Chicken cecal microbial functional gene content and resistome differ by age and barn disinfection practice
Yi Fan, Tingting Ju, Tulika Bhardwaj, Douglas R. Korver, Benjamin P. Willing

TL;DR
This study shows that the age of chickens and barn disinfection practices affect gut microbes and antibiotic resistance genes in commercial broiler chickens.
Contribution
This is the first study to evaluate sanitation practices' effects on chicken gut microbiome and resistome in a commercial setting.
Findings
Younger chickens had higher antibiotic resistance gene abundance than older ones.
Disinfectants reduced amino acid synthesis genes and enriched stress resistance genes in young chickens.
Disinfection practices affected Helicobacter pullorum and short-chain fatty acid biosynthesis pathways at day 30.
Abstract
Chemical disinfectants and water-wash methods are widely employed in sanitizing broiler chicken barns. Studies showed that disinfectants affect environmental microbial composition and antibiotic resistance genes (ARGs). However, little is known regarding how barn disinfection treatments impact the chicken gut resistome and microbial functional gene content. The current study compared the effects of disinfection and water-wash method on the gut microbiome and resistome of commercial broilers using a crossover experimental design after two production cycles at seven barns. Shotgun metagenomic sequencing performed on cecal contents collected at days 7 and 30 also allowed the evaluation of age-associated characteristics of the microbiome. The age of the chickens had the largest effects on the resistome, with younger birds having higher relative abundance of total ARGs (P < 0.05) and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
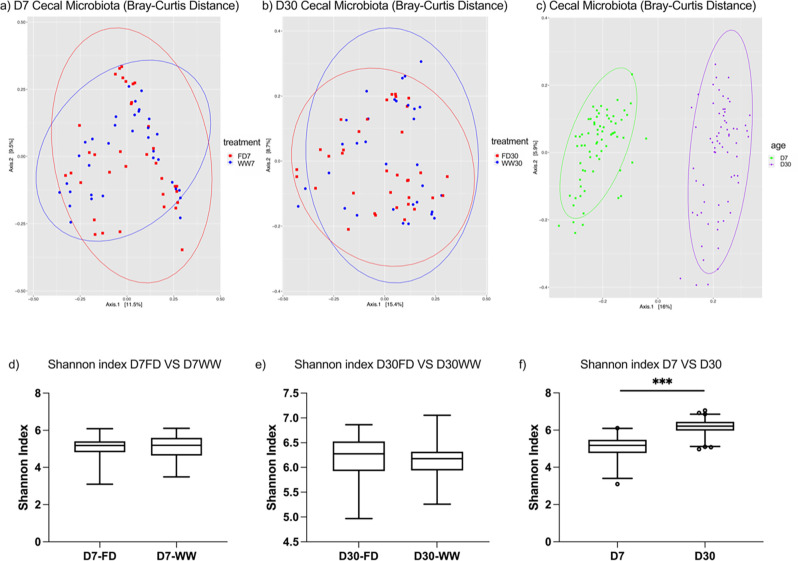 Fig 1
Fig 1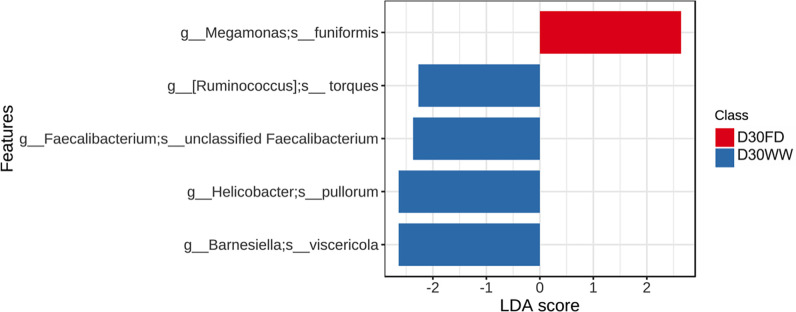 Fig 2
Fig 2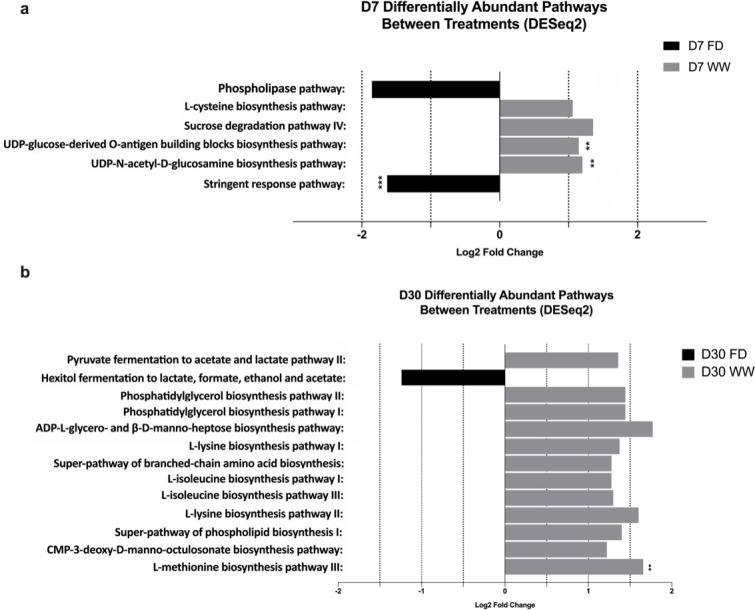 Fig 3
Fig 3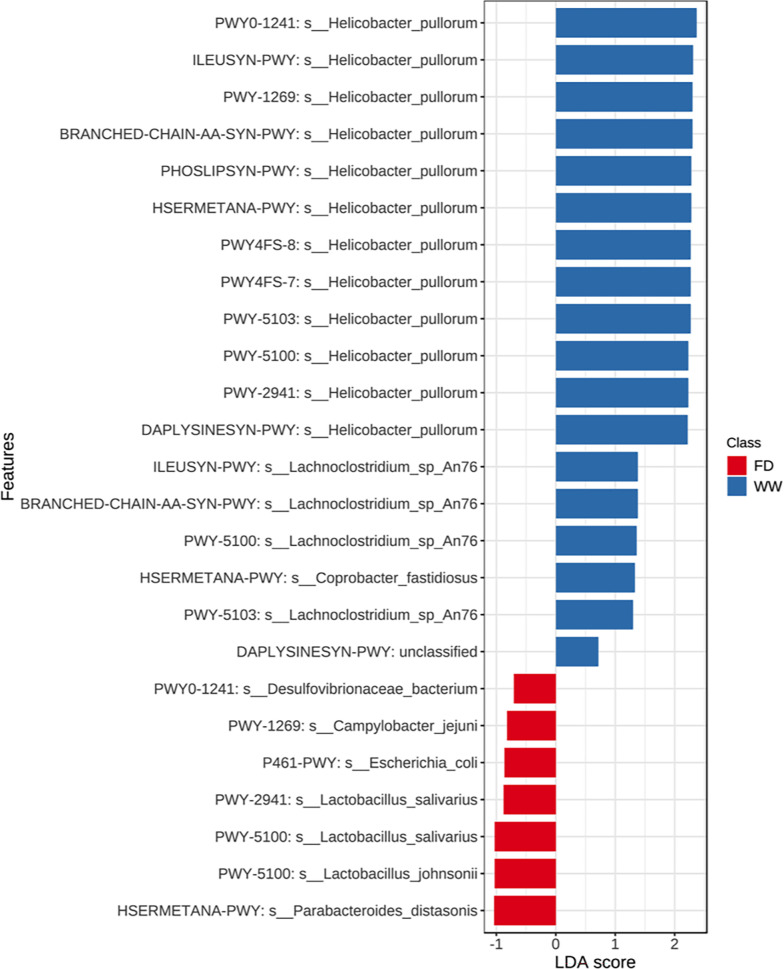 Fig 4
Fig 4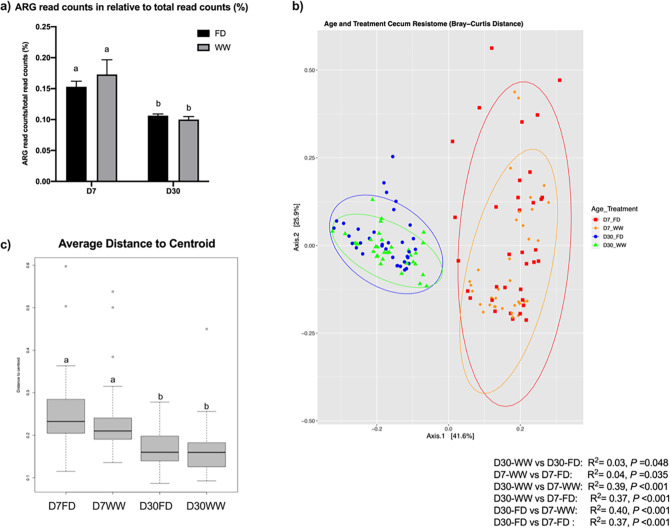 Fig 5
Fig 5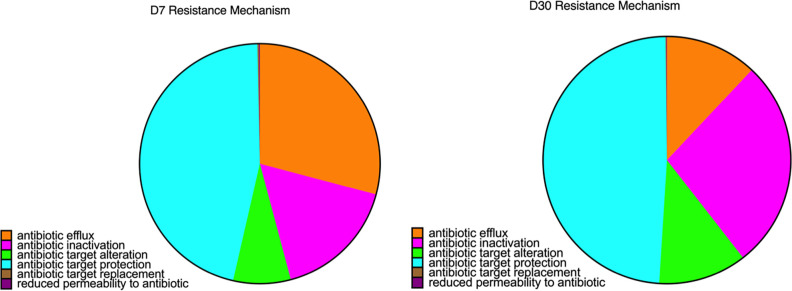 Fig 6
Fig 6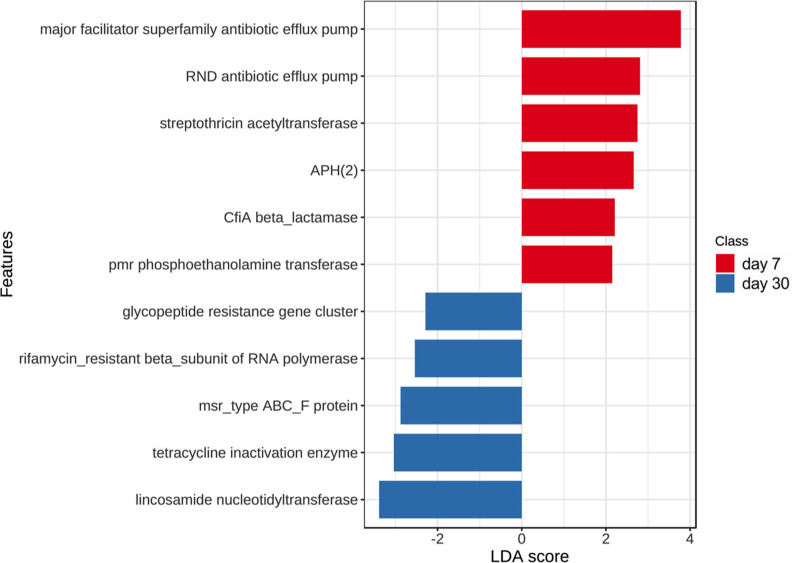 Fig 7
Fig 7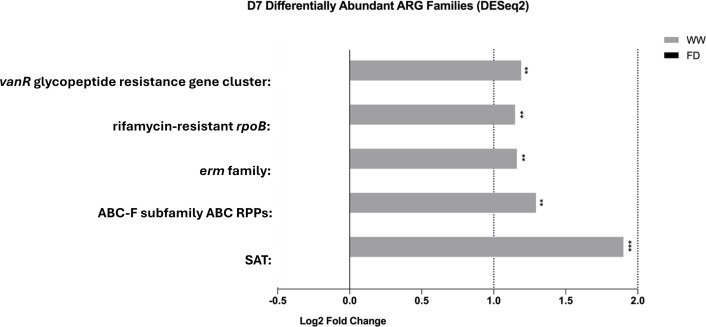 Fig 8
Fig 8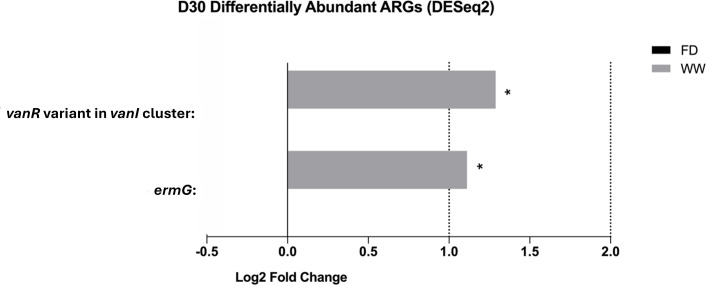 Fig 9
Fig 9- —Natural Sciences and Engineering Research Council of Canadahttp://dx.doi.org/10.13039/501100000038
- —Agriculture Funding Consortiumhttp://dx.doi.org/10.13039/100013585
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPharmaceutical and Antibiotic Environmental Impacts · Salmonella and Campylobacter epidemiology · Listeria monocytogenes in Food Safety
INTRODUCTION
