A modified targeted culturing approach provided a snapshot into interdependencies and resistome among core anaerobic bacteria of the healthy human gut
Mahnoor, Riaz Ullah, Sonyia, Rawaiz Khan, Ali Bahadar, Ibrahim Elbatel, Imran Khan, Alaa Abdulaziz Alnahari, Adnan Haider, Syed Babar Jamal, Raees Khan

TL;DR
This study used a new culturing method to explore interactions and antibiotic resistance among key gut bacteria, revealing complex relationships and potential roles in gut health.
Contribution
A modified culturomics approach enabled targeted isolation and characterization of core anaerobic gut bacteria, uncovering their interdependencies and resistome.
Findings
Bacteroides thetaiotaomicron showed dual roles in inhibiting pathogens and promoting probiotic growth.
Whole genome sequencing linked observed phenotypes to potential genetic mechanisms.
The method successfully isolated 21 core genera, including some reported for the first time.
Abstract
The core anaerobic component of the human microbiome plays a critical role in preserving the overall structure and function of the microbiome. However, how these core strains interact with each other and with other gut microbes is largely unknown. We employed a modified culturomics approach to selectively culture these core genera from the human gut and provided a snapshot into their ecological interactions, clinically relevant resistome, and whole genome sequence (WGS)-based insights into their observed phenotypes. This strategy successfully isolated representatives of the targeted 21 core genera, with some isolates reported for the first time. Cross-interactions and resistome-based profiling revealed important insights. Out of the 440 interaction assessments, a significant proportion exhibited either cooperative or competitive effects on other core genera. Interestingly, Bacteroides…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
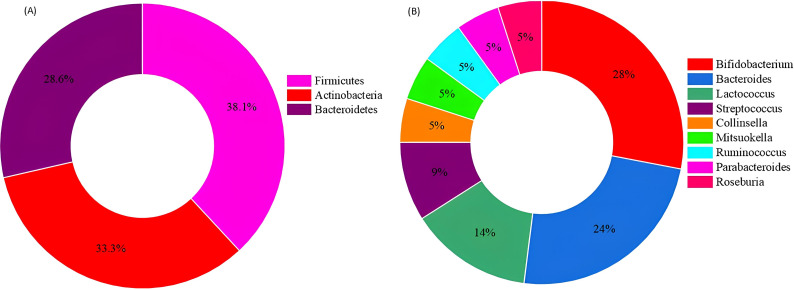 Fig 1
Fig 1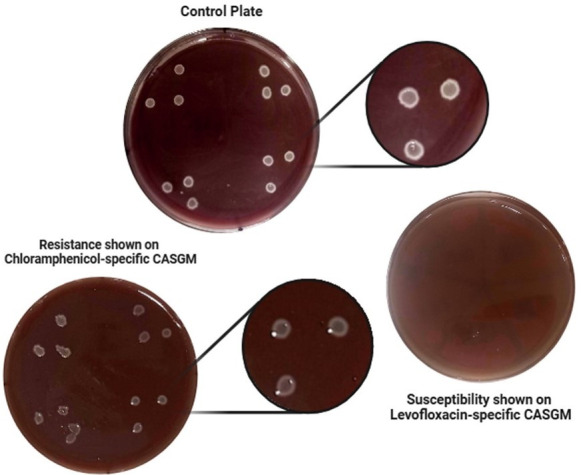 Fig 2
Fig 2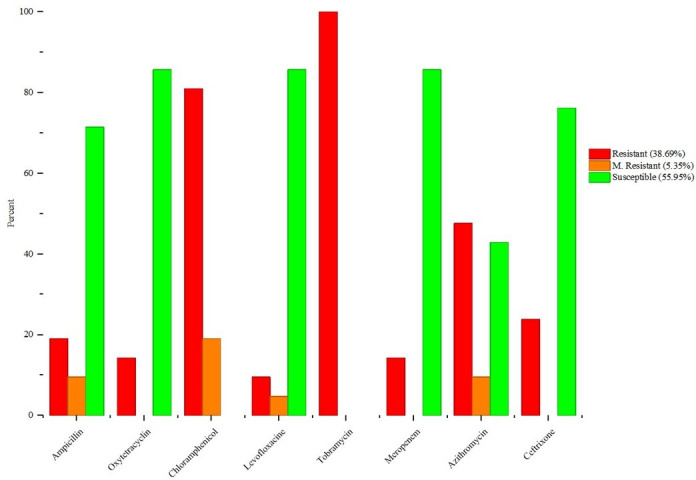 Fig 3
Fig 3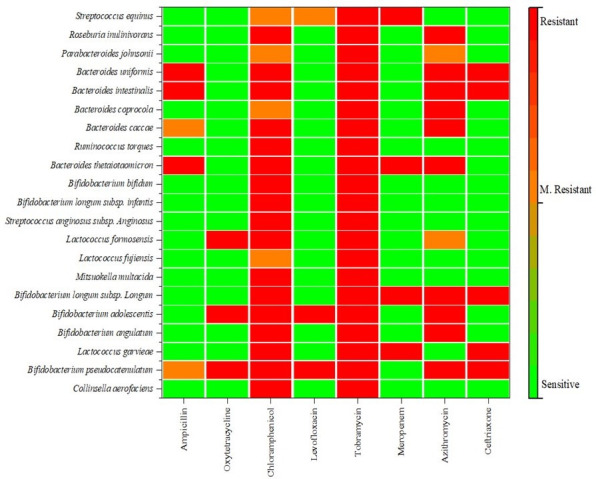 Fig 4
Fig 4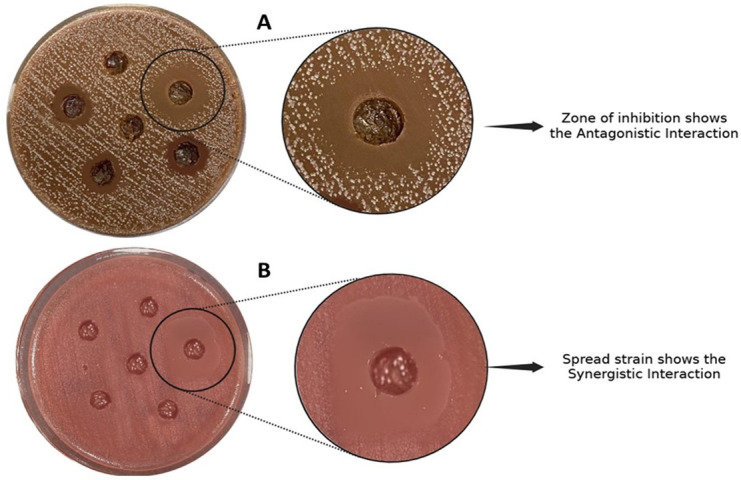 Fig 5
Fig 5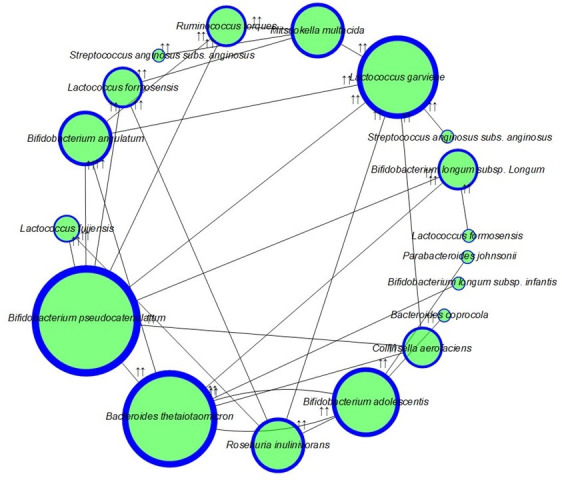 Fig 6
Fig 6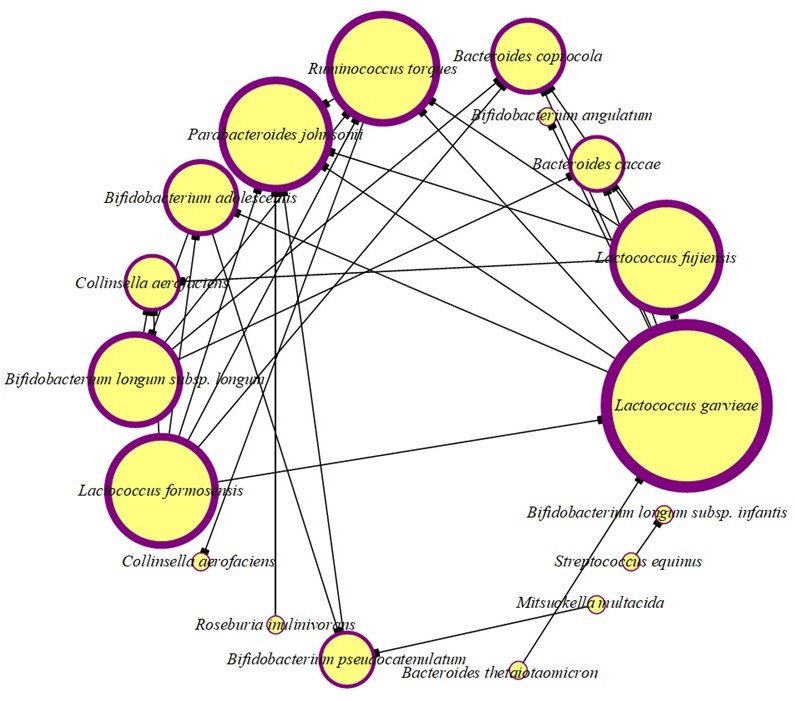 Fig 7
Fig 7 Fig 8
Fig 8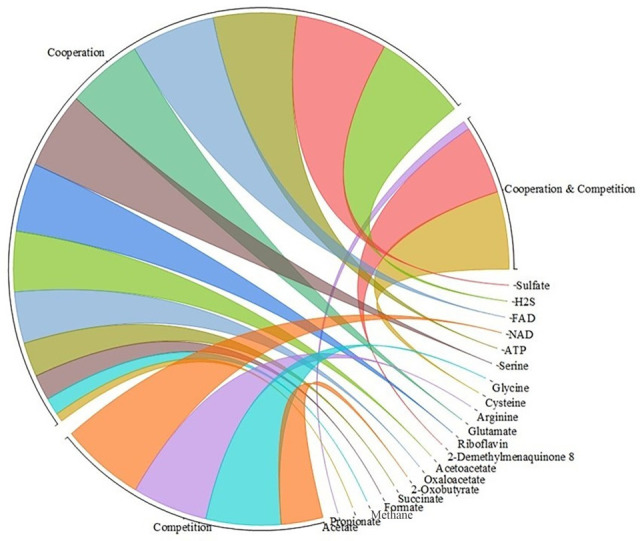 Fig 9
Fig 9| Sr. no. | Ingredients | Amount |
|---|---|---|
| 1 | Lab-Lemco Powder (Oxoid) | 2.4 g |
| 2 | Protease Peptone No. 3 (BD-Difco) | 10.0 g |
| 3 | Yeast Extract (BD-Difco) | 5.0 g |
| 4 | Sodium phosphate dibasic (Na2HPO4) | 4.0 g |
| 5 | Glucose (dextrose) | 1.5 g |
| 6 | Soluble starch (succinic anhydride) | 0.5 g |
| 7 | L-cysteine | 0.2 g |
| 8 | 1 N HCL | 50.0 mL |
| 9 | Agar | 15.0 g |
| 10 | L-cysteine·HCl·H2O | 0.5 g |
| 11 | Horse blood | 50.0 mL |
| 12 | Distilled water | Add to reach a final volume of 1-L |
| 13 | Ascorbic acid | 10.0 mL |
| S. no. | Core anaerobe ( | Cultured via CASGM in our study |
