Molecular epidemiology and population structure of Providencia stuartii obtained from humans and other sources
Nayeli Estefania Sánchez-Casiano, Nadia Rodríguez-Medina, Edgar Aguilar-Vera, Alejandro Aguilar-Vera, Luisa María Sánchez-Zamorano, Enrique Delgado-Suárez, Erendira Cervantes-Caballero, Rayo Morfi-Otero, Eduardo Rodríguez-Noriega, Esteban Gonzalez-Diaz, Carlos Antonio Couoh-May

TL;DR
This study analyzes the genetic diversity and global spread of the antibiotic-resistant bacterium Providencia stuartii using a new MLST method and identifies high-risk clones.
Contribution
A novel non-PCR-based MLST scheme for P. stuartii and a public web platform for species classification and sequence typing.
Findings
104 distinct sequence types were identified, revealing high genetic diversity in P. stuartii.
Multidrug-resistant global clones like ST72 and ST12 were found in both human and non-human sources.
ST19 and ST90 in Mexico are linked to NDM-1 and IncA/C2 plasmid replicon.
Abstract
Providencia stuartii is an emerging opportunistic pathogen in humans and belongs to a genus that has shown great genomic diversity. P. stuartii has shown relevant traits in antimicrobial resistance because of its intrinsic resistance to multiple antibiotics and its ability to acquire resistance genes that limit therapeutic options. The present study describes the molecular epidemiology of P. stuartii based on the multilocus sequence typing (MLST) scheme developed for this purpose. This non-PCR-based scheme uses seven complete housekeeping genes. In total, 518 genomes obtained from public databases and Mexican isolates were evaluated. These genomes were from different sources, such as human infections, followed by insects, wastewater, animals, and unknown origins. The molecular epidemiology showed 104 distinct sequence types (STs). We identified global clones that are multidrug-resistant…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
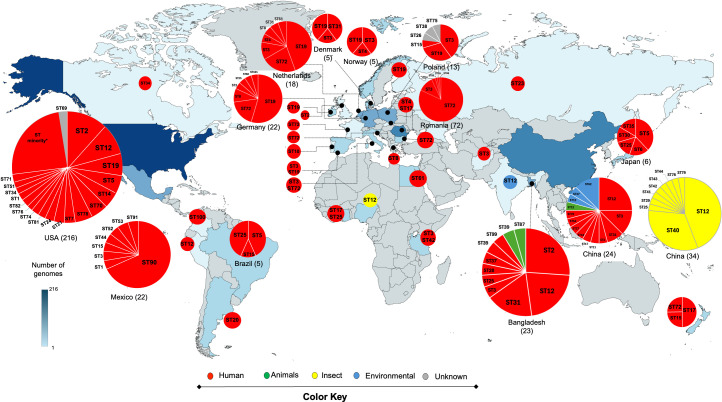 Fig 1
Fig 1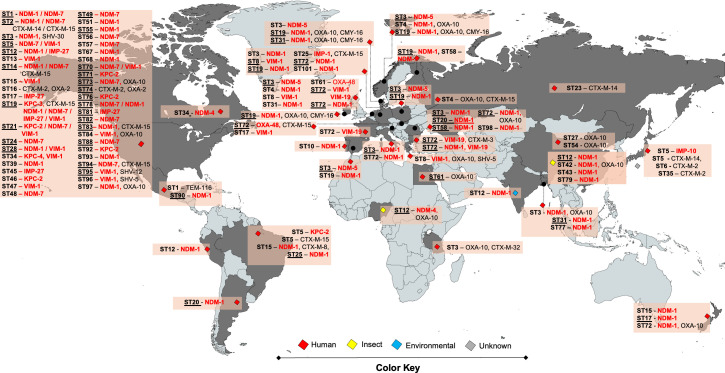 Fig 2
Fig 2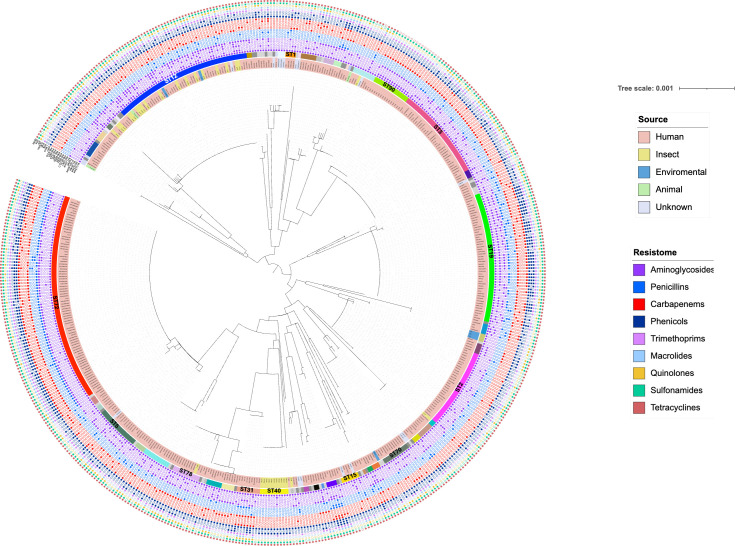 Fig 3
Fig 3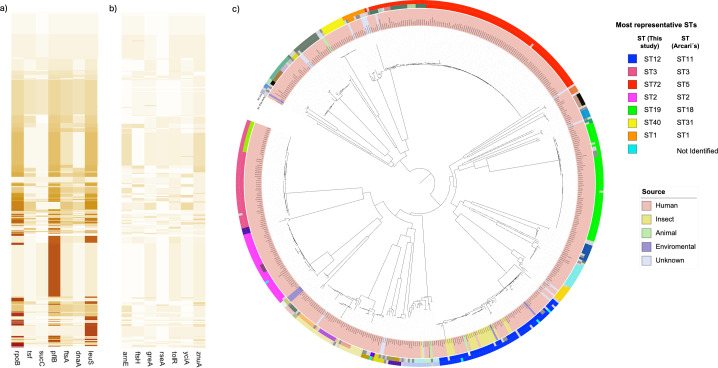 Fig 4
Fig 4| Gene | Function | Size (bp) | No. of alleles | Nucleotide diversity |
|---|---|---|---|---|
|
| RNA polymerase subunit β | 4,029 | 58 | 0.00282 |
|
| leucyl-tRNA synthetase | 2,583 | 48 | 0.00529 |
|
| Formate acetyltransferase 1 | 2,283 | 49 | 0.00412 |
|
| Chromosomal replication initiator protein DnaA | 1,392 | 20 | 0.00336 |
|
| Cell division protein FtsA | 1,257 | 19 | 0.00362 |
|
| Succinyl-CoA synthetase subunit β | 1,167 | 13 | 0.00283 |
|
| Elongation factor Ts | 852 | 16 | 0.00386 |
- —Consejo Nacional de Ciencia y Tecnologíahttp://dx.doi.org/10.13039/501100003141
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntibiotic Resistance in Bacteria · Pharmaceutical and Antibiotic Environmental Impacts · Antimicrobial Resistance in Staphylococcus
INTRODUCTION
