is a part of a wider K. pneumoniae species complex (KpSC) including seven phylogroups: K. pneumoniae sensu stricto (Kp1), K. quasipneumoniae subsp. quasipneumoniae (Kp2) and subsp. similipneumoniae (Kp4), Klebsiella variicola subsp. variicola (Kp3) and subsp. tropica (Kp5), K. quasivariicola (Kp6) and K. africana (Kp7) [5,912].
Studies of KpSC genomics have shown a highly diverse, but structured population consisting of deep-branching lineages of clonal groups (CGs) [5]. Two subsets of CGs are of particular clinical importance; those associated with the development of multidrug resistance and hypervirulence (HyV). For both subsets, the human gut ecosystem is considered an important reservoir.
However, we have limited knowledge on the diversity and dynamics of Kpn in human gut colonization. Culture-based population studies of healthy adults in Asia have shown a wide range in Kpn gut carriage rates (18–87%) [1314]. Similar culture-based studies of hospitalized patient in Australia, the UK and the USA have revealed Kpn colonization rates between 11 and 23% [61516]. A cross-sectional culture-based study of KpSC gut carriage in a Norwegian community-based large human adult general population cohort (n=2975; The Tromsø seven study; T7) showed a colonization prevalence of 16.3% and a high Kpn population diversity [17].
Standard culture-based detection methods in the study of Kpn gut carriage and ecology have limitations [3]. The gut bacterial communities contain not only complex mixtures of species but also a mixture of strains within species [1819]. Interestingly, recent progress in whole metagenomic sequencing (WMS) and bioinformatics has shown promising results in the ability to resolve bacterial microbiota complexity beyond the level of genus and species, supporting sequence type (ST) and strain-level detection [1819].
The primary aim of this study was to explore and compare two recently developed bioinformatic tools, mSWEEP [18] and Strain Genome Explorer (StrainGE) [19], in their abilities to identify the presence and diversity of Kpn strain STs in culture-enriched sweep mixed bacterial communities. We take advantage of Kpn culture-positive sample material and single-colony whole-genome sequencing (WGS) data from the previous T7 study [17].
Methods
Human faecal samples and corresponding whole-genome sequenced Kpn strains
The faecal samples (n=52) in this study were selected from KpSC culture-positive specimens within the 2975 KpSC SCAI (Simmons citrate agar with inositol) culture-screened faecal samples obtained during the cross-sectional T7 study [17]. Briefly, faecal samples were plated onto SCAI media (Sigma-Aldrich, Darmstadt, Germany) and incubated for 48 h at 37 °C [20]. Large, yellow, glossy colonies were selected as likely Klebsiella spp. and identified to the species level using MALDI-TOF MS (Bruker Daltonics, Bremen, Germany). The first colony identified as KpSC was kept for further analysis, except for three samples in which both K. pneumoniae and K. variicola isolates were identified in the same sample and kept for further analyses. DNA extraction, WGS and bioinformatic analysis were performed as described [19].
SCAI-sweep samples for WMS
The selected KpSC culture-positive samples (n=52), with corresponding WGS single KpSC isolates from the T7 study, were selected for SCAI-sweep enrichment-based WMS analysis as described [3]. Briefly, 50 µl from thawed ESwab samples was plated on SCAI media (Sigma-Aldrich). Following incubation at 37 °C for 48 h, all growth on plates was scraped, and one loaded 10 µl inoculation loop was used as input for DNA extraction using the Purelink Microbiome DNA purification kit (Thermo Fisher Scientific, Waltham, USA) with minor modifications as described [3]. DNA quality control was performed using NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific) and Qubit 3.0 fluorometer (Thermo Fisher Scientific).
WMS and data processing
Briefly, purified SCAI-sweep DNA was individually fragmented before library preparation and WMS on a NovaSeq 6000 platform (Illumina, San Diego, USA) with the 150 bp pair-end reads as described [3]. The FASTQ files were subjected to optical duplicates removal, adapter removal, sequence cleaning, human DNA removal and synchronization of unpaired reads as described [3].