|---|---|---|
| 1 |
|
|
| 2 |
| No |
| 3 |
| No |
| 4 |
|
|
| 5 |
| No |
| 6 |
| Yes |
| 7 |
|
|
| 8 |
| No |
| 9 |
| No |
| 10 |
| No |
| 11 |
| No |
| 12 |
|
|
| 13 |
|
|
| 14 |
|
|
| 15 |
|
|
| 16 |
| No |
| 17 |
|
|
| 18 |
|
|
| 19 |
|
|
| 20 |
| No |
| 21 |
|
|
| Species name | Isolated for the first time | Previous isolation niches | References |
|---|---|---|---|
|
| No | Oral microbiome, human fecal sample | ( |
|
| No | Human fecal samples | ( |
|
| Yes | Affected rainbow trout fish, dairy products, meat products, vegetables, | ( |
|
| No | Breast-fed infants’ fecal sample/human feces | ( |
|
| No | Infant fecal sample, human milk, bovine rumen | ( |
| No | 25-year-old healthy Korean female, milk-based drink, yogurt, infant formula, | ( | |
|
| No | Endocarditis patient fecal sample, piglet feces, human feces/swine feces | ( |
|
| Yes | Chinese cabbage | ( |
|
| Yes | Yan-tsai-shin (fermented broccoli stem), fermented soybean meal | ( |
| No | Human body | ( | |
| No | Healthy fecal sample/infant fecal sample | ( | |
|
| No | Fermented food, i.e., yogurt, human fecal sample | ( |
|
| No | Healthy adult fecal sample, cow, pig, goat, mouse | ( |
|
| No | Initially from human gut, bovine rumen, cecum of the feral chicken | ( |
|
| No | Human fecal sample, blood sample | ( |
|
| No | Human fecal sample | ( |
|
| No | 32-year-old Japanese female fecal sample | ( |
|
| No | Human wound, healthy human colon, stool of healthy breast-fed infants | ( |
|
| No | Japanese human fecal sample | ( |
|
| No | Human fecal sample in an M2 medium-based culture system | ( |
|
| No | Fresh horse dung, cattle, and human feces | ( |
| Species | Potential | References |
|---|---|---|
|
| Potential probiotic | ( |
|
| Potential pathogen | ( |
|
| Probiotic | ( |
|
| Probiotic | ( |
| Probiotic | ( | |
| Probiotic | ( | |
|
| Probiotic | ( |
|
| Probiotic | ( |
|
| Non-pathogen | ( |
|
| Zoonotic pathogen | ( |
|
| Pathogen | ( |
|
| Not known |
- —King Salman Center for Disability Research (KSCDR)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGut microbiota and health · Clostridium difficile and Clostridium perfringens research · Probiotics and Fermented Foods
INTRODUCTION
The human gut is home to a diverse community of microorganisms, the microbiome that plays an important role in maintaining the host’s health. This host-associated microbiome impacts the health of the host by taking part in various metabolic processes, including nutrient metabolism, regulation of the host immune system, and providing direct or indirect protection against various pathogenic organisms (1). The bacterial component of the gut microbiome is dominated by anaerobic bacteria, which may constitute up to 99% of the community depending upon the location of the gastrointestinal tract (2). This anaerobic portion of the gut microbiome has been known to facilitate the host in various metabolic functions as well as retaining eubiosis (3–6).