Providencia is a genus of Gram-negative bacteria belonging to the Enterobacterales order, the Morganellaceae family. It forms part of the gut microbiota of humans and some animals (1). Recently, advances in genomic studies have revealed remarkable diversity within the genus, creating major challenges for its taxonomic classification (2, 3). These difficulties are reflected in the misidentification of strains using conventional methods, such as MALDI-TOF, which were subsequently recognized as new species (4–6) A clear example is P. thailandensis, which years later was confirmed through genomic analyzes to correspond to P. stuartii (4). In addition, a comprehensive genomic analysis by Dong et al. (4) integrated average nucleotide identity (ANI) with phylogeny to reassess the taxonomy of the genus Providencia. This study reclassified 545 genomes into 20 species, including 13 previously described taxa and seven new unnamed taxa (Taxa 1–7) that could not be assigned to existing species. This analysis revealed extensive inconsistencies in species labeling, biochemical misidentification, and overall genomic diversity within the genus, underscoring the need to reevaluate the current classification system. Subsequently, the same group described additional novel species that further expanded the genus (5–7).
Currently, the genus includes 16 main species publicly validated and corrected under the International Code of Nomenclature of Prokaryotes (ICPN) according to the list of prokaryotic names (8) consulted in August 2025 (Table S1). Among these, P. stuartii is one of the most clinically relevant species, being a major causative agent of healthcare-associated infections and rarely with community-acquired cases. It predominantly affects immunocompromised patients and can cause pneumonia, soft tissue infections, wound infections, bloodstream infections, endocarditis, meningitis, and peritonitis. P. stuartii is notably linked to urinary tract infections, where it contributes significantly to morbidity and mortality often requiring prolonged antimicrobial treatment (9, 10). Beyond the clinical setting, Providencia species have been isolated from diverse ecological niches, facilitating their interaction with humans, animals, and the environment, and promoting their dissemination. Of particular concern is their intrinsic resistance to colistin and tigecycline combined with their ability to acquire plasmid-borne carbapenemase genes (11) representing a growing threat to public health.
Genomic analyses provide a valuable reference for precise taxonomic resolution within Providencia. Multilocus sequence typing (MLST) is the gold standard molecular tool for establishing the epidemiology of isolates across different geographic regions enabling the identification of potential relationships and emergence of pandemic or regionally dominant clones within these species (12). In the present study, a regional epidemiological surveillance was carried out to determine the regional and global molecular epidemiology of P. stuartii, providing the basis for a new MLST scheme and public database for this bacterial species (http://mlstps.insp.mx).