Livestock farming accounts for over 50% of antibiotic usage globally (1). Compared with other livestock species, chickens were reported to have the highest density of antibiotic resistance genes (ARGs) due to the high stocking density and short production cycle (2). High ARG abundance in chicken production can lead to reduced sensitivity to antibiotic treatments on infectious disease. Furthermore, it also increased the risk of ARG transmission, often using bacteria as a vessel, to humans through the food chain or environmental routes. The transmission of ARGs to human-associated bacteria can complicate the treatment of human infections, contributing to the global antimicrobial resistance crisis, with an estimated 1.27 million deaths directly attributed to antibiotic resistance in 2019 (3). Currently, to reduce bacterial load in poultry farming, biocidal agents such as benzalkonium chloride (BAC), hydrogen peroxide, glutaraldehyde, ethanol, and sodium hypochlorite have been widely applied for facility disinfection purposes (4). While chemical disinfectants can inhibit antibiotic-resistant bacteria and destruct ARGs through oxidation, they may also induce bacterial adaptation, potentially through promoting antibiotic resistance through co-selection (5). The impact of chemical disinfectants on ARG proliferation remains uncertain, with some studies suggesting that they may act as stressors, stimulating the proliferation and transfer of microbial ARGs (6–8). For example, BAC, a commonly used quaternary ammonium compound, has been associated with increased resistance to ampicillin, cefotaxime, and sulfamethoxazole in various food-related bacterial isolates (6, 9), along with co-selected ARGs (10). Some studies, on the other hand, suggested that chemical disinfectants may contribute to controlling antibiotic resistance by reducing the abundance of ARGs. For instance, quaternary ammonium compounds and sodium hypochlorite used in treating swine manure have been reported to decrease the abundance of selected ARGs [erm(B), erm(C), erm(F), intI1, tet(Q), and tet(X)] (11). Oxidants like chlorine and hydroxyl radicals have also demonstrated potential in eradicating ARGs presented in both E. coli cells and plasmids (12). With the controversial effect of chemical disinfectants on ARGs, there is limited information available regarding the effects of chemical disinfectant-treated rearing environments on the resistome in the gut of animals. In this sense, barn cleaning practices, which involve chemical disinfectants, may impact the presence and persistence of ARGs throughout production cycles. Consequently, from the perspective of food safety and environmental sustainability, it is important to explore the influence of chemical disinfectant usage in barn cleaning.