KpSC quantification and taxonomy
Quality controlled (QC) reads were subjected to Kpn abundance quantification and taxonomy profiling. These steps were performed by Kraken2 (version 2.1.2) [21] and Bracken (version 2.6.1) [22], respectively. The Kraken2 default MiniKraken DB_8GB v202003 database was enlarged by the addition of 484 KpSC genomes from the T7 study [17] including Kp1 (n=303), Kp2 (n=31), Kp4 (n=16) and Kp3 (n=134), as well as 2109 KpSC genomes from the SpARK study [23] including Kp1 (n=427), Kp2 (n=62), Kp4 (n=38) and Kp3 (n=170). Moreover, six non-KpSC Klebsiella outliers were added: Klebsiella aerogenes, Klebsiella grimontii, Klebsiella huaxiensis, Klebsiella michiganensis, Klebsiella pasteurii and Klebsiella spallanzanii. We used the Bayesian Gaussian mixture model of PopPUNK version 2.0.2 to cluster the Kp1, Kp2, Kp4 and Kp3 genomes [24]. Furthermore, the python script (extract_kraken_reads.py) from the KrakenTools package [25] was used to extract FASTQ reads assigned to the taxonomy ID: 570 (Klebsiella genus) with ‘children’ based on the Kraken2 taxonomy classification output files.
KpSC ST typing using mSWEEP and StrainGE
We used the mSWEEP (version mSWEEP_linux-v1.5.2) and StrainGE (version 1.3.7) toolkits to analyse the Kpn ST presence and diversity in the described WMS Kpn-positive SCAI sweeps. Briefly, mSWEEP is designed to estimate the relative abundance of bacterial lineages using an extensive reference genome database [18]. mSWEEP clusters the reference genomes in biologically relevant groups, performs read pseudoalignment to the references, infers probabilistic cluster relative abundances and includes a step that controls the false-positive detections [18]. The Strain Genome Search (StrainGE) toolkit has two main components [19], the Strain Genome Search Tool (StrainGST) and the Strain Genome Recovery (StrainGR). Briefly, StrainGST is a k-mer-based tool that indicates the closest reference genome(s) to the strain(s) in a sample. The StrainGR pipeline builds a concatenated reference based on the reference strains reported by StrainGST, aligns reads and analyses alignments to variant calls (SNPs and large deletions) [19]. The StrainGST can be used by itself as a standalone tool to report the presence of closely related genomes to the reference database and their relative abundance.
To identify the Klebsiella strain level and ST diversity, the extracted reads obtained from the Kraken2 output were subjected to the mSWEEP default database [18] and a custom StrainGE [19] database consisting of 1,305 Kpn NCBI refseq genomes downloaded on 18 October 2022 using NCBI Genome Downloading Scripts version 0.3.1 (https://github.com/kblin/ncbi-genome-download). Both databases were enriched by KpSC genomes from T7 (n=484) and SpARK collection (n=2109). ST detection was performed for both methods with a cut-off of 5% relative abundance to decrease possible false-positive Kpn ST-type detections.
Genomic characterization of Kpn mSWEEP bins
As mSWEEP supports the read binning algorithm by mGEMS, we further evaluated mSWEEP bins for downstream analysis [26]. Specific Kpn ST bins were assembled using SPAdes (version 3.15.5) [27]. Contigs below 1,000 bp and 2× coverage were removed during the assembly to reduce the number of low-quality contigs that might interfere with downstream analysis. Validation of species detection, K-locus, O-locus, AMR and virulence genes was performed on the assembled genomes using Kleborate (version 2.3.2) [2829].
Evaluation of StrainGR for phylogenetic relationship analysis
Phylogenetic analysis was performed using the StrainGR pipeline of the StrainGE toolkit. Briefly, StrainGR builds neighbour-joining trees from pairwise distances between strains based on genomic distances calculated from SNP rates via the Jukes–Cantor model. Phylogenetic relationships were constructed between SCAI-sweep WMS samples for three different STs (ST20, ST25 and ST26) which had been detected in at least three samples each (Table 1). A single-colony WGS isolate ST corresponding to one of the WMS samples was used as reference (Table 1).
A core SNP alignment was performed for single isolates (ST20) using Snippy (version 4.6.0) (https://github.com/tseemann/snippy). SNP distance matrix was calculated by snp-dists (version 0.8.2) from the core alignment file (https://github.com/tseemann/snp-dists). Maximum likelihood phylogeny of the core genome alignment was constructed using RAxML (RAxML, version 8.2.12) with 1,000 bootstrap [30]. All the phylogenetic trees were visualized by FigTree (version 1.4.4) (http://tree.bio.ed.ac.uk/software/figtree/).
Statistical analysis
All statistical analysis was performed in R version 4.3.2. Statistical differences between two groups were analysed using a one-sided Wilcoxon signed-rank test (unpaired). Samples in which only a single ST was detected were included to compare concordant and non-concordant groups for mSWEEP and StrainGE and to evaluate the effect of read numbers on mSWEEP performance and p-kmers on StrainGE performance. Analysis of differences between three or more groups was performed by Kruskal–Wallis test followed by Wilcoxon test with Bonferroni correction. Concordance between ST detection by WGS and each metagenomic method (mSWEEP and StrainGE) was quantified using Cohen’s kappa (κ), which measures agreement between categorical classifications beyond chance, using the R package irr (version 0.84.1). P-values ≤0.05 were considered significant.