MATERIALS AND METHODS
Regional Providencia spp. epidemiological surveillance
This work was part of a surveillance study of Providencia spp. that was conducted in two periods; the first period was from 2012 to 2018 and the second from 2019 to 2024. Fig. S1 details the workflow carried out in both periods. The surveillance study was in collaboration with several institutions and hospitals in Mexico that provided the strains (Table S2). A total of 160 clinical isolates were obtained and identified by automatized systems using VITEK 2 (BioMérieux, Marcy l'Etoile, France) or MALDI-TOF MS (Bruker Daltonics, Bremen, Germany) as P. rettgerii (90 isolates) and P. stuartii (70 isolates). The clinical characteristics of P. stuartii are described in Table S3.
Characterization of P. stuartii clinical isolates
The 70 clinical isolates of P. stuartii were evaluated using the Carba NP test following the recommendations of the Clinical and Laboratory Standards Institute (CLSI; 2023) (13). Carbapenemase-encoding genes (NDM, VIM, IMP, and OXA-like families) were screened by polymerase chain reaction (PCR) (Table S4).
All isolates were genotyped by pulsed-field gel electrophoresis (PFGE) (14). Genomic DNA was digested with SfiI, and clonal relatedness was assessed according to Tenover’s criteria. Clusters were defined as DNA banding patterns sharing >80% similarity. Dendrograms were generated using Dice and UPGMA coefficients implemented in GelCompare Software II, v.6.6.11 (Applied Maths, Inc.; Sint-Martens-Latem, Bélgica).
Plasmid DNA from all strains was extracted using alkaline lysis protocol described by Kiesser (15), and plasmid profiles were visualized by agarose gel electrophoresis. Escherichia coli NCTC50192, which carries plasmids of known size, served as the molecular weight reference. Conjugation assays were performed following the method described by Miller (16) to assess the transferability of plasmid-borne NDM gene. Sodium azide-resistant E. coli J53 was used as the recipient strain. Transconjugants were selected on LB agar containing 100 μg/mL sodium azide and 2 μg/mL imipenem. PCR amplification for NDM and plasmid extraction in transconjugants was performed to confirm plasmid transfer.
MLST scheme development
Whole-genome sequencing, assembly, and annotation
Isolates selected for whole genome sequencing (WGS) were chosen based on PFGE fingerprint patterns, sample origin, year of collection, isolation site, resistance genes, and plasmid content (Fig. S2). One and 26 P. stuartii isolates were selected from the first and second surveillance periods, respectively (Fig. S1). WGS of these 27 isolates was performed using the Illumina NextSeq platform. Read quality was assessed using FastQC v0.12.1, and adapter trimming and quality filtering were performed with TrimGalore v0.6.5 (15). Genomes were assembled using Unicycler v3 in bold mode. The species assignation was confirmed by ANI (17) using pyani v0.2.x, and by maximum likelihood (ML) phylogeny reconstruction using core-genome single-nucleotide polymorphisms (SNPs) identified with Snippy v2 (18). Genomes that showed ANI values between 84% and 85% and formed a different clade were excluded from further analysis (5 genomes) (Fig. S1).
The P. stuartii genomes were deposited in GenBank under the BioProject “PRJNA1272119”. The genome of the strain 15,300 was included in the development of the MLST scheme, and the remaining 21 genomes served for MLST validation (Fig. S1).
Selection and identification of P. stuartii genomes from public databases for MLST development
In January 2024, a total of 638 Providencia spp. genomes were obtained from RefSeq database, which were selected and subjected to quality control using CheckM v2. The taxonomy classification was defined jointly by ANI analysis and ML phylogeny of the core genome SNPs using Snippy v2. We used snippy-multi and snippy-core options to generate the core genome SNP alignment. ML phylogenetic reconstruction was carried out using IQ-TREE under the GTR + I + G substitution model with 1000 bootstrap replicates and visualized in iTOL v6 web platform (Interactive Tree of Life) (19). A total of 95 genomes that displayed ANI values >99% relative to P. stuartii reference genome and the strain 15300 were used to develop the MLST scheme (Fig. S1b).
In addition, a pan-genome analysis was performed for genomes labeled as P. stuartii. Gene annotation was performed using Prokka v.1.13.4 (20) with the --prodigaltf option, and the resulting GFF files were used as input for Panaroo v.1.2.8 (21), which was run with –clean-mode strict and –remove-invalid-genes options. The core alignment generated was used to construct a ML phylogeny with IQ-TREE (GTR + I + G model). The phylogeny and the gene content data were then visualized with Phandango v. 1.3.1 (22).