Some members of the microbiome that play an important role in maintaining the overall structure and function of the microbial community are termed core microbiome components. This core microbiome component is gaining popularity in the scientific community as these core microbes are commonly found in healthy individuals (7–9). The core microbiome component is comparable to the queen in a bee colony, and the absence of these core microbes has been found to significantly delay the restoration of the gut microbiome or even lead to dysbiosis (10). For example, an analysis of large-scale microbiome data from four discovery cohorts across three continents—comprising over 500 profiles from 117 individuals—identified 21 bacterial species strongly associated with microbiome recovery following antibiotic therapy (10). However, the clinically relevant resistance and interactions among these core candidate genera are largely unexplored.
Understanding the co- and cross-interactions among core anaerobes and other gut microbes is crucial for identifying the factors that shape the overall community structure. The core microbiome has been explored in various review articles and metagenomics-based meta-analysis studies and declared as core based on their prevalence, stability, and functional importance (7, 11). Furthermore, targeting the core microbiome component has significant implications for synthetic microbiome transplants as it minimizes the bias and limitations associated with traditional microbiome transplantations, including pathogen transfer, transient results, and sustainable production (12, 13). Various anaerobic genera dominate the core microbiome component, including Bacteroides, Clostridium, Faecalibacterium, and Prevotella, and these are of particular interest due to their essential role in maintaining gut health by playing a role in many critical processes, such as fiber fermentation, short-chain fatty acid (SCFA) production, and immune modulation (14, 15). However, how these core genera are functionally dependent on each other, the nature of their co- and cross-interactions, and their clinically relevant antimicrobial resistance (AMR) remains poorly understood. This highlights the need for targeted approaches to explore their ecological and functional roles (16).
Recent advances in sequencing technologies have identified and defined the core taxa. However, sequencing technologies alone are unable to give valid insights into the overall functional capabilities of these core taxa, as they need to be validated in vitro. One of the major limitations so far in the characterization of the cross-interactions and clinically relevant resistome of these organisms is the culturing limitations. Since a major fraction of these core anaerobic bacteria is uncultured, this leads to a significant gap in our understanding of the human gut microbiome and the interactions within the microbial community (17). Microorganisms within a community interact with each other via various mechanisms, and these interactions are of immense importance for their survival within the community, as well as to modulate the overall microbial community dynamics (18). One of the traditional approaches for culturomics is the use of many different culture media types to selectively culture the targeted gut microbes. This non-economical and labor-intensive approach further necessitates the need for the development of unique representative media types, which can be utilized for the targeted culturing of the core bacteria. One ideal approach to design such media is mimicking the nutrient-rich conditions of the human gut. This specially designed media can then be utilized to investigate the interdependencies and cross-interactions among core microbes as well as among other anaerobes (19, 20).
Bacterial ecological interactions are broadly classified into several categories, with the most common being synergistic, antagonistic, and bifunctional. Synergistic interactions occur when co-cultured species mutually enhance each other’s growth and proliferate, often through cross-feeding of metabolites such as SCFAs, vitamins, or amino acids (21). Conversely, antagonistic interactions involve inhibitory effects that occur due to the secretion of bacteriocins or secondary metabolites, which suppress the proliferation of competitors and help define niche boundaries (22). Bidirectional interactions show context-dependent relationships where strains can have dual nature by promoting or inhibiting other’s growth due to relative abundances or nutrient-level dependencies (23).
Exploring the interdependencies in terms of co- and cross-interactions among the core members of the gut microbial community is of significant importance due to many reasons. Primarily, having an idea of how the core microbiome responds to clinically relevant antibiotics will further guide us regarding the impact of those antibiotics on the core taxa as well as on overall community structure (24). Additionally, such profiling will help clinicians in informed decision-making prior to prescribing antibiotics and selected alternatives with potential minimal impact on the gut microbiome. Moreover, the knowledge of co- and cross-interactions among community members will help us gauge which types of interactions govern the community dynamics and are responsible for maintaining the overall structure and function of the community (25).
Here, we have developed a unique culturomics strategy by utilizing a core anaerobe-specific growth medium (CASGM). Using CASGM, we targeted the core anaerobic fraction of the healthy human gut microbiome. This approach has the advantage as it bypasses the need for using diverse culture media types. Furthermore, using CASGM, we gave a snapshot into the cross-interactions among these core anaerobes and also determined their interactions with other members of the community. Additionally, using this approach, we explored the clinically relevant resistome of these organisms. In addition, we have correlated the observed phenotype with potentially responsible genetic elements by using a combinatorial approach of CASGM and whole-genome sequencing (WGS), which provided valuable insights into observed interdependencies (26, 27). In summary, our CASGM-based modified culturomics approach can be utilized to selectively culture core anaerobes and provide a snapshot of their resistome and interdependencies, thus shedding light on this largely unexplored research area, which is in its embryonic stage and needs to be explored.