Rarefaction analysis
A simulation-based rarefaction analysis was performed to estimate the effective Klebsiella genus read sequencing depth required for reliable ST detection. For each sample with a WGS-concordant ST detected, the vector of mSWEEP-assigned read counts to the concordant ST was down-sampled to a series of target Klebsiella genus read depths using multinomial sampling. Target depths began at the lowest Klebsiella genus read count observed in our sample collection (SWE93) and increased in intervals up to 20 million Klebsiella genus reads (175,101; 3×10⁵; 1×10⁶; 2×10⁶; 5×10⁶; 1×10⁷; and 2×10⁷ reads). At each depth, down-sampling was repeated 100 times to account for stochastic variation, and an ST was considered detected if the down-sampled ST-assigned read count exceeded an empirically derived threshold of 1.5×10^5^ reads. This threshold was defined as the lowest number of mSWEEP-assigned reads associated with an ST that was concordant with WGS (SWE71), compared to the highest read count among non-concordant ST detections not attributable to colony picking of a low-abundance ST (SWE8). Rarefaction analysis was conducted using mSWEEP outputs only, due to the high concordance between mSWEEP and StrainGE in both ST detection and agreement with WGS results.
Computational resources
The computations were performed on resources provided by Sigma2 – the National Infrastructure for High Performance Computing and Data Storage in Norway (https://www.sigma2.no/) (project number: nn9794k).
Ethics statement
Collection and analysis of samples for this study was approved by the Regional Committee for Medical and Health Research Ethics, North Norway (REC North reference: 2016/1788, 2014/940 and 67006).
Results
Detection Kpn ST by mSWEEP and StrainGST in SCAI-sweep metagenomes compared to single-colony WGS
A total of 52 Kpn culture-positive faecal samples underwent SCAI-sweep enrichment WMS, followed by analysis by mSWEEP and the StrainGST component of StrainGE for Kpn ST presence and diversity and compared to ST detection by single-colony WGS. The selected SCAI-sweep samples, total read counts, percentage of Kraken2 extracted Klebsiella genus reads, single-colony WGS Kpn-isolate ST results, the WMS Kpn ST-typing results and ST-matching read and k-mer numbers from analysis by mSWEEP and StrainGE, respectively, are given in Table 2. Notably, 49 samples had a single Kpn ST detected by single-colony WGS, whereas three samples (SWEs 21–23) had a Kp1 and Kp3 ST detected within the same sample. Processed WMS samples ranged from 32 to 107×10^6^ PE reads (mean: 62×10^6^; median: 58×10^6^; sd: 22×10^6^) (Table 2). Kraken2 extracted Klebsiella (genus) reads varied from 0.3 to 96% relative abundance of sweep metagenomes (mean: 39.2%, median: 28%, sd: 33.2%).
The overall concordance in Kpn ST detection between mSWEEP and StrainGST compared to the single-colony WGS is presented in Table 3. Briefly, mSWEEP-detected STs were concordant with the reference single-colony WGS ST in 46/49 single-ST and 2/3 double-ST samples, respectively. The corresponding results for StrainGST were 44/49 and 2/3, respectively. This gave very strong agreement in ST detection with WGS for both methods, with Cohen’s κ 0.93 (P<0.001) for mSWEEP and 0.89 (P<0.001) for StrainGE, respectively.
Discrepancies to single-colony WGS STs were observed in samples SWE8, SWE11 and SWE93 in both mSWEEP and StrainGST metagenome analysis. Both mSWEEP and StrainGST detected the same alternative and dominant ST (ST4096) in SWE8 compared to the single-colony WGS-detected ST14, suggesting that the colony picking for WGS analysis had selected a minority ST in this sample. In SWE93, mSWEEP analysis detected an alternative ST (ST159 vs single-colony WGS ST20), whereas no Kpn-ST was identified by StrainGST, suggesting that this was not due to the detection of low-abundance ST by the colony picking strategy. No ST was detected by either mSWEEP or StrainGE in SWE11 (single-colony WGS ST4255). A discrepancy to single-colony WGS ST was also observed in SWE103 in StrainGST analysis only (ST2042 vs single-colony WGS ST4660). In this sample, StrainGST identified an alternative ST (ST2042) which was also identified by mSWEEP as the second most abundant Kpn-ST and is likely a true-positive finding. Similarly, StrainGE did not detect the single-colony WGS-detected ST4657 in SWE98; however, it detected three additional STs which were also detected by mSWEEP.