Development of MLST scheme for P. stuartii
Core genes were extracted from the 96 genomes identified as P. stuartii from the previously described pan-genome analysis. A total of 2,814 core genes were identified, and after filtering out gene duplications, hypothetical or putative proteins, and genes smaller than 600 bp, a total of 987 core genes were obtained. These genes were analyzed using DnaSP v6.12.03 (23) to determine genetic diversity and identify housekeeping genes evolving under negative selection. Seven single-copy genes, rpoB, leuS, pflB, dnaA, ftsA, sucC, and tsf, were selected to be considered in the P. stuartii MLST scheme. The number of alleles and nucleotide diversity are detailed in Table 1.
Sequence type determination and validation of MLST scheme
The sequence types (STs) were determined for the 96 P. stuartii genomes using the MLST scheme developed in this study. The human strain P. stuartii 15300 obtained in the first surveillance period (2018) was assigned as the ST1. Subsequent STs were assigned chronologically based on isolation dates available in the BioSample metadata for each genome in RefSeq genome (Data set S1).
Validation of the MLST scheme incorporated 422 additional public genomes: 401 downloaded from RefSeq and GenBank between February 2024 and 31 August 2025, and 21 genomes from this study (as detailed above) (Fig. S1b).
Implementation of ANI for accurate identification of P. stuartii
The MLST platform for P. stuartii (http://mlstps.insp.mx) incorporates the ANI tool to ensure the correct identification of P. stuartii from other species of Providencia genus. This tool enables users to verify the correct taxonomic assignment of candidate genomes prior to ST designation. The reference genomes for each bacterial species analyzed using ANI were P. stuartii BML2537 (NCBI RefSeq assembly number GCF_010320365.1), P. rettgeri FDAARGOS_1450 (NCBI RefSeq assembly number GCF_019048105.1), P. alcalifaciens DSM_30120 (GCF_000173415.1) and P. hangzhouensis PR-310 (GCF_029193595.2).
Molecular epidemiology of P. stuartii and metadata analysis
The molecular epidemiology of P. stuartii was determined using a total of 518 genomes (Fig. S1; Data set). The resistome was determined in silico using ABRicate software (24), which contains the Resfinder database (25). Moreover, plasmid replicon typing was determined using PlasmidFinder (26) and Mobile Element Finder (MEF) (27).
For the phylogenetic analysis of the seven concatenated MLST genes, IQ-TREE was used starting with the model finder to determine the substitution model that best fitted the data; therefore, an ML phylogeny was performed under the TIM3 +F + I + G4 substitution model.
goeBURST analysis
The goeBURST-1.2.1 program was used to analyze STs of P. stuartii isolates and to assign isolates to a clonal complex (CC). A CC is defined as groups of STs that have recently diversified into single-locus variants (SLVs) from a common founder (28). A CC was built of at least three STs related to a single locus variant.
Comparison of MLST schemes for P. stuartii
During the development of the P. stuartii MLST system described in this article, we noted that Arcari et.al (29) developed simultaneously a P. stuartii MLST system. To compare both schemes, we extract each of the gene fragments described in the Arcari system (arnE, ftsH, gerA, rseA, tolR, yciA, and znuA), and the STs were assigned among the 518 P. stuartii genomes. We designated our strain 15300 as ST1 and followed the same chronological assignment approach described in our scheme. A basic heatmap of the allelic profiles from both MLSTs was generated to visualize differences in locus variation. We evaluated both allele profiles using Mantel’s test, allele diversity per locus, and Simpson’s index. In addition, we also constructed an ML phylogenetic tree based on the core genome from the 518 genomes of P. stuartii that are part of the MLST, under the GTR + I + G model and assigned ST metadata to each scheme.
RESULTS
P. stuartii regional epidemiological surveillance study, carbapenemase identification and plasmid profile
Between 2017 and 2024, 70 clinical isolates of P. stuartii were recovered from seven institutions in Mexico. Of these, 37.1% (26/70) produced carbapenemases with NDM being the only variant identified (Table S2). Isolates were obtained from tracheal/bronchial secretions (24.4%), blood cultures (15.7%), wound samples (15.7%), secretion (14.3%), urine cultures (12.9%), expectoration (2.8%), stool culture (2.8%), tissue (1.4%), and unknown sources (10%) (Table S3). PFGE analysis revealed four major clonal groups, which were carriers of NDM associated with the Hospital Civil de Guadalajara. Plasmid profiling showed that most isolates harbored a 160-kb plasmid encoding NDM (Fig. S2). Conjugation assays confirmed the transfer plasmid-borne NDM to E. coli J53Azi^R^.
Genome characteristics of P. stuartii clinical isolates
The 22 isolates confirmed as P. stuartii were sequenced (Fig. S1), which were obtained from bronchial/tracheal secretions (33.3%), blood cultures (33.3%), urine cultures (19.1%), wound swabs (9.5%), and secretion (4.8%) (Fig. S2). P. stuartii species classification was confirmed with ANI >99%. ResFinder analysis identified the NDM-1 in 14/22 genomes with 100% coverage and identity, consistent with PCR-based characterization. These strains carried plasmids of the IncA/C2 incompatibility group and mobile genetic elements including Tn7, IS5075, and ISCfr1. Genomic features of these isolates are detailed in Table S5.