MATERIALS AND METHODS
Screening and selection of healthy human donors
Healthy human donors (age range: 18–40 years old) were selected on the basis of standardized questionnaires, and their fecal samples were taken with informed consent. Some of the main inclusion criteria included no use of antibiotics and probiotics by the donor in the last 6 months, with no current or previous history of any gastrointestinal inflammatory disorders. Donors meeting all the criteria were included in the study, and their fecal materials were collected in sterile containers, immediately aliquoted, and processed for bacterial isolation. These aliquots were also stored in 40% glycerol at −80°C for backup and future processing.
Isolation of core anaerobic bacteria
In this study, two biological replicates were processed at different time points for isolation and culturing. We designed a specialized phosphate buffer solution (PBS) by adding ascorbic acid (0.01 g/L), which is a strong antioxidant. We named this buffer as the “anaerobe viability-supportive PBS (AVSPBS)” and will be referred to as AVSPBS hereafter. The collected stool samples were suspended in AVSPBS and were homogenized to make a uniform suspension. In summary, we suspended a total of 1 g of fresh fecal material in 9 mL of AVSPBS in a sterile Falcon tube. This mixture was then homogenized using a vortex to make a uniform suspension and was serially diluted. Specifically, successive 10-fold dilutions, ranging from 1 × 10⁻¹ to 1 × 10⁻¹⁰, were prepared by transferring 100 µL of the suspension mixture into 900 µL of fresh AVSPBS. Each dilution was carefully mixed to ensure uniform bacterial distribution. We isolated anaerobic bacterial species by handling the samples in biosafety level 2, which is the recommended biosafety level for handling human gut microbiota. Contaminant measures, such as autoclaving media and spreaders at 121°C for 15 min, filtering antifungal and antibiotic solutions, sterilizing the biosafety cabinet environment with UV light, pouring media inside the biosafety cabinet, wearing lab coats and gloves, and sterilizing hands with 70°C ethanol, were taken. These measures were adapted during the isolation and purification processes to avoid contamination. Furthermore, to minimize or exclude potential cross-contamination of stool samples, each individual’s fecal samples were processed in a separate biosafety cabinet along with other containment measures as mentioned above.
Preparation of CASGM
To target the culturing of core anaerobes from healthy donors’ fecal samples, we designed a modified complex growth medium, termed as CASGM. The CASGM was formulated by adding ascorbic acid (1 g/L) and L-cysteine (0.2 g/L) as additional antioxidants. In summary, the culture media were prepared by adding all the ingredients (Table 1) in a sterile flask. L-cysteine (0.2 g) was added to 50 mL of 1 N HCl and mixed thoroughly. Then, the remaining components and 950 mL of distilled water were added. The pH was adjusted to 7.6–7.8, and the media were sterilized by autoclaving at 121°C for 15 min. Upon cooling (at ≈50°C), 5% horse blood (50.0 mL) and 1% ascorbic acid (10.0 mL) were added aseptically (Table 1).
Screening and shortlisting of core anaerobic bacteria
Anaerobic serial dilutions in AVSPBS were further proceeded using the spread plate method by evenly spreading 100 µL from each dilution onto CASGM culture media plate, and the plates were kept in an anaerobic jar with Oxoid AnaeroGen 2.5L packs (Thermo Scientific) and Oxoid Resazurin Anaerobic indicator (Thermo Scientific) to ensure anaerobic conditions. We used Resazurin redox dye strip because of its low toxicity toward microorganisms and its high efficiency even at low concentrations (1–2 µg mL^−1^) (28). An anaerobic jar system was used to isolate core anaerobic bacteria, with suitable positive and negative control bacteria to ensure that the jar was providing the required environment for culturing anaerobic bacteria. The jar was then tightly closed to avoid oxygen exposure during the incubation period. The jar was incubated at 37°C for 48 h, but additional incubation of 24 h was continued to support the growth of slow-growing core anaerobes. Pure isolates were shortlisted on the basis of colony morphology, and a single unique colony was processed further to avoid duplication.
Optimizing conditions for cryopreservation of the core anaerobes
Obligate anaerobes are highly sensitive to any available oxygen; therefore, to ensure their viability during cryopreservation, we used a modified glycerol solution. Cryoprotectants and their sterilization conditions include 40% glycerol (autoclaved at 121°C for 20 min), amended with 0.2-micron syringe-filtered ascorbic acid (1 g/L) and glutathione (0.1 g/L). The purified colonies were preserved in this modified 40% glycerol stock and stored at −80°C. The glycerol stocks were checked regularly every month to ensure the viability of anaerobes.
DNA extraction, 16S rRNA gene amplification, and identification of core anaerobes using Sanger sequencing
DNA extraction was performed using two different methods: the boiling method and a commercial DNA extraction kit (Solarbio). The 16s rRNA gene (V3–V4) region was amplified by using universal primers 341-F (5′ CCTACGGGNGGCWGCAG 3′) and 805-R (5′ GACTACHVGGGTATCTAATCC 3′). The PCR conditions included an initial denaturation at 95°C for 15 min, followed by 25 amplification cycles of denaturation (1 min at 95°C), annealing (25 s at 47.8°C), and extension (30 s at 72°C), with a final extension at 72°C for 5 min. The PCR products were analyzed on a 1.5% agarose gel in 1× TBE buffer (45 mM Tris-borate and 1 mM EDTA) at 120 V for 90 min using the BIORAD ChemiDoc XRS system. The amplified 16S rRNA gene products were sequenced using Sanger sequencing technology (Macrogen, South Korea). The sequence data were analyzed using BioEdit, EzBioCloud, and NCBI databases for the identification of core anaerobic bacterial strains. Initially, several hundred isolates were identified via 16S ribosomal RNA V3–V4 region. Later on, selected isolates were processed for WGS, and along with species-level identification validation, genomes were annotated for metabolites implicated in antagonistic and synergistic interactions.
Clinically relevant AMR profiling of core anaerobes
To evaluate the impact of clinically relevant antibiotics on representative core anaerobic genera, we conducted AMR profiling. The rationale for selecting these eight antibiotics was their clinical relevance and their representation of eight different antibiotic categories, which are broadly used against a variety of infections. Using the drop method, a set concentration of 15 µg/mL for eight classes of antibiotics—oxytetracycline, meropenem, ampicillin, chloramphenicol, ceftriaxone, levofloxacin, azithromycin, and tobramycin—was prepared in CASGM. Core anaerobic strains were suspended in AVSPBS to achieve an optical density (OD) ranging from 0.1 to 0.12 at 600 nm. From this suspension, 1.5 µL of each strain was carefully spotted onto plates with and without antibiotic supplementation. Incubation was carried out at 37°C for 48 h, with regular inspection of the plates to monitor zones of inhibition. Control plates, devoid of antibiotics, served as baselines for comparative analysis. Each AMR assay was conducted in three technical replicates, with positive and negative controls.