Regarding sweep samples with two detected STs from single-colony WGS (SWE21-23), both STs were identified by mSWEEP and StrainGST in 2/3 samples (SWE22 and -23). Only one of the WGS-detected STs (ST151) was detected by both mSWEEP and StrainGST in SWE21. The secondary WGS-detected ST in SWE21, ST1209-3LV (Kp3 phylogroup), was not detected by either mSWEEP or StrainGST. In this sample, two alternative Kp3 STs were detected by StrainGST (ST355 and ST1562), while no additional STs were detected by mSWEEP.
The relative abundance of Klebsiella genus reads estimated by Kraken2 in sweep samples with concordant results to single-colony WGS ranged from 0.6 to 96 % (mean: 41.5%, median: 28.8% and SD: 32.9%). Despite sample SWE71 having the second lowest abundance of Klebsiella genus reads of all sweep samples (0.6%), this sample was still concordant with the single-colony WGS ST. The Kraken2 estimated Klebsiella genus abundances in sweep samples that were non-concordant with single-colony WGS by both mSWEEP and StrainGST (SWE8, -11 and -93) were low (6.1%, 0.9% and 0.3% respectively).
We next investigated whether the observed non-concordance of the STs between single-colony WGS detection and mSWEEP/StrainGE was related to a lower number of reads assigned to STs by these tools. Comparing the number of reads assigned by mSWEEP between concordant and non-concordant STs demonstrated that concordant STs had a significantly higher number of assigned reads than the non-concordant STs (P=0.002) (Fig. 1a). Similarly, comparing the total number of unique k-mers in samples that matched the database (pk-mers) between concordant and non-concordant samples by StrainGST demonstrated that concordant samples had significantly higher number of k-mers than the non-concordant samples (P=0.006) (Fig. 1b) .
*Comparison of (a) read numbers assigned to STs by mSWEEP and (b) k-mers in samples assigned to the KpSC database by StrainGST (pk-mers) between samples with concordant and non-concordant STs to single-colony WGS ST detection. *P<0.01 (single-tailed Wilcoxon signed-rank test).
Detection of within-sample ST diversity
Within-sample Kpn ST diversity (≥2 STs) was detected in 19 and 15 samples by mSWEEP and StrainGST analysis, respectively (Table 2). Twelve sweep metagenomes with multiple STs identified overlapped between mSWEEP and StrainGST, of which the same additional STs were identified by both tools in ten samples. The maximum number of STs identified per sample was four detected by mSWEEP in sample SWE98 (Table 2).
Estimation of minimum required genome coverage and rarefaction analysis
Minimum genome coverage for reliable ST detection was estimated from sample SWE71, as this sample had the lowest number of mSWEEP assigned reads and StrainGE assigned pk-mers that resulted in ST detection concordant with WGS (n=150,136 reads; Table 2). Assuming an average genome size of 5.5 Mbp, this equated to ~4.1× genome coverage at 150 bp reads [31]. Compared to sample SWE93, which had the highest number of mSWEEP assigned reads to a non-concordant ST that was not detected by StrainGE, thus could not be explained by colony picking of a low-abundance ST (n=73,494 reads; Table 2), this approximated to 2.0× genome coverage. Thus, under the experimental conditions used, we consider a threshold of ~150,000 assigned reads – corresponding to ~4× genome coverage – to be a practical minimum requirement for reliable ST detection.
Using this threshold, we performed rarefaction analysis on all samples with a concordant ST detected by mSWEEP (n=49) to investigate the influence of sequencing depth on detection of concordant STs as well as within-sample ST diversity. Rarefaction was performed on the total number of reads assigned to the Klebsiella genus, as this was the input to both mSWEEP and StrainGE, and showed large variation between samples (median: 1.73×10^7^ reads, range: 1.73×10^5^–9.85×10^7^; Table 2). When samples were rarefied to the lowest number of Klebsiella genus reads observed across our dataset (SWE93; n=175,101 reads; Table 2), this analysis predicted that mSWEEP would have detected the most dominant ST only 65.3% of samples (n=32/49). In all cases, this would have corresponded to the concordant ST detected by WGS. Detection of at least one ST in 100% of samples, however, was not predicted until there were at least 500,000 Klebsiella genus reads (Fig. 2a). Similarly, of the samples with multiple STs detected by mSWEEP (n=19), rarefaction analysis predicted that even at depths of up to 1×10^6^ Klebsiella genus reads, mSWEEP would have failed to detect more than one ST in 63.2% of samples, and detection of full ST diversity in 100% of samples would occur only at depths above 20×10^6^ Klebsiella genus reads (Fig. 2b).