MLST scheme for P. stuartii and validation
Prior to the development of the MLST scheme, a curation of the 638 genomes of the Providencia genus was performed due to the identification of several inconsistencies in species assignment. Notably, genomes deposited under the name P. thailandensis clustered within P. stuartii clade with ANI >99%, confirming that both names correspond to the same species (Fig. S3). In contrast, another clade with genomes labeled as P. stuartii displayed ANI values well below the accepted species delineation threshold (83%–85%). Furthermore, pangenome analysis showed that although these groups shared most core genes, their accessory genomes were clearly distinct, supporting their classification as separate species (Fig. S4).
Based on ANI and SNP phylogeny, 95 genomes out of 638 were confirmed as P. stuartii, and together with the strain 15300 were used to identify complete single-copy genes for MLST. The scheme is composed of rpoB (RNA polymerase β-subunit), tsf (elongation factor Ts), sucC (succinyl-CoA synthetase β-subunit), pflB (formate acetyltransferase 1), ftsA (cell division protein FtsA), dnaA (chromosomal replication initiator protein DnaA), and leuS (leucyl-tRNA synthetase) (Table 1).
We determined 34 distinct STs among the 96 genomes used for the MLST development, whereas the MLST validation, using a dataset of 422 P. stuartii genomes, identified 70 additional STs. This resulted in a total of 104 STs among the 518 evaluated genomes.
Molecular epidemiology of P. stuartii
The genomes of 518 P. stuartii isolates from various countries were primarily obtained from humans (88.2%), followed by insects (7%), environmental (1.2%), animals (0.6%), and unknown sources (3%) (Data set S1).
The global distribution of the STs is shown in Fig. 1 and Supplementary Data set S1. Some sequence types stand out, such as ST72, which is the most frequently identified (15.4% [n = 80/518]) and detected across several European countries and New Zealand. In addition, ST12 accounted for 10.4% (54/518) and was identified in countries from different continents and it was detected in all sample categories: humans, insects, environmental, animal, and one of unknown origin. ST3 had a worldwide distribution, being present in 14 different countries.
Molecular epidemiology of 518 P. stuartii isolates. The map illustrates the global distribution of each ST. The STs were distributed in pie charts according to the proportions of genomes from each of the corresponding countries. The origin of the isolates is indicated using color codes. ST72 exhibits the highest number of isolates, primarily in Romania, followed by Bulgaria, Netherlands, Germany, Slovenia, France, Italy, and New Zealand.
The United States, Romania, and China contributed the largest number of genomes. However, China exhibited the greatest diversity in isolation sources, including humans, insects, and wastewater. In contrast, genomes from the United States and Romania originated exclusively from human clinical samples. With respect to Mexico, ST1 derived from clinical samples, was also detected in the United States, whereas ST90 was the most frequent ST among Mexican human isolates.
Molecular epidemiology of ESBL- and Carbapenemase-producing P. stuartii
Of the 518 P. stuartii genomes analyzed, 66.4% contained at least one carbapenemase gene (Fig. 2; Fig. S5). The USA contributed the highest number of genomes (n = 216), assigned to different STs with substantial variability in carbapenemase gene content. In contrast, most human clinical samples from China lacked carbapenemase genes, except for two isolates encoding OXA-10, a β-lactamase with weak carbapenemase activity. Notably, ST72, one of the most common human-associated STs, showed a high prevalence of NDM-1, predominantly in Romania and other European countries. The ST19 (48 isolates) exhibited a notable diversity of carbapenemase genes and ESBLs, including members of the CTX-M family, and was distributed across nine countries. The ST90 (14 isolates), harboring NDM-1 and IncA/C2, was detected exclusively in Mexico.
Molecular epidemiology of P. stuartii isolates producing ESBLs and carbapenemases distributed across 104 STs. The map illustrates the global distribution of 518 P. stuartii. Underlined and highlighted STs correspond to those with two or more isolates. The origin of the isolates is indicated using color codes. The USA, China, Brazil, Romania, Germany, Netherlands, and Bangladesh exhibited greater diversity of STs with various ESBLs and carbapenemases.
A notable finding was the high proportion of isolates from flies that carried the NDM-1 or NDM-4 genes, primarily within ST12, with additional isolates belonging to ST42, ST43, and ST79. These genomes originated in China and Nigeria (Fig. 2), highlighting their potential epidemiological relevance.
In general, NDM-1 was the most prevalent carbapenemase gene (37.8%, 196/518), followed by NDM-7 (5.4%, 28/518). These NDM alleles were predominantly associated with ST72, ST12, ST19, ST3, and ST90 (Fig. S5). The ST19 (50 isolates) harbored NDM-1, NDM-7, KPC-3, IMP-27, and VIM-1. ST3 (36 isolates) comprised exclusively human clinical isolates and was associated with NDM-1 and NDM-5, while ST5 (13 isolates) carried primarily VIM-1 and NDM-7. Carbapenemases from the VIM, IMP, and KPC families were detected at lower frequencies compared with NDM.