Assessment of cross-interactions among anaerobes
To investigate interspecies growth interactions among core anaerobic strains, we utilized the agar overlay method. Each strain was prepared in a homogeneous suspension using AVSPBS, with an OD standardized between 0.1 and 0.12 at 600 nm. A 100 µL aliquot of each suspension was evenly spread onto CASGM using sterile cotton swabs to create a uniform bacterial lawn. Wells were then formed in the same CASGM plates, and 100 µL of other core anaerobic strains was introduced into these wells. Control plates, containing only the wells with bacterial strains and no overlay, were prepared similarly. All plates were carefully placed in an anaerobic jar to prevent any splashing or spills, followed by incubation at 37°C for 48 h. Each strain was processed in three technical replicates. Upon completion of incubation, results were evaluated by comparing test plates with controls: an enhancement in bacterial growth around the wells indicated a synergistic interaction, while the presence of clear inhibition zones around the wells signified antagonistic activity.
Analysis of Bifidobacterium angulatum WGS reads
Purified isolate of the selected anaerobe, B. angulatum, having a unique interaction profile, was sequenced in collaboration with GetGenome (UK) using third-party sequencing services. A web-based pipeline of the DOE Systems Biology Knowledgebase (Kbase) was used for analysis. Raw reads were processed for quality control assessments using the FastQC tool. PRINSeq, Trimmomatic, and Bloom Filter Corrector were applied for filtering low-complexity reads, trimming adapters, and removing sequencing artifacts, respectively. High-quality contiguous sequences were processed for de novo assembly using SPAdes with multi-kmer strategy. The genome assembly was evaluated for completeness and contamination using QUAST and CheckM, respectively. Complete (>95%) genome with <5% contamination was taxonomically classified through GTDB and annotated via RAST. An annotated assembly was processed for metabolic modeling using the Build Metabolic Model tool and KEGG pathways to elucidate potential genetic determinants of microbe-microbe interactions.
RESULTS
Targeted culturing of core anaerobes
To culture the targeted core anaerobes, we initially isolated 49 anaerobic strains from healthy fecal donor samples using our CASGM-based targeted culturing approach. To optimize resources and reduce sequencing costs, we performed preliminary differentiation based on colony morphology, allowing us to identify and retain only unique isolates. This screening resulted in 24 distinct anaerobic isolates, which were subsequently sent for identification via Sanger sequencing and further analysis.
Successful culturing of core anaerobes with CASGM-based approach as confirmed by 16S rRNA-based identification
Using the CASGM-based culturing approach, we successfully isolated differentially abundant species, including 21 anaerobes, of which 11 were targeted core anaerobic species (Table 2), representing 52.4% of the species we aimed to culture. The complete list of isolates is provided in Table 2. These isolates represented a range of phyla, including Firmicutes (38.09%), Actinobacteria (33.3%), and Bacteroidetes (28.57%) (Fig. 1A). The isolates were further classified into nine distinct genera, with Bifidobacterium constituting the highest abundance at 28.57%, followed by Bacteroides and Lactococcus, each at 23.8%. Other identified genera included Streptococcus (9.52%) and Collinsella, Mitsuokella, Ruminococcus, Parabacteroides, and Roseburia, each contributing 4.76%.
Percentage distribution of identified anaerobic isolates according to (A) phylum, (B) genus.
The majority of these isolates, 66.67%, were obligate anaerobes, followed by 19.04% facultative anaerobes, 9.52% microaerophilic anaerobes, and 4.76% microaerotolerant anaerobes. Specifically, the obligate anaerobes included species such as Collinsella aerofaciens, Bifidobacterium pseudocatenulatum, B. angulatum, Bifidobacterium adolescentis, Mitsuokella multacida, Bifidobacterium bifidum, B. thetaiotaomicron, Ruminococcus torques, Bacteroides caccae, Bacteroides coprocola, Bacteroides intestinalis, Bacteroides uniformis, Parabacteroides johnsonii, and Roseburia inulinivorans. Facultative anaerobes included Lactococcus garvieae, Lactococcus fujiensis, Lactococcus formosensis, and Streptococcus equinus, while microaerophilic anaerobes included Streptococcus anginosus subsp*. anginosus* and Bifidobacterium longum subsp*. infantis*. Finally, Bifidobacterium longum subsp*. longum* was identified as a microaerotolerant anaerobe.
This selective culturing successfully cultured core anaerobic genera, underscoring the method’s capacity to support diverse yet targeted bacterial communities and to promote insights into the composition and functional potential of anaerobic microbiomes.
Expansion of the cultured gut anaerobes via CASGM approach
Our targeted culturing approach successfully isolated 11 core anaerobic species along with other important anaerobes from the healthy human gut microbiome. Interestingly, 19.04% of these anaerobes—namely, L. fujiensis, L. formosensis, S. anginosus subsp. anginosus, and L. garvieae—have not been previously isolated from the human gut. This study marks the first successful isolation of these strains from healthy individuals using the CASGM approach (Table 3).
For instance, L. fujiensis was initially isolated from Chinese cabbage, while L. formosensis was first identified in Yan-tsai-shin (fermented broccoli stem) and subsequently in fermented soybean meal (38). L. garvieae was originally isolated from diseased rainbow trout and has since been found in dairy products, meat products, and vegetables. In contrast, there are limited isolation data available for S. anginosus subsp. anginosus. To date, no prior studies have confirmed the isolation of these strains from the healthy human gut (Table 3).
Functional classification based on their potential roles indicated that the majority of the isolated core anaerobes possess probiotic potential with some potential pathogens
To classify the isolated core anaerobes based on their potential roles within the gut microbiome, we conducted a comprehensive review of previously published, well-established studies to categorize each isolate as either a potential probiotic or a potential pathogen. This classification took into account each isolate’s physiological functions and any associations with disease etiology.
Our analysis revealed that 66.6% of the isolated anaerobes potentially exhibit probiotic potential. Among the probiotic candidates, strains such as C. aerofaciens, B. pseudocatenulatum, B. angulatum, B. adolescentis, L. formosensis, B. longum subsp*. infantis*, B. bifidum, R. inulinivorans, B. coprocola, B. intestinalis, B. uniformis, and M. multacida demonstrate significant probiotic attributes. P. johnsonii also exhibits probiotic potential; however, it was concurrently classified as a potential rare pathogen due to its increased prevalence in patients with alopecia.