In addition to environmental factors like disinfection practices, the age of chickens plays a crucial role in the development of the gut microbiota. Studies have shown that the chicken gut microbiome undergoes successional changes as they mature (13, 14). The microbial abundance of the microbiome increases during the first week of life, with compositional shifts of important taxa occurring over several weeks (15). Understanding these age-dependent microbiome dynamics is essential, as they can play an important role in shaping the microbial functional gene content and the overall ARG profile within the gut.
In broiler chicken production, barn sanitation has been used with the goal of enhancing biosecurity and preventing disease transmission between flocks. Both full sanitation with chemical disinfectants (FD) and water-wash (WW) method are widely employed in the Canadian poultry production system (16). To date, large-scale metagenomic studies have advanced understanding of the poultry gut resistomes by characterizing the ARG carriage, relative abundance, and diversity of ARGs in the gut of commercial broiler chickens (17–23). These works have elucidated important dynamics of ARG profiles linked to factors such as different production modes (21, 22), antibiotic administrations (20, 22, 23), geographic location (17, 18), and bacterial community composition (19). However, despite these valuable insights, the present work represents the first research to directly assess how chemical disinfectants applied for barn sanitation impact the chicken gut resistome at commercial production scale. Our previous study showed that FD resulted in an undesired increased carriage of Campylobacter jejuni accompanied by alterations in the cecal microbial composition, revealed by 16S rRNA gene amplicon sequencing (24). Full disinfection also resulted in lower cecal short-chain fatty acids (SCFAs) when compared to the WW (24). Additionally, as age has been identified as an influencing factor in the gut microbiota composition, understanding how the successional changes in microbiota affect the microbial functional gene content and the ARG profile is essential for a comprehensive understanding of the chicken gut microbiome. In the current study, we sought to gain greater insight into the effects of barn sanitation and age on the functional gene content of the gut microbiome, specifically in terms of microbial metabolic capacity and the profile of antibiotic resistance genes.
MATERIALS AND METHODS
Animals
The current study was performed according to the guidelines of the Canadian Council on Animal Care with approval of the University of Alberta Animal Care and Use Committee (AUP00002377). Broiler chicken management and barn cleaning practices were described previously (24). Briefly, the animal study was conducted on seven commercial broiler chicken barns owned and managed by the same producer in Alberta, Canada, between the months of June to September. All barns were similarly engineered single-story production houses with cement floors (Fig. S1). During each production cycle, samples were collected from barns that had undergone two consecutive rounds of repeated cleaning treatments, including both FD and WW. For FD treatment, manure and litter were completely removed from the barn after chickens were depopulated. Subsequently, chemical disinfection was performed using foam containing 7% sodium hydroxide, 7% 2-(2-2-butoxyethoxy) ethanol, 6% sodium laureth sulfate, 5% sodium N-lauroyl sarcosinate, and 5% tetrasodium ethylenediaminetetraacetic acid on all surfaces within the facilities, followed by high- and low-pressure water rinse with water temperature set at 35℃. After the facilities were air-dried, foam containing 10% glutaraldehyde, 10% benzalkonium chloride, and 5% formic acid was applied to surfaces of the facilities for 60 min, followed by high-pressure water rinse, overnight air-dry, and fresh litter placement. For WW treatment, manure and used litter were removed, followed by low-pressure water rinse with the water temperature set at 35°C for all facility surfaces, air-dry overnight, and fresh litter placement (wood shaving, 10–15 cm deep).
A cross-over design was performed, which resulted in a total of 14 production flocks with seven flocks assigning to each treatment to ensure that every barn had gone through both FD and WW treatments. The study included a total of 140 chickens with 35 chickens sampled from each treatment at day 7 (D7) and day 30 (D30) to analyze their cecal microbiota. Sampling at D7 was intended to represent the starter phase, whereas sampling at D30 was chosen to be as close as possible to slaughter (day 32) while avoiding the fasting period prior to processing. In each production flock, Ross 308 broiler chicks were placed within 12 h post-hatch and confined to half of the house, then allowed access to the entire house starting at D7. The flock size was approximately 14,000 birds, with a final stocking density of 30 kg/m^2^. All chickens were fed the same diet without antibiotics ad libitum and sent for processing at 32–35 days of age when the average target live weight of 1.8 kg was reached. At D7 and D30 of age, five broilers per flock were randomly selected from five different areas within each barn (Fig. S1) and euthanized using cervical dislocation. Approximately 300 mg of cecal contents were collected in autoclaved 1.5 mL Fisherbrand Microcentrifuge tubes (catalog # 05-408-129) using sterile technique, placed on dry ice until being transported to the lab, and stored at −80°C.
Shotgun metagenomic sequencing
The total DNA extraction process was conducted as previously outlined (24). In summary, DNA extraction was carried out from homogenized cecal contents using the QIAamp Fast DNA Stool Mini Kit (Qiagen, Valencia, CA, USA), including an extra bead-beating step utilizing approximately 200 mg of garnet beads at a speed of 6.0 m/s for 60 s (FastPrep-24 5G instrument; MP Biomedicals Inc., Santa Ana, CA, USA). DNA samples were quantified using a determined by using a Quant-iT PicoGreen dsDNA assay kit (Invitrogen, Waltham, MA) and assessed for purity using a Nanodrop 2000c spectrophotometer (Thermo Fisher Scientific). For each sample, approximately 300 ng of extracted DNA meeting purity criteria (OD 260/280 ratio of 1.8, OD 260/230 ratio between 2.0 and 2.2, and concentration between 20 and 100 ng/μL) was sent to Genome Quebec Innovation Centre for shotgun metagenomic sequencing. Library preparation and shotgun sequencing were performed by the Genome Quebec Innovation Centre (Montreal, Canada). Prior to library preparation, the Genome Quebec Innovation Centre performed an additional quality control step, reassessing the DNA quantity using their own Qubit instrument to ensure having at least 150 ng of DNA for successful library construction. Libraries for all samples including the ZymoBIOMICS Gut Microbiome Standard (cat. # D6331, Zymo Research Corp. CA, USA) were prepared using the Nextera DNA Flex Library Prep Kit (Illumina Inc., San Diego, CA, USA), and the subsequent shotgun sequencing was executed on a NovaSeq 6000 system (Illumina Inc., San Diego, CA, USA). Read quality was assessed using FastP v0.23.2, and low-quality reads, sliding windows, adaptors, polyG, and duplicated sequences were removed (25). Kneaddata v0.10.0 was used to remove host DNA contaminants (https://github.com/biobakery/kneaddata). Briefly, a chicken host reference database was built using bowtie2 v2.4.1 with genome Gallus_gallus 105 release from Ensembl (26). Subsequently, reads aligned to the host genome were removed as host contaminants. Microbial taxonomic classification was profiled using Kraken2 (v2.1.2) (27), and the relative abundance estimation was conducted by Bracken2 (v2.6) (28). Bacterial taxa that appeared in less than 5% of the samples were filtered out for subsequent analyses. Genome assembly was performed via megahit (v1.2.9) with default parameters (29). The abundance of functional genes and enriched pathways was estimated using HuMAnN3 (v3.0.1) based on the UniProt 90 database followed by annotation using the Metacyc database (30, 31). The relative abundance of aligned genes and pathways was normalized to copy numbers per million reads using the HuMAnN3 utility scripts (31).