Plasmid replicon typing identified different incompatibility groups, with IncA/C2 being predominant (60.4%, [n = 313/518]). This Inc group is known to have a broad host range among Enterobacterales and frequently carries multidrug-resistance modules, including NDM alleles. Consistent with this, IncA/C2 was strongly associated with isolates that carried carbapenem resistance genes in our dataset (Fig. S6).
The phylogenetic analysis using the seven concatenated genes from the proposed MLST scheme revealed six main clusters, each corresponding to the predominant STs: ST72, ST12, ST19, ST3, ST2, and ST90 (Fig. 3). The metadata associated with these genomes indicated that P. stuartii commonly harbors resistance genes to multiple classes of antimicrobial, including fluoroquinolones, aminoglycosides, and β-lactams. Notably, genes associated with intrinsic resistance were present in more than 90% of the analyzed genomes, specifically aac(2′)-Ia (99.2%), tet(B) (92.3%), and catA3 (94.4%). These highlight the resistome of P. stuartii, on which additional acquired genes, particularly NDM alleles, are frequently observed.
ML phylogenetic tree based on concatenated gene sequences (13,563 bp) from the MLST scheme for P. stuartii. Isolates are color-coded according to the sample origin. STs are highlighted and labeled with their respective names. The resistome present in each isolate is indicated by colored boxes, the presence of NDM is indicated by red boxes.
Clonal complex and founder ST
The goeBurst analysis revealed a population structure characterized by the presence of several clonal complexes. The largest complexes were centered on ST72, ST1, ST12, and ST78, identified as founders, which showed multiple SLVs. CC1 dominated by ST72 corresponded to the most frequent ST in the total set of genomes analyzed. This complex included numerous closely related descendants (ST5, ST94, ST71, and ST103), which all were isolated from humans. Additionally, smaller clonal complexes were identified comprising those founded by ST25 and ST19 (Fig. S7).
Comparison of MLST schemes
In 2024, Arcari et al. proposed a MLST scheme using seven gene fragments (greA, ftsH, tolR, arnE, znuA, yciA, and rseA) and a core genome MLST (cgMLST) based on 2,296 loci for the identification and typing of P. stuartii. However, we found some challenges in the implementation of their scheme, including the absence of a publicly accessible database for ST assignment; furthermore, it did not include the core genes used in their cgMLST. Their analysis was developed using 71 P. stuartii genomes.
To compare both systems, we extracted the gene fragments proposed by Arcari et al. and assigned STs to our collection of 518 P. stuartii genomes. During this process, we identified “N” insertions, which complicate allele identification, in the provided greA and znuA sequences. Since MLST is based on allelic variation, and “N” insertions represent four distinct variations. Therefore, to ensure consistency, we extracted the gene fragments using our reference genome (strain 15300) such as ST1.
Our MLST system assigned 104 STs among the 518 genomes, while Arcari’s scheme identified 55 STs among 513 genomes. Five genomes were excluded due to truncated or missing loci. The heatmap of allelic profiles showed greater heterogeneity with our MLST scheme compared with Arcari’s. This finding was supported by Simpson’s diversity index, which approached 1, suggesting sufficient genetic variation in our genes (Table S6). Mantel’s test revealed that both MLST systems captured similar patterns of strains, but the concordance is low (r = 0.1792, P = 1 × 10⁻⁴). However, the phylogenetic analysis based on core genome alignment showed that Arcari’s scheme was unable to capture slight differences in allele sequence from closely related isolates (Fig. 4).
Heatmap and phylogenetic analysis showing allelic profile variation in P. stuartii. (a) Allelic profile variation of 518 P. stuartii genomes using the MLST scheme of this study. (b) Allelic profile variation of 513 P. stuartii genomes using the scheme of Arcari et al. (c) Phylogenetic analysis based on the core genome and compared with the ST distribution assigned with the two MLST schemes of P. stuartii.
DISCUSSION
P. stuartii has emerged in recent years as an opportunistic pathogen with increasing reports of infections worldwide (30–33). In contrast, reports in Mexico have been scarce. In this study, clinical isolates from different institutions in the country were integrated to strengthen regional epidemiological surveillance of Providencia spp. We identified a high proportion of clinical isolates (37.1%) carrying the NDM-type carbapenemase, with clonal dissemination and persistence in the Hospital Civil de Guadalajara as demonstrated by PFGE. The presence of the NDM gene agrees with several reports worldwide that identified it as the main carbapenemase in P. stuartii (30, 32, 34), followed by IMP (35) and VIM (31). The relevance of this species lies in its intrinsic resistance to colistin and tigecycline, together with the acquisition of NDM and other carbapenemases, which confers resistance to carbapenems, limiting therapeutic options for these infections (33, 35–37).