Rarefaction analysis estimating Klebsiella genus read depth required for reliable ST detection. (a) Percentage of samples in which detection of the dominant ST would be predicted to occur at varying Klebsiella genus read depths. (b) Percentage of samples with multiple STs detected in which all STs were predicted to be detected at varying Klebsiella genus read depths. The threshold for confident ST detection (n=150,000 ST-assigned reads by mSWEEP) was derived from the sample with WGS concordant ST detection that had the lowest observed Klebsiella genus reads (n=150,136, Table 2), compared to the sample with the highest observed Klebsiella genus reads that resulted in non-concordant ST detection (n=73,494, Table 2).
Comparison of computational resources
We also compared computational resources required for ST detection and abundance by mSWEEP and StrainGE. In our computational setup, mSWEEP required on average 11 h per sample (number of tasks: 1, CPUs per task: 15, memory per CPUs: 66 GB), while StrainGE required on average 1 h per sample (number of tasks: 4, CPUs per task: 8, memory per CPUs: 38 GB). Additionally, while mSWEEP ST analysis required access to high-performance computing, StrainGE ST analysis could be run on a laptop.
Downstream analysis of the mSWEEP bins
The assembled Kpn binned reads from mSWEEP were further analysed for species, STs, virulence genes, K-locus, O-locus and AMR genes using the Kleborate/Kaptive pipeline (Table S1, available in the online Supplementary Material). We observed species match in 47 assembled genomes with single-colony WGS reference genomes. The five exceptions included SWE-8 (no KpSC match) as well as SWE-11, -71, -90 and -93 (KpSC not detected). We observed 100% concordance in STs between assembled and single-colony WGS genome for the remaining 47 assemblies. Assembly analysis of two samples (SWE-14 and −19) with two STs detected by mSWEEP revealed only one of the reference STs (ST20) in SWE-14 and the reference ST (ST17) plus a single-locus variant of the reference ST (ST17-1LV) in SWE-19.
We also compared virulence genes, K-locus, O-locus and AMR genes between the assembled genomes and single-colony WGS reference genomes for samples that matched STs with corresponding reference STs. Virulence score differed in nine assembled genomes (SWE-34, -47, -72, -92, -94, -100, -101, -102 and -103). Most of the virulence profile matched with a few exceptions: virulence factors were not detected either in reference (yersiniabactin: SWE-44) or in the assembled genome (yersiniabactin: SWE-13, -21, -35, -46, -47, -70, -92 and -94; colibactin: SWE-101, -102 and -103; aerobactin and salmochelin: SWE-100). Hypermucoviscosity rmp gene profiles matched across all assembled Kpn bins and reference genomes. K-locus was consistent in 45 of the assembled genomes with references (no match for SWE-42 and -95). We observed differences in O-locus for three samples (SWE-40, -75 and -99).
Overall, we observed no differences in Kleborate resistance scores between the assembled bins and their corresponding single-colony WGS reference genomes. Acquired fluoroquinolone resistance (Flq_acquired) gene profiles matched for all the assembled bins with their reference genome. However, acquired aminoglycoside resistance genes (AGly_acquired) were undetected in SWE-35, -40, -45 and -47 assembled bins but detected in their corresponding reference genomes. Similarly, there were no mismatches in detectable acquired resistance genes against sulphonamide (Sul_acquired), tetracycline (Tet_acquired) and trimethoprim (Tmt_acquired), but several bin assemblies lacked detection (Sul_acquired: SWE-35, -40, -43, -45 and -46; Tet_acquired: SWE-35, -40, -45 and -46; and Tmt_acquired: SWE-35 and -40). Acquired β-lactamase genes (Bla_acquired) and SHV mutation profiles also matched very well between assembled and reference genomes with few exceptions (Bla_acquired: SWE-47, -84 and -88; SHV_mutations: SWE-47 and -88).
Assembly metrics (total genome length and N50) did not show any significant differences (P>0.4) in total assembly length between genomes that matched features with single-colony WGS reference genomes (median=5,700,622, IQR=5,563,627–6,390,470), genomes that had mismatch features with single-colony WGS reference genomes (median=6,304,149, IQR=5,902,887.75–7,408,573.5) and genomes where the features were undetected/unknown (median=6,032,838.5, IQR=5,411,485.75–7,035,193). But there was a significant reduction in N50 values (P=0.035) of assembled genomes where some features were undetected/unknown (median=101,011, IQR=15,815.5–150,611.25) than the genomes where all the features matched with reference genomes (median=159,384, IQR=92,876–180,565).