Antibiotic resistance-encoding genes were annotated against the Comprehensive Antibiotic Resistance Database (CARD, version 3.1.4) via Resistance Gene Identifier (RGI, version 5.1.0) with a cutoff set at 95% identity (32). To reveal the distributions of microbial taxa, functional genes, and ARGs across samples, principal coordinate analysis (PCoA) based on Bray-Curtis distance metric was performed via vegan package (1.11-0) in R (version 3.6.1). To evaluate dispersion, the “betadisper” function in the vegan package (1.11-0) was used to calculate distance to centroid. Due to potential biases introduced by variations in the relative abundance of bacterial taxa, 16S rRNA copy numbers harbored by different bacterial species as well as fluctuation caused by ARGs located on mobile genetic elements, identified ARGs per sample were normalized to reads per million total ARG reads for subsequent comparisons.
Statistical analyses
Permutational multivariate analysis of variance (PERMANOVA) tests were used to determine clustering significance using the adonis function in the vegan package (1.11-0) in R (version 3.6.1, false discovery rate (FDR) adjusted P < 0.05), setting housing as a blocking effect. Differentially abundant microbial taxa, gene pathways, and ARGs associating with treatments or age were identified using LDA effect size (LEfSe) implemented in the lefser R package (Bioconductor version 3.15) with housing as “blockCol” (blocking effect). A significant cut-off was set at LDA score > 2 and FDR-adjusted P < 0.05. Differential abundance analysis of profiled gene pathways and ARGs between groups was performed using the DESeq2 package in R (Bioconductor version 3.15) (33). Significance of differential abundance was determined with FDR-adjusted P < 0.05 and log2 fold change > 1. Subsequently, the effect of housing factor was assessed by the likelihood ratio test in DESeq2 with an adjusted P-value threshold of 0.1. Except for the statistical analyses mentioned above, GraphPad Prism 8 (Graphpad Software, San Diego, CA, USA) was used to conduct statistical analyses. To determine differential significance of the microbiome alpha diversity indices, the Kruskal–Wallis test was used with significance set at P < 0.05. Two-way ANOVA was used to assess total ARG reads between different barn sanitation practices and sampling time points. The Spearman correlation was used to correlate ARG abundance and the relative abundance of bacterial species. Correlation significance was determined by the corr.test function with FDR-adjusted P < 0.05, and a moderate association was determined by ∣R∣ > 0.4. Correlation was visualized using the corrplot package in R (version 3.6.1).
RESULTS
Cecal microbial structures and functional capacities were impacted by both barn cleaning methods and age
In this study, shotgun metagenomic sequencing was used to provide a comprehensive profile of the cecal microbial community to further assess the effects of barn cleaning methods, yielding an average read depth of 52,133,725.06 ± 1,265,988.36 quality-controlled reads per sample (mean ± SEM) for downstream analyses. These reads were processed using Kraken2, resulting in 34,252,401.09 ± 6,541,062.17 reads per sample aligning to the RefSeq bacteria database. At the phylum level, Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria made up the majority cecal microbial communities both at days 7 and 30. However, there were shifts in the relative abundance of these phyla from D7 to D30 microbiota (D7 vs. D30: Firmicutes, 62.14% vs. 24.59%; Bacteroidetes, 25.57% vs. 53.27%; Proteobacteria, 8.21% vs. 15.59%; Actinobacteria, 0.76% vs. 5.92%; and other phyla, 3.32% vs. 0.63%). PERMANOVA analyses based on Bray-Curtis distance dissimilarity matrix revealed that barn cleaning methods had limited impact (adonis P = 0.23, Fig. 1a) on the D7 microbial community structure and a modest impact on D30 microbiota (R^2^ = 0.02, adonis P < 0.01, Fig. 1b). However, there were distinct separations between D7 and D30 cecal microbiota irrespective of the barn cleaning method (Fig. 1c, adonis P < 0.001). The D30 microbiota had greater distance to the centroid compared with the D7 microbiota (distance to centroid = 0.48 and 0.56 for D7 and D30, respectively, FDR-adjusted P < 0.01), indicating that the D7 microbiota had greater homogeneity. In addition, the Shannon index indicated that barn cleaning methods had minimal impact on alpha diversity at D7 (P = 0.96) and D30 (P = 0.25); however, differences were observed between D7 and D30 microbial communities as reflected by increased Shannon diversity with age (P < 0.05) (Fig. 1d through f). In terms of differentially abundant taxa, LEfSe analysis revealed no taxa with differences in the relative abundance between FD and WW groups at D7 (FDR adjusted P > 0.05). However, at D30, Ruminococcus torques, Faecalibacterium prausnitzii, Barnesiella viscericola, and Helicobacter pullorum were enriched in the WW group, whereas Megamonas funiformis was higher in the FD group (FDR-adjusted P < 0.05, LDA >2, Fig. 2). In addition, successional changes on the cecal microbial composition were demonstrated by LEfSe analysis. Briefly, 11 and 8 bacterial families were associated with the D7 and D30 chicken cecal microbiota, respectively (Fig. S2).
*Broiler chicken cecal microbial structure affected by the barn cleaning practices and age. (a–c) Factors impacting the cecal microbial structures. Age had a major impact on microbial compositions, whereas the cleaning methods had a modest effect on the D30 cecal microbiota. (d–f) Factors affecting the cecal microbial alpha-diversity as indicated by the Shannon index. At D30, the richness and evenness of the cecal microbial species significantly increased compared to that at D7. FD, full disinfection; WW, water-wash; **, P < 0.001.
Differentially abundant bacterial species between barn cleaning practices at day 30 suggested by LEfSe analysis (LDA score > 2 and FDR-adjusted P < 0.05). At day 30, Ruminococcus torques, Barnesiella viscericola, Helicobacter pullorum, and Faecalibacterium prausnitzii were more abundant in the ceca of the chickens from the WW treatment group, whereas Megamonas funiformis was more abundant in the chicken cecal microbiota of the FD group. FD, full disinfection; WW, water-wash.