Others were classified as potential pathogenic strains, i.e., S. anginosus subsp*. anginosus* and L. garvieae, the latter being a known zoonotic pathogen. Moreover, R. torques, a species known for its dual nature, potentially acts as both a probiotic and exhibits pathogenic effects. However, current data are insufficient to conclusively support its pathogenicity. Due to inadequate studies, L. fujiensis and S. equinus were considered non-pathogenic strains.
Snapshot of clinically relevant AMR indicates high susceptibility to clinically relevant antibiotics
Antimicrobial susceptibility was evaluated by comparing bacterial growth on antibiotic-supplemented plates to that on control plates (Fig. 2). Strains whose growth was completely inhibited are termed susceptible, whereas those showing slight growth were deemed moderately resistant, and the strains whose growth was not inhibited are referred to as resistant.
Representative picture of AMR profiling of core anaerobes against different tested antibiotics.
AMR profiling revealed that 55.95% were susceptible to the eight tested antibiotics. This indicates that over half of the core anaerobic bacterial population can be targeted by standard clinically relevant antimicrobial treatments and may cause dysbiosis. Meanwhile, 38.69% of the isolates demonstrated resistance, suggesting the presence of adaptive mechanisms that facilitate survival despite antibiotic exposure. Additionally, 5.35% of the anaerobic strains exhibited moderate resistance (MR), which may become susceptible at high antibiotic dosages (Fig. 3).
The combined clustered column chart illustrates the susceptibility profiling of isolated bacterial strains categorized as resistant, MR, and susceptible to antibiotics. The red color presents resistance, and the orange color shows MR, while the green color denotes sensitivity.
Detailed analysis revealed that 100% of the 21 strains were resistant to tobramycin, followed by 80.95% resistance to chloramphenicol. A notable proportion of isolates also exhibited resistance to azithromycin (47.61%), a member of the macrolide class. In contrast, the highest susceptibility rates of core anaerobes were shown against antibiotics such as meropenem, levofloxacin, and oxytetracycline (Fig. 3).
Chloramphenicol (19.09%) depicted the highest ratio of MR, followed by azithromycin and ampicillin (9.53% each) (Fig. 3). B. pseudocatenulatum and B. caccae were MR to ampicillin, L. formosensis and P. johnsonii are MR to azithromycin, whereas L. formosensis, B. coprocola, P. johnsonii, and S. equinus exhibited MR to chloramphenicol (Fig. 4).
Heatmap of antibiotic susceptibility, resistance, and MR of the anaerobic isolates, including core anaerobes against eight different classes of antibiotics. Red color shows resistance, orange color shows MR, and green color shows sensitivity.
Some of the organisms showing resistance to at least one agent in three or more antimicrobial categories were considered multidrug resistant. These organisms include B. pseudocatenulatum, L. garvieae, B. angulatum, B. adolescentis, B. longum subsp*. longum*, B. thetaiotaomicron, L. formosensis, B. caccae, B. coprocola, B. intestinalis, B. uniformis, P. johnsonii, R. inulinivorans, and S. equinus (Fig. 4).
Snapshot of cross-interactions among core anaerobes revealed synergistic, antagonistic, and bifunctional impacts on probiotic and pathogenic strains
To have an idea of the synergistic and antagonistic activities of all isolated anaerobes, cross-interaction studies were performed. A total of 440 microbial interactions were studied, where 31 synergistic and 35 antagonistic responses were noticed. Most importantly, 33.33% of anaerobes showed both synergistic and antagonistic behaviors. This dual role indicates the complexity of microbial interactions, where certain strains can promote the growth of some taxa while simultaneously inhibiting others. Overall, 80.93% of the isolated anaerobes revealed either synergistic or antagonistic interactions.
Isolates showing synergistic potential (Fig. 5B) were termed as helper strains, which include L. formosensis, S. anginosus subsp*. anginosus, M. multacida, B. longum* subsp*. longum, B. pseudocatenulatum, C. aerofaciens, B. adolescentis, B. angulatum, R. inulinivorans,* and B. thetaiotaomicron. B. pseudocatenulatum showed the highest number of synergistic interactions impacting the growth of seven other anaerobic strains. Other major helper strains that enhanced the growth of ≥3 other strains include M. multacida, R. inulinivorans, B. thetaiotaomicron, and B. adolescentis. Out of these 12 helper strains, 7 strains showed “both” synergistic and antagonistic interactions. These helper strains enhanced the growth of important gut bacteria, such as L. formosensis, which enhanced the growth of the multifunctional probiotic strain B. longum subsp*. longum. B. pseudocatenulatum* promoted the growth of several probiotics, while inhibiting P. johnsonii. Similarly, B. pseudocatenulatum enhanced the growth of L. fujiensis, B. longum subsp*. longum, and R. torques* (Fig. 6 and 7). Interestingly, the potential pathogenic bacteria B. thetaiotaomicron showed a synergistic effect on the growth of four probiotic bacteria, including B. longum subsp*. longum, B. adolescentis, B. angulatum,* and C. aerofaciens (Fig. 6).
Representative pictures of antagonistic and synergistic interactions among gut anaerobes. The figure demonstrates antagonistic and synergistic interactions between isolated anaerobes. (A) Antagonism is illustrated by the presence of a zone of inhibition, which indicates the suppression of growth in the surrounding areas. (B) Synergism is evidenced by enhanced growth around the wells.
Interaction network showing species growth enhancement by helper species among anaerobic isolates. Size varies with the number of interactions, i.e., larger size demonstrates more interactions, while smaller size shows one or two interactions. The largest size source node shows interactions with eight other species, and the smallest size source node attributes a single interaction. Double upward arrows near the target node present the species whose growth was enhanced, while the emerging edge link from the source node denotes the corresponding helper species.
Interaction network showing species growth inhibition by antagonistic species among anaerobic isolates. The size of the circle represents the number of interactions: the largest size shows the maximum, and the smallest size illustrates the minimum number of interactions among the species. The inhibition symbol at the end of the link indicates the species whose growth was inhibited, while the emerging link without the inhibition symbol denotes the antagonistic species responsible for inhibition.