Evaluation of StrainGR in analysing phylogenetic relationships between identical Kpn STs from different metagenome samples
To investigate the ability of the StrainGR component of StrainGE to accurately analyse relationships between closely related Kpn strains, phylogenetic analysis was performed on all sweep samples in which the same ST had been identified [ST20 (n=3 samples), ST25 (n=3 samples) and ST26 (n=4 samples)], compared to a single-colony WGS isolate from one of the samples as reference (Table 1). Phylogenetic analyses of ST20 samples (SWE14, SWE36 and SWE72) confirmed that ST20 detected in the SWE14 sweep metagenome was the same strain as the corresponding single-colony WGS isolate SWE14 ST20, localizing within the same clade with no estimated genetic distance between these two STs (Fig. S1, Table S2a). The SWE36 and SWE72 WMS-detected ST20 strains belonged to a different clade than both the SWE14 WMS-detected ST20 strain and corresponding single-colony WGS ST20 strain (Fig. S1). Similarly, analysis of the ST26-containing sweep samples (SWE20, SWE26, SWE23 and SWE85) confirmed the presence of SWE85 WMS-detected ST26 and corresponding single-colony WGS ST26 in the same clade, although estimated a genomic distance of 0.000001 between these strains (Fig. S2, Table S2c). The SWE20, SWE26 and SWE23 were more distantly related (Fig. S2, Table S2c). Analysis of ST25 samples (SWE46, SWE45 and SWE35), however, demonstrated the presence of SWE35 WMS-detected and single-colony WGS ST25 strains in different clades with estimated genomic distance of 0.00001 (Fig. S3, Table S2d). Notably, this ST25 was identified as the minority ST in this sweep sample, which was dominated by ST10, with relative abundances 12 and 78.7% respectively. Moreover, the unique k-mers assigned to ST25 in this sweep sample (n=11,042) were considerably lower than the average unique k-mers assigned to all STs in the phylogenetic analysis (n=47,641). The single-colony WGS SWE35 ST25 strain was placed in the same clade as the SWE46 WMS-detected ST25 despite not corresponding to this sweep sample (Fig. S3). The SWE45 WMS-detected ST25 was assigned to a different clade than the other three ST25 strains (Fig. S3).
The accuracy of the phylogenetic relationships between the WMS-detected ST20 strains created by StrainGR was also compared to the core genome phylogeny of the corresponding ST20 isolates from these samples. The core genome phylogeny of ST20 isolates demonstrated similar phylogeny structures compared to that of ST20 metagenome samples created by StrainGR (Fig. S4, Table S2b). As was identified by StrainGR, the single-colony WGS STs from SWE36 and SWE72 were more closely related to each other than single-colony isolate from SWE14, depicted from the location of the clades (Fig. S4). The ST25 SWE35 single-colony WGS isolate, which was included as a distantly related outlier, was located furthest away in the tree. The pairwise SNP distance matrices of the ST20 isolates also had similar relative values to the corresponding pairwise estimated genomic distances between WMS-detected ST20 strains (Table S2a, b). Thus, phylogenetic relationships of the ST20 single isolates using core genome alignment corresponded to the phylogenetic relationships of the WMS-detected ST20 strains predicted by StrainGR.
Discussion
The global spread of AMR by Kpn is tightly linked to the clonal dissemination of a relatively small number of high-risk lineages [532]. Moreover, HyV in Kpn is strongly associated with lineages within CG23 [33]. The ability to accurately detect and analyse Kpn at the subspecies level is therefore essential for public health surveillance and to better understand Kpn colonization dynamics and ecology. Here, we have taken advantage of our previous study of SCAI-sweep faecal samples with identified Kp1 and Kp3 STs using single-colony WGS [317] to evaluate and compare the performance of two newly developed WMS-based bioinformatic tools to identify Kpn ST diversity in culture enriched bacterial metagenome samples.
Both the mSWEEP and StrainGST analysis of culture-enriched SCAI-sweep metagenomes showed a high concordance with the expected Kpn STs from the previous single-colony WGS data [17]. The non-concordance observed in STs was directly proportional to the lower number of PE reads and k-mers assigned by mSWEEP and StrainGE, respectively. Moreover, the agreement between mSWEEP and StrainGE in alternate STs detected in non-concordant samples SWE8, SWE98 and SWE103 suggests that these were true STs present in these samples that were not detected by WGS due to the single-colony picking strategy used.