Cecal microbial functionalities were subsequently profiled and annotated, resulting in an average of 64.63% ± 1.18% of the total reads per sample (mean ± SEM) being mapped to the UniProt 90 database and further annotated. At D7, six pathways were altered by the barn cleaning methods (Fig. 3a, log2 fold-change > 1, FDR-adjusted P < 0.05), including the sucrose degradation pathway IV (PWY-5384), the L-cysteine biosynthesis pathway VI (PWY-I9), the super-pathway of UDP-glucose-derived O-antigen building blocks biosynthesis (PWY-7328), the UDP-N-acetyl-D-glucosamine biosynthesis pathway (UDPNAGSYN-PWY), the phospholipase pathway (LIPASYN-PWY), and the stringent response guanosine 3′-diphosphate 5′-diphosphate metabolism pathway (PPGPPMET-PWY). Among these altered pathways, PWY-5384 was mainly harbored by Escherichia coli, Lactobacillus spp., and Bifidobacterium spp. At D30, a series of pathways were altered by barn cleaning methods (Fig. 3b). Specifically, 12 pathways were enriched in the WW group, including the pyruvate fermentation to acetate and lactate pathway II (PWY-5100), the L-lysine biosynthesis pathway I (DAPLYSINESYN-PWY), the L-lysine biosynthesis pathway II (PWY-2941), the L-isoleucine biosynthesis pathway I (ILEUSYN-PWY), the L-methionine biosynthesis pathway III (HSERMETANA-PWY), the L-isoleucine biosynthesis pathway III (PWY-5103), the phosphatidylglycerol biosynthesis pathway I (PWY4FS-7), the phosphatidylglycerol biosynthesis pathway II (PWY4FS-8), the ADP-L-glycero- and β-D-manno-heptose biosynthesis pathway (PWY0-1241), the CMP-3-deoxy-D-manno-octulosonate biosynthesis pathway (PWY-1269), the super-pathway of phospholipid biosynthesis I (PHOSLIPSYN-PWY), and the super-pathway of branched chain amino acid biosynthesis (BRANCHED-CHAIN-AA-SYN-PWY), whereas the FD group had an enriched pathway involved in hexitol fermentation to lactate, formate, ethanol, and acetate (P461-PWY) (Fig. 3b). The total of 13 altered pathways at D30 were attributed to 62 different bacteria species. The enriched PWY-5100 in the WW cecal microbial community could be considered a consequence of an increased level of H. pullorum (Fig. 4, FDR-adjusted P < 0.05, LDA >2), whereas the contribution of Lachnoclostridium sp. An76 was relatively smaller (FDR adjusted P < 0.05, LDA = 1.36). Indeed, the observed pathway enrichment linked to amino acid syntheses at the WW group was mainly attributed to H. pullorum. Compared with the modest impact on cecal microbial functional gene content by barn cleaning methods, age was shown as a strong factor affecting cecal microbial functionalities. A total of 89 pathways were significantly different between D7 and D30 cecal microbiome (Table S1, log2 fold-change > 1, FDR adjusted P < 0.05, DESeq2 analysis with sanitation treatments as blocking factor).
*Microbial functional pathways that were significantly impacted by the barn cleaning treatments at days 7 (a) and 30 (b) revealed by DESeq2. The graph shows differentially abundant genetic pathways harbored by the chicken cecal microbial communities at days 7 and 30 suggested by DESeq2, respectively (FDR P < 0.05, log2 fold-change >1). (a) At day 7, the FD-derived chicken gut microbiome had enriched stringent response pathway coupled with decreased abundance of pathways linked to amino acid synthesis, saccharide degradation, and bacterial cell wall synthesis. (b) At day 30, the FD-derived chicken gut microbial functional capacity had decreased abundance of genetic pathways linked to multiple amino acid syntheses. D7, day 7; D30, day 30; FD, full disinfection; WW, water-wash; **, FDR P < 0.01; **, FDR P < 0.001.
LEfSe results of metabolic pathways harbored by specific bacterial species and the association with treatments at day 30 (FDR-adjusted P < 0.05). LEfSe result suggested that the increased abundant pathways in the WW group were mainly contributed by Helicobacter pullorum. FD, full disinfection; WW, water-wash.
The effect of barn cleaning practices and age on the cecal resistome
An average of 0.133% ± 0.012% of total reads per sample (mean ± SEM) were mapped to the CARD database. Both barn cleaning methods and age had an impact on the resistome (Fig. 5b, FDR P <0.05). Beta-dispersion analyses revealed that the cecal resistome of the 7-day-old broiler chickens had greater distance to centroid compared to that in D30 broiler chickens (P < 0.01), indicating more variations in cecal antimicrobial resistance at D7 (Fig. 5c). The relative abundance of total ARGs was higher at D7 compared with D30 (Fig. 5a). Overall, a total of 496 ARGs from 60 gene families and 386 ARGs from 52 gene families were identified from D7 and D30 chicken cecal microbiome, respectively. Among the detected ARGs, the tetracycline-resistant gene (tet) tetW was most abundant, followed by lincosamide nucleotidyltransferase genes (lnu) lnuC, tet(34), tet(W/N/W), tetQ, the erythromycin ribosomal methylation 23S ribosomal RNA methyltransferase (erm) ermB, aminoglycoside resistance gene (APH) APH(3)-IIIa, tetO, and tet32. In terms of gene families, tetracycline-resistant ribosomal protection proteins (RPPs) family was most abundant, followed by the lnu family and the major facilitator superfamily (MFS) antibiotic efflux pump (multi-drug-resistant) family. These ARG families collectively accounted for over 90% of the broiler chicken cecal resistome (92.08% ± 11.03% mean ± SEM). At D7, the most abundant ARG families in the cecal resistomes were the tetracycline-resistant RPP family, the MFS antibiotic efflux pump family, the resistance-nodulation-cell division (RND) antibiotic efflux pump (multi-drug resistant), the erm family, and the lnu family. Although at D30, the top five abundant gene families were the tetracycline-resistant RPP followed by the lnu family, the MFS antibiotic efflux pump family, the aminoglycoside nucleotidyltransferase (ant) family ant(6) (aminoglycoside-resistant), and the erm family. When characterizing the resistance mechanism and distribution of the ARGs, we discovered that antibiotic target protection, which mainly consisted of tetracycline resistance via tetracycline-resistant RPPs, was the most common mechanism of antibiotic resistance. In addition, antibiotic efflux and antibiotic inactivation were the second most common resistance mechanism found at D7 and D30, respectively (Fig. 6). LEfSe results supported the successional change of the chicken cecal resistome (Fig. 7, FDR P < 0.05, LDA > 2). In accordance with the different antibiotic mechanisms between D7 and D30, LEfSe analysis identified that 11 ARG gene families were associated with the age of the broiler chickens (Fig. 7). Particularly, the MFS antibiotic efflux pumps and the RND antibiotic efflux pump showed the highest LDA score at D7, indicating that antibiotic efflux plays a key role in the early life cecal resistome. Although on D30, AMR gene families conferring antibiotic inactivation, such as the lnu family and the tetracycline inactivation enzyme, had increased predominance.
The chicken cecal resistome was affected by the barn sanitation practices and sampling time points. (a) ARG read counts relative to total read counts. Generally, at D7, a higher percentage of ARG read counts was detected in the chicken cecal microbiota compared to D30. (b) PCoA plots based on Bray-Curtis similarity distance matrix showing resistome clusters by treatments or age. Barn cleaning methods had a modest effect on the distribution of microbial resistome. Age was the main driver of the resistome pattern. (c) The average distance to centroid based on beta-disperse showed that the variation between resistomes was greater among the D7 chickens in comparison to the D30 chickens. ARG, antibiotic-resistant genes; D7, day 7; D30, day 30; FD, full disinfection; WW, water wash.
Antibiotic resistance mechanism conferred by detected ARGs in the cecal microbial resistomes of D7 and D30 chickens. Differences in the major antibiotic-resistant mechanisms harbored by the 7-day and 30-day chicken cecal microbial resistomes were observed. With the mechanism of antibiotic target alteration being dominant on both ages, genes conferring antibiotic efflux and antibiotic inactivation were representative of the days 7 and 30 chicken cecal resistome, respectively.