In this context, the present study not only performed the molecular characterization of Mexican clinical isolates but also incorporated comparative analyses of P. stuartii genomes deposited in public databases to determine the molecular epidemiology worldwide; for this purpose, we developed a robust MLST system for P. stuartii, with an online public database for this bacterial species (http://mlstps.insp.mx). In this repository, a preliminary ANI analysis is performed to ensure accurate species identification.
During the process of development and genomic curation, we noted substantial taxonomic inconsistencies in RefSeq. Several genomes labeled as P. thailandesis correspond to P. stuartii (ANI >99%), a finding consistent with the recent taxonomic reclassification by Dong et al. (4). Conversely, genomes deposited as P. stuartii but belonging to recently described P. zhejiangensis (taxon 7) exhibited ANI values of 83%–85%, confirming misclassification. Although an ANI cut-off point of 98.21% was previously proposed to define P. stuartii (38), our analyses revealed that genomes truly belonging to this species consistently exceeded 99%, providing an even more precise delimitation. This highlights the need for continuous taxonomic curation in public repositories, particularly for genera undergoing rapid expansion and reclassification.
The traditional MLST, based on fragments of constitutive genes, requires labor-intensive and costly laboratory procedures, including DNA extraction, PCR amplification, purification, and Sanger sequencing for each locus (12). As the cost of WGS continues to decline, the MLST system proposed in this study based on the assignment of STs using seven full-length gene that allows capturing greater allelic variation, which enhances the robustness of the analysis and enables greater standardization and global comparability. Furthermore, access to complete genomes enables the simultaneous recovery of additional genomic features, such as the pangenome, resistome, and virulome that extend well beyond conventional ST assignments. Notably, we did not adopt a cgMLST approach due to variability in the core genome defining a fixed set of loci beings challenging because the number of shared genes can fluctuate depending on the size of the pangenome, genomic divergence (39), and assembly quality, which would affect reproducibility and comparison between studies. Using this scheme, we identified 104 STs among 518 genomes, the most comprehensive P. stuartii MLST to date, now accessible through an online public database (http://mlstps.insp.mx).
Our global epidemiological analysis revealed STs of relevance. ST72 emerged as the dominant clone and founder of CC1, mostly from human clinical isolates in Europe and frequently associated with NDM-type carbapenemases. ST12, in contrast, showed the broadest ecological distribution, present in human, insects, animal, and environmental samples. This wide ecological distribution underscores its potential for dissemination across ecological niches, reflecting its clonal expansion. In addition, ST19 and ST3 were associated with different carbapenemases and ST90, restricted to Mexico, was associated with NDM-1.
Notably, a several proportion of P. stuartii genomes carried plasmids with the IncA/C2 incompatibility group, which represents 60.4% of plasmid types. This plasmid family is widely recognized for its ability to disseminate NDM enzymes across species of Enterobacterales, and for its broad host range (40). The IncA/C2 is strongly associated with carbapenemase-positive isolates reinforcing its epidemiological importance.
Although most genomes originated from human sources, a significant subset (7%) belonged to house flies from China and Nigeria, which were associated with ST12 and carried the NDM gene. It has previously been reported that P. stuartii and other Providencia species are commonly found in the gut microbiota of house flies (9). Regarding the Nigerian flies, the carbapenemase NDM-4 was identified; these resulted from a pilot surveillance study of antimicrobial-resistant bacteria in Nigerian hospitals (41). The authors found NDM in 8% of the sampled flies, and it was primarily carried by Providencia species, such as P. hangzhouensis, P. huaxiensis, P. manganoxydans, P. rettgeri, and P. stuartii, along with Enterobacter spp., E. coli, and K. pneumoniae, among others. Furthermore, a national surveillance study conducted by Zhou et al. in China identified high rates of carbapenem-resistant Enterobacterales, of which Providencia spp. predominated with 90.6% attributed to the dissemination of NDM-1 (42). The detection of carbapenemase-producing P. stuartii in insects in China and Nigeria warrants further attention and highlights its relevance from a One Health perspective. This stems from the role of insects om the role of insects as a reservoirs and vehicles for the dissemination of resistance genes among various Enterobacterales and different ecological niches due to its frequent contact with wastewater, hospital environments, and decomposing organic matter. This threat is heightened in low- and middle-income countries, particularly in tropical climates, and may reflect the level of environmental contamination by carbapenem-resistant bacteria (41–43).
The resistome showed that nearly all genomes carried intrinsic resistance determinants, such as aac(2')-Ia, tet(B), and catA3 (44, 45) and multiple acquired resistance genes, including ESBL and carbapenemase families (NDM, IMP, KPC, and VIM). The high diversity and prevalence of these genes emphasize the substantial genomic plasticity and acquisition potential of P. stuartii. Selective pressure in hospitals including prolonged use of tigecycline and colistin can that its emergence (46), especially considering the global expansion of NDM-1 and the presence of variants with greater carbapenem-hydrolyzing activity (47), such as NDM-7.