Our findings further indicate the importance of sufficient read coverage in making accurate ST identification by these tools. The minimum coverage required for accurate strain identification by mSWEEP has been reported as ~0.30× for Kpn, corresponding to at least 16,000 reads from a specific lineage in a dataset of 1,000,000 reads (100 bp read length) [18]. Similarly, StrainGE reports as low as 0.1 × coverage for accurate strain detection [19]. Based on our results, under the experimental conditions used, we estimated that sufficient read coverage to ~4×, corresponding to approximately 150,000 ST-specific reads (150 bp read length). This increased requirement may have been related to the SCAI-culture enriched faecal samples retaining a complex assortment of closely related Enterobacterales, as well as the high diversity and close relatedness of the KpSC phylogeny itself, making reliable differentiation between STs challenging [2031]. Indeed, rarefaction analysis predicted that if all samples had received the same number of Klebsiella genus reads equal to that of the lowest sample, mSWEEP would have only detected 65.3% of concordant STs. Similarly, detection of the full within-sample ST diversity achieved by mSWEEP was not predicted to occur until the Klebsiella genus reached 20×10^6^ reads. As only 46% of all samples (n=24/52) reached this depth, it is therefore possible that our analysis may have underestimated true Kpn ST diversity of our samples.
In the three samples with two STs detected by single-colony WGS, both mSWEEP and StrainGST correctly identified both STs in two samples and one of the two STs in the third sample. Furthermore, we observed good concordance between mSWEEP and StrainGST in detection of within-sample Kpn ST diversity in 10 out of 12 samples. According to the nutrient-niche hypothesis, closely related strains and species are potentially important competitors within the gut microbiome due to large overlap in metabolic requirements [34]. Indeed, Klebsiella oxytoca has been suggested to be an important inhibitor of Kpn within the gut microbiome in vivo [35]. Within-sample Kpn strain diversity and co-carriage of Kpn with other Klebsiella genera, however, has been previously reported [3638]. Moreover, horizontal gene transfer (HGT) of carbapenemase genes between Kpn strains has been observed within a co-colonized gut microbiome [39]. Studying co-colonization of multiple Kpn strains within a single gut microbiome is therefore important to better understand within-species competition and cooperation, and the extent and conditions under which HGT occurs in this setting. The ability of WMS-based tools to accurately detect and study within-sample Kpn ST diversity is therefore an important advantage of this technology over single-colony WGS, in which such detection is more laborious.
Genomic analysis beyond ST detection, such as identifying determinants of AMR and virulence, has significant impact on treatment decisions and infection outcomes. mSWEEP is designed for metagenomic binning which allows for genome assembly-based downstream analysis of such determinants. However, it is important to firstly evaluate the reliability of Kpn read-binning by mSWEEP in order to perform accurate downstream analysis. The majority of genomes assembled from the mSWEEP bins reported the same species as the single-colony WGS reference genome with only two mismatches. Although there was no discrepancy in detected STs between mSWEEP-assembled genomes and the corresponding single-colony WGS reference genomes, we found additional STs detected from direct mSWEEP analysis in two samples that did not match either the mSWEEP-assembled genomes or the single-colony WGS reference. These additional STs differed from the mSWEEP-assembled genomes by only a single allele, suggesting that direct mSWEEP detection had assigned a single strain to two very closely related genomes in its database. These findings indicate that ST assignment based on assembly of the mSWEEP bins gives better resolution of ST detection compared to direct mSWEEP analysis. We also screened the assembled Kpn genomes from mSWEEP bins for Kpn serotype as well as AMR and virulence determinants and compared these with single-colony WGS reference genomes. Most of the assembled genomes had the same K-locus, wzi allele, O-locus and yersiniabactin as the single-colony WGS reference genome. Most of the virulence and resistance profiles of the assembled genomes also matched the corresponding reference genomes. Assembly metrics indicated that fragmentation or lack of contiguity measured by reduced N50 might affect successful downstream analysis. These suggest that mSWEEP bins can be useful for downstream analysis; however, data generated from mSWEEP bins needs to be evaluated carefully to prevent misinterpretation.
During clonal outbreaks, it is crucial to identify infection sources and characterize transmission dynamics to support targeted containment efforts [40]. Phylogenetic analysis reconstructs the evolutionary relationships between strains, which helps to track transmission between individuals or communities and identify the sources of infection or reservoirs [41]. StrainGR phylogenetic analysis showed that, for two of the three STs tested (ST20 and ST26), isolate and sweep samples from the same individual were more closely related than sweep samples of the same ST from different individuals. Core genome alignment of ST20 isolates also validated this finding. However, StrainGR failed to identify the single-colony WGS ST25 isolate from sample SWE35, and the corresponding WMS-detected ST25 was in fact the same strain. ST25 in this sample was detected as a minority ST with comparatively lower k-mer coverage by StrainGST. This indicated that the accuracy of phylogenetic relationships predicted by StrainGR may be compromised at lower coverage levels. Although our findings demonstrate that StrainGR shows promise in recreating phylogenetic relationships between closely related Kpn strains, further studies involving a larger number of samples containing closely related strains are required to better define the accuracy and limitations of StrainGR in this setting.