Differentially abundant cecal microbial antibiotic-resistant gene families between days 7 and 30 suggested by LEfSe. Graph showing antibiotic gene families associated with different ages (FDR P < 0.05, LDA > 2). At day 7, genes encoding antibiotic efflux pumps were representative of the chicken cecal microbial resistome. At day 30, antibiotic-resistant genes conferring antibiotic inactivation (e.g., the lincosamide nucleotidyltransferases and tetracycline inactivation enzymes) were more predominant. RND, resistance-nodulation-cell division; APH, aminoglycoside resistance gene; pmr, polymyxin resistance; msr, macrolide resistance; ABC, ATP binding cassette.
Compared with the age effect, barn cleaning methods had a modest impact on the chicken resistome profile. Specifically, five AMR gene families including the erm family, rifamycin-resistant beta-subunit of RNA polymerase (rpoB), streptogramin vat acetyltransferase, ATP-binding cassette (ABC)-F subfamily RPPs (macrolide- and lincosamide-resistant), and vanR glycopeptide resistance gene cluster were found to be more abundant in the D7-WW treatment compared with the D7-FD treatment (Fig. 8, FDR P < 0.05). At the gene level, cprR (peptide antibiotic-resistant), ermB (macrolide-, lincosamide-, and streptogramin-resistant, MLS-resistant), lnuA and lnuB (lincosamide-resistant), lsaE (pleuromutilin-, streptogramin-, and lincosamide-resistant), oleB (macrolide-resistant), tet(L) and tetM (tetracycline-resistant), vatE (streptogramin-resistant), Vibrio anguillarum chloramphenicol acetyltransferase gene (phenicol-resistant), the Bifidobacterium bifidum ileS (mupirocin-resistant), the Bifidobacterium adolescentis rpoB (rifampicin-resistant), and vanR variants in the vanA, vanG, and vanL clusters (vancomycin-resistant) were more abundant in the D7-WW group. At D30, the impacts of cleaning methods on the chicken gut microbiome were subtle, indicating that the barn cleaning practices had greater impacts in younger chickens. No differentially abundant ARG gene families were identified between FD and WW by DESeq2. However, genes including ermG (MLS-resistant) and vanR variant in vanI cluster (vancomycin-resistant) were enriched in the D30-WW group (Fig. 9).
*Differentially abundant antibiotic-resistant gene families between FD and WW at D7 suggested by DESeq2. The graph shows differentially abundant antibiotic-resistant gene families between barn sanitation practices at day 7 suggested by DESeq2 (FDR P < 0.05, Log2 fold change > 1). Some persistent ARG gene families (e.g., erm family) were enriched in the WW-derived chicken cecal microbiome at day 7. ARG, antibiotic-resistant gene; vanR, vancomycin-resistant gene R component; rpoB, gene encoding β-subunit of bacterial RNA polymerase; erm, 23S ribosomal RNA methyltransferase; ABC-F subfamily ABC RPPs, ABC-F ATP-binding cassette ribosomal protection protein genes; SAT, Streptogramin A acetyltransferase genes, FD, Full disinfection; and WW, Water-wash; ***, P < 0.001; *, P < 0.01.
*Differentially abundant antibiotic-resistant gene between FD and WW at D30 suggested by DESeq2. The graph shows differentially abundant antibiotic-resistant genes between barn sanitation practices at day 30 suggested by DESeq2 (FDR P < 0.05, Log2 fold change > 1). Compared to the impact of barn cleaning practices at day 7, the effects on the 30-day chicken gut microbial resistome were relatively smaller. On the gene level, ermG and vanR variants in the vanI cluster were enriched by the WW treatment. ARG, antibiotic-resistant gene; vanR, vancomycin-resistant gene; erm, 23S ribosomal RNA methyltransferase genes; FD, Full disinfection; and WW, Water-wash; , P < 0.05.
DISCUSSION
To date, limited information is available regarding how barn cleaning methods affect the chicken cecal microbiota. Using 16S rRNA sequencing technique, we previously reported that at D30, the genus Helicobacter was enriched in the WW group (24). Due to initial resource limitations, in-depth shotgun metagenomic sequencing could not be performed alongside our earlier phenotypic and amplicon-based sequencing. This study, therefore, builds on those findings, enabled by additional funding obtained after our prior publication (24). In the current study, functional genetics analyses suggested that the barn cleaning methods altered the functional gene content of the chicken cecal microbiota. The exposure to FD at 1 day of life may impact the assembly of the early cecal microbiota of chicks and thereby lead to altered microbial functional gene content observed later in life, which possesses decreased genetic potential for amino acid and SCFA synthesis. In addition, we confirmed that chicken cecal H. pullorum harboring genes linked to SCFA and amino acid production was higher in the WW treatment. In the present study, the sample size of each production flock was set to five broiler chickens at each age. While increasing the sample size within each flock may strengthen the conclusion, it is also important to recognize that previous microbiome studies observed cohousing effects, which can lead to misinterpretation of data (34). In the current study, we also observed a flock effect of the microbiome data (data not shown). In this case, increasing the number of observations of barns could help eliminate bias introduced by flock effect. Furthermore, our experimental design included both FD and WW treatments for each barn, which helps control for biases related to cohousing and management practices. Shotgun metagenomic sequencing of the samples provided taxonomic information to the species level with confidence. In the current study, the WW group exhibited an increased relative abundance of H. pullorum, F. prausnitzii, B. viscericola, and R. torques. Conversely, there was a notable decrease in the relative abundance of M. funiformis in the WW group compared to the FD group*.* While H. pullorum has been suggested to be an opportunistic pathogen (35), its precise role in poultry and its pathogenicity to human and chickens remains unclear. Importantly, H. pullorum is increasingly recognized as an emerging zoonotic organism that can colonize poultry and may be transmitted to humans through contaminated meat when undercooked, where it has been associated with human colitis and may exacerbate hepatitis C virus infection (36, 37). However, direct causal links between H. pullorum and other pathogens and diseases in poultry or humans remain limited and not fully elucidated. Given its enrichment in the WW group, this finding raises a potential food safety concern that warrants caution. Future studies are needed to investigate the prevalence of viable H. pullorum in poultry products, its persistence through processing, and the actual risk it may pose to human health. Similar to the current study, a previous study showed that the prevalence of Faecalibacterium increased in chicken ceca from the group treated by recycled litter compared with the fresh litter group, which was concluded as a beneficial effect of recycled litter (38). As a commensal member also found to be enriched in conventionally raised chickens (39), F. prausnitzii was identified as a promoter of epithelial health for its ability to produce metabolites such as butyrate (40). Moreover, it was found to show anti-inflammatory activity in broilers (41). B. viscericola has also been characterized as an important commensal in barn-raised chickens (42–44). In addition, members from Ruminococcus have been characterized as important butyrate producers in the gut that degrades mucins (40, 45); however, there is limited information available concerning the role of R. torques in the chicken gut.
The current study unveiled alterations in a range of microbial metabolic pathways within the chicken ceca as a result of barn cleaning practices. For example, at D7, the stringent response pathway (PPGPPMET-PWY) was enriched in the cecal microbiome of the FD group, which was primarily harbored by E. coli. The stringent response regulates genes involved in response to nutrient starvation or environmental stresses (46). It is important for bacterial virulence and persistence in the environment (e.g., resistance to antimicrobials) for a variety of taxa (46) and is used by Bacteroides to shift from growth to stasis (47). Furthermore, a previous study revealed that the stringent response can induce microbes to the viable but nonculturable state, a state which has strong tolerance to environmental stresses with minimum nutrient requirement (48). In this sense, it is reasonable to speculate the enrichment of the stringent pathway might indicate that the chemical disinfection selects for bacteria harboring PPGPPMET-PWY to tolerance harsh environmental conditions (49).