Core anaerobic isolates facilitated the growth of potential pathogenic taxa through cross-feeding interactions
Interestingly, several core anaerobes traditionally regarded as probiotic or potentially probiotic, such as Bifidobacterium strains*, M. multacida, R. inulinivorans,* and C. aerofaciens, were found to significantly enhance the growth of pathogenic, potentially pathogenic, and zoonotic species (Fig. 6 and Table 4). For example, all of the mentioned strains supported the growth of L. garvieae. Additionally, various B. adolescentis, B. pseudocatenulatum, and C. aerofaciens enhanced the growth of the potential zoonotic pathogen B. thetaiotaomicron (Fig. 6 and Table 4). Moreover, M. multacida also enhanced the growth of the pathogenic bacteria S. anginosus subsp*. anginosus*. This observation highlights an important yet underappreciated duality, where beneficial strains, through metabolic cross-feeding or environmental modulation, may inadvertently promote the growth of undesirable or harmful microbes. Such findings challenge the conventional view of probiotics as unilaterally beneficial and underscore the necessity of evaluating microbial interactions at the strain level within defined community contexts. These results carry critical implications for the rational design of microbiome-targeted therapies and raise caution against generalized assumptions in probiotic applications.
Synergistic interactions revealed interesting interdependencies when the growth media playing field was changed
A notable bidirectional growth-promoting interaction was observed between B. thetaiotaomicron and B. adolescentis. When the playing field was altered—by pre-spreading one strain across the agar surface and inoculating the other into a central well—each strain enhanced the growth of the other in both configurations. B. thetaiotaomicron facilitated the growth of B. adolescentis when used as the background strain, and reciprocally, B. adolescentis promoted B. thetaiotaomicron under reversed conditions (Fig. 6). This mutualistic interaction suggests a potential cross-feeding or environmental modulation mechanism that is robust across positional contexts, underscoring their ecological interdependence within the gut microbiota.
Antagonistic interactions revealed pathogen-pathogen, probiotic-pathogen, and pathogen-probiotic antagonism potential, along with other interdependencies
Isolates showing antagonism against ≥3 strains were termed as major antagonists, and this group was dominated by Lactococcus species. Of these, L. garvieae, L. formosensis, and L. fujiensis showed the highest number of antagonistic interactions by inhibiting the growth of seven, six, and five strains, respectively (Fig. 7 and Table 4). Some of the potential pathogenic isolates inhibited the growth of other potential pathogens, such as potentially pathogenic L. garvieae, which inhibited the growth of B. caccae and P. johnsonii. L. garvieae also inhibited the growth of various potentially probiotic genera, including Bifidobacterium, Lactococcus, and Bacteroides. Other isolated strains showing inhibition of potential pathogens include L. formosensis, L. fujiensis, B. longum subsp*. longum, B. pseudocatenulatum, R. torques,* and R. inulinivorans. Some probiotic potential isolates antagonized the growth of other potentially probiotic strains (Fig. 7 and Table 4). Strains against which high antagonism was observed include B. caccae, B. coprocola, R. torques, P. johnsonii, and C. aerofaciens.
Genomic insights into B. angulatum focusing on microbe-microbe interactions
B. angulatum not only acted as a helper strain, promoting the growth of L. garvieae and R. torques, but it also exhibited growth enhancement when co-cultured with several other strains, including B. thetaiotaomicron, B. longum subsp*. longum*, and Bifidobacterium pseudocatenulatum. Interestingly, while its growth was antagonized by L. garvieae, the latter’s growth was, in turn, enhanced by B. angulatum, suggesting a dynamic shift in the microbial interactions at play. These intriguing reciprocal relationships prompted us to have a look into the genomic profile of B. angulatum through WGS. The main aim of the WGS was to look for potential genetic elements that might be responsible for the observed co- and cross-interactions, as well as the resistome. The WGS analysis revealed that the genome size of B. angulatum was 2.09 Mbp having guanine to cytosine ratio (G + C) of 59.64 (Fig. 8).
Various regions of the genome of Bifidobacterium angulatum, such as confirmed coding region (CDS) annotation, tRNA, rRNA, possible coding region (ORFs) prediction, AT contents, and GC contents. The outermost circle represents the CDS, and inner circles show AT and GC contents.
Genome-wide analysis revealed that this organism carries genes encoding various enzymes that are important for the biosynthesis of various metabolites and may potentially be involved in the observed interactions. Some of the representative examples include the presence of genes encoding acetate kinase and pyruvate formate-lyase, which are involved in the synthesis of acetate and formate and are known to influence microbial behavior. Additionally, the presence of genes encoding adenylosuccinate lyase and argininosuccinate lyase, which are indirectly involved in the production of succinate, is further reported to be involved in complex microbial interactions, such as aiding in the growth of dormant bacteria. Moreover, genes encoding another enzyme, fumarylacetoacetate hydrolase, were present in the genome. This enzyme has a direct role in acetoacetate production, which is considered a key metabolite playing a role in microbial symbiosis as well as competition between members of the bacterial community.
Other genes, the products of which can potentially be involved in the observed interactions, include the biosynthesis of glutamate, transcription repressor ArgR, phosphoserine aminotransferase, and phosphoserine phosphatase-producing genes, which seem to have important roles as per previous reports in cooperative interactions among microbes.
The metabolic model, constructed using the RAST-annotated genome, highlighted 529 genes and 783 compounds, with 20 metabolites identified as potentially involved in inter-strain interactions. Notably, 65% of these metabolites were associated with cooperative interactions, while 20% exhibited competitive behavior. A remarkable 15% of the metabolites were implicated in both cooperative and competitive roles, mirroring the reciprocal nature of B. angulatum’s interactions observed in the phenotypic assays (Fig. 9). This genomic analysis not only provides insights into the metabolic underpinnings of these interactions but also strengthens our understanding of B. angulatum’s versatile role within the microbial ecosystem.
Various metabolites potentially involved in microbe-microbe interactions. The metabolites, as per their functions, are classified into three categories: cooperation, competition, and cooperation/competition.
DISCUSSION
The primary objective of this study was to develop a targeted culturomics strategy for isolating core anaerobic bacteria from the healthy human gut and investigate their resistome and interspecies interaction potential. To this end, we successfully optimized a CASGM, formulated to mimic the gut’s unique microenvironment, including its low redox potential, oxygen limitation, and specialized nutrient requirements. Using CASGM, we successfully cultured 21 unique anaerobic bacterial species from the gut, and 11 of these were the core anaerobes. Thus, our CASGM approach successfully targeted the culturing of more than 50% of the core anaerobic species, making it a valuable approach for overcoming the limitations of previous anaerobe-specific culturing approaches (10).