Both tools performed well in terms of identifying the Kpn ST that corresponded to single-colony WGS detection. mSWEEP required more CPU hours and more computing resources compared with StrainGST, which was related to the binning process of mSWEEP. Despite this, the ability to bin reads is an important advantage of mSWEEP, allowing recovery and assembly of strain genomes for in-depth downstream analysis. While StrainGE does not have this function, it nonetheless demonstrated the ability to create accurate phylogenetic relationships between closely related strains. In addition, within the StrainGR component of StrainGE is also the ability to perform strain-aware variant calling at low coverages which was not explored as part of this study [19]. Both algorithms have no limitations considering the input size (sample size). Outputs for both tools were easily interpreted. Overall, this study suggests the preferred use of mSWEEP when the research goal involves reconstructing or characterizing complete strain genomes from metagenomic data. In contrast, StrainGE is more suitable for rapid and computationally efficient detection of strain-level diversity in large datasets. Both tools produce interpretable outputs and can be effectively applied depending on the specific analytical objective and available computational resources.
The limitations of the study were the low number of samples examined and the limited number of WGS single colonies picked per sample for comparison to sweep WMS detection. Additionally, the reference database included Kpn genomes isolated from the same faecal sweep samples used in the analysis, which introduces the possibility of detection bias. However, both tools had access to the same reference database which also had unique STs from the baseline T7 study making the analysis comparable [17].
Conclusions
This study demonstrates the potential of WMS-based tools to perform accurate sub-species detection of Kpn in complex microbial communities. While StrainGE and mSWEEP both accurately detected STs when compared to single-colony WGS, both tools had differing strengths and weaknesses. These were predominantly related to differences in computing requirements and potential for downstream genome recovery and analysis. The ability to accurately detect within-sample ST diversity was a strength of both StrainGE and mSWEEP. Utilizing these tools in future studies can enhance our understanding of Kpn colonization ecology and dynamics, with potential to inform future public health surveillance and high-risk Kpn strain tracking.
Supplementary material
10.1099/mgen.0.001638Uncited Supplementary Material 1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kopotsa K Mbelle NM Osei Sekyere J Epigenomics, genomics, resistome, mobilome, virulome and evolutionary phylogenomics of carbapenem-resistant Klebsiella pneumoniae clinical strains Microb Genom 20206 mgen 00047410.1099/mgen.0.00047433170117 PMC 8116673 · doi ↗ · pubmed ↗
- 2Ramos PIP Fernández Do Porto D Lanzarotti E Sosa EJ Burguener G et al An integrative, multi-omics approach towards the prioritization of Klebsiella pneumoniae drug targets Sci Rep 201881075510.1038/s 41598-018-28916-730018343 PMC 6050338 · doi ↗ · pubmed ↗
- 3Lindstedt K Buczek D Pedersen T Hjerde E Raffelsberger N et al Detection of Klebsiella pneumoniae human gut carriage: a comparison of culture, q PCR, and whole metagenomic sequencing methods Gut Microbes 202214211850010.1080/19490976.2022.211850036045603 PMC 9450895 · doi ↗ · pubmed ↗
- 4Effah CY Sun T Liu S Wu Y Klebsiella pneumoniae: an increasing threat to public health Ann Clin Microbiol Antimicrob 202019110.1186/s 12941-019-0343-831918737 PMC 7050612 · doi ↗ · pubmed ↗
- 5Wyres KL Lam MMC Holt KE Population genomics of Klebsiella pneumoniae Nat Rev Microbiol 20201834435910.1038/s 41579-019-0315-132055025 · doi ↗ · pubmed ↗
- 6Gorrie CL Mirceta M Wick RR Edwards DJ Thomson NR et al Gastrointestinal carriage is a major reservoir of Klebsiella pneumoniae infection in intensive care patients Clin Infect Dis 20176520821510.1093/cid/cix 27028369261 PMC 5850561 · doi ↗ · pubmed ↗
- 7Vornhagen J Roberts EK Unverdorben L Mason S Patel A et al Combined comparative genomics and clinical modeling reveals plasmid-encoded genes are independently associated with Klebsiella infection Nat Commun 202213445910.1038/s 41467-022-31990-135915063 PMC 9343666 · doi ↗ · pubmed ↗
- 8Salazar C Giménez M Riera N Parada A Puig J et al Human microbiota drives hospital-associated antimicrobial resistance dissemination in the urban environment and mirrors patient case rates Microbiome 20221020810.1186/s 40168-022-01407-836457116 PMC 9715416 · doi ↗ · pubmed ↗