Furthermore, it is interesting to note that the early enrichment of PPGPPMET-PWY in the FD-derived gut microbiome at D7 coincided with a decreased capacity in amino acid synthesis, including branched-chain amino acid synthesis, observed at D30. Although causality cannot be established from the present data, this temporal pattern raises the hypothesis that early life stress responses might have long-term effects on microbiome development. For example, ppGpp (guanosine pentaphosphate and guanosine tetraphosphate) is important in modulating the activity of CodY, a DNA binding-protein that serves as an important transcription regulator in some low-GC, gram-positive bacteria (50). During stress, the accumulation of ppGpp lowers the intracellular GTP pool, leading to the inactivation of CodY (49). Meanwhile, it is also known that CodY regulates biosynthesis of branched-chain amino acids (51), leucine and isoleucine (52). We speculate that in this study, early exposure to chemical disinfectants could have triggered such stringent response-related regulatory shifts, potentially contributing to the observed later-life reduction in branched-chain amino acid biosynthesis potential. Therefore, future study focusing on transcriptional regulations will be needed to test this proposed linkage and to further elucidate the interplay between chemical disinfection, bacterial stringent responses, amino acid synthesis, and microbiome development.
At day 30, microbial metabolic pathways linked to SCFA production (acetate and lactate) and amino acid biosynthesis (L-methionine, L-lysine, L-isoleucine, and branched-chain amino acids) were enriched in the WW group. This suggests that the WW treatment may have led to an enhancement in the nutrient utilization functional gene content of the gut microbial community at day 30. In the current study, PWY-5100 was mainly harbored by H. pullorum in both the FD and WW group*,* and to a lesser extent, Lachnoclostridium sp. An76 and Lactobacillus salivarius. While the specific impact of the enrichment of the pathway in the WW group remains to be confirmed by further study, previous research has highlighted its potential relevance in chicken gut health, particularly in host pathogen defense. Previously, Gong et al. reported that chickens infected by Clostridium perfringens had a significant inhibition of the pyruvate fermentation to acetate and lactate pathway II (PWY-5100) in the cecal microbiome, which was restored by the supplementation of probiotic L. plantarum (53). It is important to note that our study did not include a bacterial challenge. Therefore, while the enrichment of PWY-5100 in the WW-derived microbiome is interesting, further studies on assessing how the increased capacity of PWY-5100 affects digesta SCFA or chicken performance with pathogen challenges would be needed.
Interestingly, PWY-1269 was shown to be mainly harbored by H. pullorum and C. jejuni in the chicken gut microbiome of the WW and FD, respectively. PWY-1269 encodes genes that produce acid sugar 3-deoxy-α-D-manno-2-octulosonate, which is a component of bacterial lipopolysaccharides (LPS) (54–56). We have previously reported that C. jejuni was decreased in the D30-WW group (24), which was consistent with the result from functional analyses in the current study as reflected by decreased *C. jejuni-*derived LPS genes in WW-cecal microbiome. Regarding the ARG profiles in the chicken cecal microbiota, in the current study, the most abundant ARGs detected confer resistance to tetracycline [tetW, tet(42), tet(W/N/W), tetQ, tetO, and tet(32)], MLS (lnuC, ermB), and aminoglycoside (APH(3)-IIIa), whereas β-lactam resistance was found in relatively low prevalence. Tetracycline-resistance genes, particularly tetW, were frequently detected in environments related to livestock farming (57–61), as well as bacteria isolated from the chicken gut (62). Previously, Munk et al. reported that the majority of ARGs in chicken fecal samples collected from multiple European countries were tetracycline-, aminoglycoside-, and MLS-resistant genes (18). Similarly, chicken fecal samples collected from China were high in aminoglycoside, tetracycline, MLS, and β-lactam resistance (63). The low abundance of β-lactam resistant genes observed in this study may link to the prohibition of prophylactic use of β-lactam in poultry farming since 2018 (64), whereas β-lactam was reported to be one of the most commonly used antibiotics in poultry production in China (65).
Our observations indicate that both barn cleaning methods and the age of the chickens influenced the microbial resistome. Age emerged as the primary factor driving alterations in the resistome. In line with previous studies (66, 67), we observed that the barn-cleaning effects on the gut resistome decreased with increasing age (66, 67).
While our initial expectation was that disinfection might enhance the selection of antibiotic resistance genes (ARG), our results indicate that disinfection is linked to a reduced abundance of ARG. This finding suggests that the impact of disinfection on ARG transmission between flocks merits further evaluation as a potential strategy for controlling ARG spread. It has been reported that poultry litter harbored high densities of AMR bacteria and antibiotic residuals (68–70). Although poultry litter was removed in the current study, without chemical disinfectants, WW might preserve bacteria carrying ARGs. Therefore, it is reasonable to assume that compared to WW, the chemical disinfectants used in the current study may be more effective in controlling ARGs. Notably, at both D7 and D30, ARGs from the erm family and the glycopeptide resistant gene cluster (vanR) were depleted by the disinfection. Currently, more than 30 erm genes have been characterized, and a number of them (e.g., ermB, ermC, ermG, ermF, and ermX) were frequently detected in livestock farming-related environments (71–73). Frequent horizontal transfer of erm genes through mobile genetic elements has been reported within the gut microbiota (74, 75), as well as between intestinal and environmental bacteria (76, 77), accounting for their distribution among diverse taxa. Additionally, erm genes were found to persist stably both in the gut and in the environment 2–3 years after the removal of antibiotic selection pressure (73, 78). Furthermore, erm genes were also known to spread through poultry dust (61, 79), indicating possibilities of persistent erm residues from the previous production cycles. The vanS/vanR, two-component regulatory system, is important in activating and regulating transcription of the glycopeptide gene cluster. Vancomycin-resistant genes have been detected in poultry farms (80) and products (81). Similar to erm genes, a recent study revealed a glycopeptide-resistant gene cluster persisting in the environment for 20 years (82). In addition, glycopeptide resistance gene clusters are also highly transferable via plasmids (81, 83), making it difficult to identify the main carriers.
In the present study, we found that the relative abundance of total ARGs was higher in the ceca of D7 chickens compared to D30. Previously, Lebeaux et al. reported similar results in human infants showing that the overall relative abundance of ARGs was higher at 6 weeks than 1 year (84). We noted a heightened proportion of genes encoding antibiotic target alteration coupled with a diminished proportion of genes encoding antibiotic efflux in the chicken gut resistome from day 7 to day 30. The successional change of the chicken gut microbiota may have contributed to the alteration of cecal ARG composition. LEfSe analysis suggested that the family Lachnospiraceae and Enterobacteriaceae were strong biomarkers of the chicken cecal microbiota at D7. In addition, Spearman correlation revealed that the relative abundance of the MFS antibiotic efflux pump family was positively correlated to Lachnospiraceae (Lachnoclostridium sp An76), further supporting the role Lachnospiraceae may play in the gut microbial resistome (Fig. S3). Juricova et al. compared ARG sequences and bacterial genomes and reported that the family Lachnospiraceae was an important reservoir for MFS antibiotic efflux pumps (62). Thus, the predominance of the MFS antibiotic efflux pumps detected in the D7-cecal resistome may be partially explained by the predominance of Lachnospiraceae in early life. In addition, Enterobacteriaceae (E. coli and E. albertii) were shown to be positively correlated with the abundance of the RND antibiotic efflux pumps (Fig. S3). Enterobacteriaceae are known to harbor the RND antibiotic efflux pumps (85, 86), and many RND genes were enriched in Enterobacteriaceae in chicken litter and cloacal samples (67). Thus, the enriched RND antibiotic efflux pump family at D7 may be a consequence of the early colonization of Enterobacteriaceae. In addition, among all taxa, E. coli was associated with the highest number of ARG families (Fig. S3). Interestingly, in the human infant resistome study, Lebeaux et al. concluded that the human early-life resistome composition was primarily driven by E. coli (84).