Although multiple MLST systems have been described for other pathogens, such as A. baumannii (48, 49) and E. coli (50–52), several authors adopt one system or include information from both MLST systems (53, 54). In the case of P. stuartii, we assume the same could occur. The P. stuartii MLST described in the present work provides greater robustness and enables comprehensive genomic insights beyond simple ST classification. Likewise, a public database update allows the determination of the bacterial species through ANI and the assignation of new sequence types.
Conclusions
The development of MLST for P. stuartii enabled a comprehensive assessment of the global genetic diversity and population structure of this emerging multidrug-resistant pathogen. The scheme identified different STs distributed worldwide and epidemiologically relevant lineages grouped into clonal complexes. The ST72 and ST19 emerged as high-risk clones due to their strong association with carbapenemase genes, particularly NDM. ST12 is a globally distributed lineage detected across multiple ecological niches, including humans, environmental, and especially insects, where it frequently carried NDM alleles. Additional clinically relevant clones include ST90 prevalent in Mexico, and ST3 both demonstrated a broad international distribution. The widespread presence of IncA/C2 plasmid-type replicons among P. stuartii genomes highlights the importance of this incompatibility group as a reservoir of plasmids carrying NDM and other resistance determinants. Furthermore, the taxonomic classification in public databases underscores the need for genome-based approaches to ensure accurate species identification within Providencia. Overall, this study provides a tool for a more robust analysis and offers a general overview of the molecular epidemiology worldwide, emphasizing the importance of continuous genomic surveillance, particularly due to the acquisition of carbapenemase genes in the P. stuartii.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adeolu M, Alnajar S, Naushad S, S Gupta R. 2016. Genome-based phylogeny and taxonomy of the “Enterobacteriales”: proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int J Syst Evol Microbiol 66:5575–5599. doi:10.1099/ijsem.0.00148527620848 · doi ↗ · pubmed ↗
- 2Yuan C, Wei Y, Zhang S, Cheng J, Cheng X, Qian C, Wang Y, Zhang Y, Yin Z, Chen H. 2020. Comparative genomic analysis reveals genetic mechanisms of the variety of pathogenicity, antibiotic resistance, and environmental adaptation of Providencia genus. Front Microbiol 11:572642. doi:10.3389/fmicb.2020.57264233193173 PMC 7652902 · doi ↗ · pubmed ↗
- 3Wang P. 2023. Genomic epidemiology and heterogeneity of Providencia and their bla NDM-1-carrying plasmids. Emerg Microbes Infect 12:2275596. doi:10.1080/22221751.2023.227559637874004 PMC 10796120 · doi ↗ · pubmed ↗
- 4Dong X, Jia H, Yu Y, Xiang Y, Zhang Y. 2024. Genomic revisitation and reclassification of the genus Providencia. m Sphere 9:e 0073123. doi:10.1128/msphere.00731-2338412041 PMC 10964429 · doi ↗ · pubmed ↗
- 5Dong X, Xiang Y, Shen P, Xiao Y, Zhang Y. 2025. Clinical emergence of Providencia zhejiangensis sp. nov. and Providencia xihuensis sp. nov.: Genomic insights into antimicrobial resistance and geographical distribution. Int J Antimicrob Agents 65:107484. doi:10.1016/j.ijantimicag.2025.10748440023453 · doi ↗ · pubmed ↗
- 6Dong X, Yu Y, Liu J, Cao D, Xiang Y, Bi K, Yuan X, Li S, Wu T, Zhang Y. 2023. Whole-genome sequencing provides insights into a novel species: Providencia hangzhouensis associated with urinary tract infections. Microbiol Spectr 11:e 0122723. doi:10.1128/spectrum.01227-2337732781 PMC 10581081 · doi ↗ · pubmed ↗
- 7Dong X, Xiang Y, Yang P, Wang S, Yan W, Yuan Y, Zhou S, Zhou K, Liu J, Zhang Y. 2024. Novel Providencia xianensis sp. nov.: a multidrug-resistant species identified in clinical infections. Eur J Clin Microbiol Infect Dis 43:1461–1467. doi:10.1007/s 10096-024-04821-y 38714595 PMC 11271419 · doi ↗ · pubmed ↗
- 8Göker M, Christensen H, Fingerle V, Kostovski M, Margos G, Moore ERB, Oren A, Patrick S, Reischl U, Vázquez-Boland JA. 2025. List of Recommended Names for bacteria of medical importance: report of the Ad Hoc Committee on Mitigating Changes in Prokaryotic Nomenclature 75. doi:10.1099/ijsem.0.006943 · doi ↗