The D30 resistome was associated with antibiotic inactivation genes, particularly genes encoding LNUs (mainly lnuC gene) and tetracycline inactivation enzymes (mainly tetX gene and its variants). Lincosamide-resistant gene lnuC has been identified in the genus Streptococcus (87) and was shared extensively between different phyla (88). Emerging evidence also showed that C. jejuni and C. coli harbor lnuC (89–92). The increased relative abundance of Streptococcaceae and Campylobacteraceae at D30 may partially explain the enriched lnu family observed in older chickens. Consistent with the effect of age, lnu genes were also more predominant in adult cattle compared to calves (66). Interestingly, Bacillus subtilis was positively correlated to the lnu family (Fig. S3). B. subtilis has exhibited an ability to naturally activate the competence state and uptake foreign DNA (93), making it a potential carrier of lnu genes. However, the degree to which microbial compositions affect ARG composition is still unclear (94), especially in the case of highly mobile ARGs such as lnuC.
The present study is subject to several limitations that should be considered when interpreting the findings. First, although a cross-over design across seven broiler barns was used for multiple production cycles, only five birds per barn per time point were sampled. While this approach allowed us to capture data from multiple barns and time points, the relatively small number of biological replicates per flock provides only a glimpse of the nature of the poultry gut microbiome within this broader environment. In particular, it may have led to underestimation of subtle differences in the functional and ARG profiles. Sampling depth in the current study was determined by constraints such as sequencing costs and challenges of field-scale sampling. We acknowledge that studies with higher per-group sample sizes would be better positioned to detect smaller effect sizes and to provide more comprehensive representation of the microbiomes. Second, due to insufficient environmental DNA extracted from environmental samples, the microbiome of the initial environmental samples, such as litter and surface swabs, was not assessed. Given poultry litter and barn surfaces can retain substantial ARG reservoirs after cleaning and disinfection, it is possible that residual ARGs carried by bacteria in the barn environment acted as sources for re-colonization or re-seeding of resistomes in subsequent flocks. The absence of these environmental data limits our ability to disentangle direct impacts of disinfection from environmental re-seeding and constrains interpretation of the long-term efficacy of the intervention. Third, the resistome analyses relied on shotgun metagenomic sequencing and in silico analyses. While this culture-independent approach provides valuable information, it is difficult to confirm whether these genes are transcriptionally active, translated into functional proteins, or confer phenotypic resistance within the gut microbial community. Likewise, metabolic pathway prediction and analyses derived from metagenomics data do not distinguish between active and inactive metabolic responses. Last but not least, while our findings suggest that FD may modestly reduce certain ARG carriage in the chicken gut, the present data does not directly demonstrate whether this translates into reduced ARG dissemination through the production chain, for example, in the meat processing plants or in final meat products.
Therefore, future work should aim to increase the number of birds sampled per barn to improve statistical power and sensitivity for the detection of less prevalent microbial or resistance determinants. In addition, increased volume of environmental sampling to obtain enough material for comprehensive environmental ARG surveillance is warranted to capture the full dynamics of resistance gene persistence and reintroduction. Furthermore, culture-based studies assessing antibiotic/disinfectant susceptibility and ARG mobility targeting isolates of keystone taxa of commercially raised broiler chickens are of great importance. Complementary functional validation using approaches such as metatranscriptomics or reverse transcription-qPCR targeting a selection of resistance genes, proteomic or metabolomics profiling will also be essential for linking genomic potential to gene expression, protein activity, and metabolic function.
Conclusion
This is the first study to report that the impact of disinfectants in broiler production on microbial functional gene content and resistome. We showed that barn chemical disinfection may alter the composition of the chicken gut microbiota and thereby lead to decreased microbial functional gene content for amino acid and SCFA metabolism. Conversely, although differences were modest, FD may be beneficial through lowering abundance and diversity of ARGs. For broiler chicken production, the current study presents a practical trade-off. While chemical disinfection may help slightly mitigate ARG carriage, especially those that are persistent, it also reduces the gut microbial functional gene content, which may have longer-term implications for flock performance. Considering current evidence, prioritizing a robust gut microbiome may outweigh the subtle benefit of ARG suppression, particularly in the current production settings where prophylactic antibiotic usage is already strictly controlled. Therefore, frequent FD treatment between every flock may not be universally justified if the main goal is to minimize ARG burden, as WW methods also support beneficial microbiome functions and do not compromise flock performance (24). Future on-farm decisions should consider both antimicrobial stewardship and the importance of a robust gut microbiome. Additional studies incorporating longer-term production outcomes and flock health monitoring are warranted to refine recommendations.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Van Boeckel TP, Glennon EE, Chen D, Gilbert M, Robinson TP, Grenfell BT, Levin SA, Bonhoeffer S, Laxminarayan R. 2017. Reducing antimicrobial use in food animals. Science 357:1350–1352. doi:10.1126/science.aao 149528963240 PMC 6510296 · doi ↗ · pubmed ↗
- 2Qian X, Gu J, Sun W, Wang X-J, Su J-Q, Stedfeld R. 2018. Diversity, abundance, and persistence of antibiotic resistance genes in various types of animal manure following industrial composting. J Hazard Mater 344:716–722. doi:10.1016/j.jhazmat.2017.11.02029154097 · doi ↗ · pubmed ↗
- 3Murray CJL, Ikuta KS, Sharara F, Swetschinski L, Robles Aguilar G, Gray A, Han C, Bisignano C, Rao P, Wool E, et al.. 2022. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. The Lancet 399:629–655. doi:10.1016/S 0140-6736(21)02724-0 · doi ↗
- 4Kampf G. 2018. Biocidal agents used for disinfection can enhance antibiotic resistance in gram-negative species. Antibiotics (Basel) 7:110. doi:10.3390/antibiotics 704011030558235 PMC 6316403 · doi ↗ · pubmed ↗
- 5Mc Namara PJ, Levy SB. 2016. Triclosan: an instructive tale. Antimicrob Agents Chemother 60:7015–7016. doi:10.1128/AAC.02105-1627736758 PMC 5118979 · doi ↗ · pubmed ↗
- 6Zeng J, Li Y, Jin G, Su J-Q, Yao H. 2022. Short-term Benzalkonium Chloride (C 12) exposure induced the occurrence of wide-spectrum antibiotic resistance in agricultural soils. Environ Sci Technol 56:15054–15063. doi:10.1021/acs.est.2c 0473036069710 · doi ↗ · pubmed ↗
- 7Zhang Y, Gu AZ, He M, Li D, Chen J. 2017. Subinhibitory concentrations of disinfectants promote the horizontal transfer of multidrug resistance genes within and across genera. Environ Sci Technol 51:570–580. doi:10.1021/acs.est.6b 0313227997135 · doi ↗ · pubmed ↗
- 8Kim M, Weigand MR, Oh S, Hatt JK, Krishnan R, Tezel U, Pavlostathis SG, Konstantinidis KT. 2018. Widely used Benzalkonium chloride disinfectants can promote antibiotic resistance. Appl Environ Microbiol 84:e 01201-18. doi:10.1128/AEM.01201-1829959242 PMC 6102991 · doi ↗ · pubmed ↗