Previously used traditional culturomics approaches, which usually relied on generic anaerobic media types or single batch culture systems, often resulted in limited success as well as did not capture major fractions of the core anaerobes (60). Our CASGM-based approach, on the other hand, utilized a modified media capable of providing stringent anaerobic conditions, which enabled the growth and targeted culturing of a major fraction of the anaerobic organisms. In addition, this strategy resulted in the culturing of previously uncultured taxa from the human gut, thus expanding our knowledge and bioresource of the gut microbiome. To our knowledge, this is the first study that targeted and successfully cultured a major fraction of the core anaerobes by utilizing a single modified growth medium. Thus, our CASGM approach addresses the limitations of the utilization of labor-intensive and costly multi-media approaches and offers a sustainable alternative for targeted culturing of the core anaerobes.
Our comprehensive AMR profiling of the targeted core anaerobes revealed that the majority (55.95%) were susceptible to the tested panel of eight widely used, clinically relevant antibiotics. These include oxytetracycline, meropenem, ampicillin, chloramphenicol, ceftriaxone, levofloxacin, azithromycin, and tobramycin. This is a very valuable finding, suggesting that the majority of the cultured anaerobic strains, both including core and non-core, are vulnerable to the standard antibiotic prescriptions. However, all core anaerobes also revealed resistance to at least two of the tested antibiotics, suggesting that these organisms harbor intrinsic and/or acquired mechanisms that allow them to withstand these antibiotics upon exposure (61).
An interesting observation was that all core isolates were resistant to tobramycin, an aminoglycoside antibiotic. This resistance likely reflects a known pharmacodynamic limitation, as aminoglycosides require oxygen-dependent transport systems for cellular uptake—rendering them ineffective under strict anaerobic conditions such as those present in anaerobic jars (62–66). This AMR snapshot is instrumental in guiding informed antibiotic prescriptions and also reinforces the need for careful selection of antimicrobials in clinical settings to preserve beneficial core taxa and avoid unnecessary microbiome disruption.
Comprehensive cross-interaction profiling among the cultured gut anaerobes revealed an intricate network of ecological behaviors, including synergistic, antagonistic, and bifunctional interactions. The majority of the isolates exhibited measurable effects on growth, highlighting the prevalence of inter-strain influence within the gut ecosystem. Interestingly, 33.33% anaerobic isolates revealed a bifunctional role as they promoted the growth of some taxa but inhibited the growth of others. This further suggests that the cross-interaction behavior of a microbe is not fixed but is context dependent, such as the nutrient availability and the nature of cross-feeding (67, 68). Moreover, various strains showed the role as key facilitators or helper species, each promoting the growth of three or more other strains and termed as major facilitators. These major facilitators include B. pseudocatenulatum, R. inulinivorans, M. multacida, and B. adolescentis. One of the strains, B. pseudocatenulatum, showed interesting behavior that supported the growth of beneficial strains, including B. longum and L. fujiensis, while it inhibited the growth of the potentially gut pathogenic strain P. johnsonii (3). The potentially pathogenic L. garvieae showed antagonistic activities, thereby inhibiting the growth of both various potentially beneficial and harmful strains, suggesting its competitive niche occupant nature (32).
Antagonism (competition) and synergism (cooperation) among the gut microbes in vivo within the human gut microbiome have been predicted using modeling, metagenomics, and ecological studies, although not directly observed (8, 69, 70). One of the notable cross-interactions observed was the mutually synergistic relationship between B. thetaiotaomicron and B. adolescentis. This positive interaction held steady even when the experimental setup was flipped—regardless of which strain was plated first or added to the central well—pointing to a stable mutualism, possibly driven by cross-feeding or shared environmental shaping. Such bidirectional interactions echo previous findings where B. thetaiotaomicron modulates gut community structure by liberating polysaccharides and other metabolites utilized by neighboring microbes (71). Interestingly, although B. thetaiotaomicron is sometimes associated with disease states, it enhanced the growth of several probiotic taxa, including B. longum, B. angulatum, B. adolescentis, and Collinsella aerofaciens, highlighting the limitations of taxonomic classification in inferring functional roles (72).
In a self-contradictory way, various potentially beneficial anaerobes, including B. pseudocatenulatum, M. multacida, and R. inulinivorans, supported the growth of potentially pathogenic bacteria, including L. garvieae and S. anginosus. This observation further cautions against the traditional binary labeling of strains as probiotic or pathogenic and further suggests that the ecological behavior of a strain in a community strongly depends on the community context (25). Furthermore, L. formosensis exhibited both cooperative and antagonistic behaviors—promoting B. longum subsp. longum while inhibiting six other strains, including B. coprocola, R. torques, and B. adolescentis—demonstrating its dualistic ecological nature.
In summary, our results suggest that a variety of complex interactions exist among the core gut bacteria and within the community, which, in most instances, are position sensitive, and these interactions are governed more by metabolic function and environmental compatibility rather than taxonomy. These findings further support a shift in microbiome-related research from assessment based solely on community structure to microbial interaction-driven frameworks, which can better explain overall community resilience, dysbiosis, and community assembly (67, 72, 73).
Our WGS analysis of the B. angulatum strain gives useful insights regarding the potentially responsible genetic element for the observed phenotype. Since this strain displayed a dual role, where, on one hand, it promoted the growth of L. garvieae and R. torques, on the other hand, some other strains, such as B. thetaiotaomicron and B. longum, enhanced its growth. Furthermore, WGS analysis revealed a variety of genes and pathways that could potentially be involved in this observed dual behavior. For instance, the presence of genes encoding enzymes that are involved in the synthesis of SCFA (acetate via acetate kinase and formate via pyruvate formate-lyase). Other genes involved in the production of carboxylic acids, such as succinate, oxaloacetate, and several amino acids such as glutamate, serine, arginine, cysteine, and glycine. The above-mentioned metabolic products are well known to be involved in microbial cross-feeding and facilitate and/or affect cooperative behaviors among gut microbial members (14, 74).
Some of the observed synergistic interactions may potentially arise from the secretion of readily available carbon and nitrogen sources by helper bacteria that may be utilized, upon which the neighboring bacteria can thrive. Furthermore, WGS analysis also revealed the presence of genetic elements that might be responsible for the observed competitive behavior. For instance, the presence of genes involved in sulfur metabolism and NAD biosynthesis gives a hint about possible mechanisms through which this strain can monopolize nutrient resources under constrained environments, potentially leading to the inhibition of certain strains. Additionally, the presence of regulatory elements such as ArgR transcription factor (linked to arginine metabolism), along with additional important enzymes such as phosphoserine aminotransferase and phosphoserine phosphatase, indicates the capacity and advantage of this strain for fine-tuned responses based on environmental cues.
Together, these findings suggest that the ecological role of a strain in a community could potentially be predicted from a microbe’s genome (67, 72). B. angulatum in our study acts as an interesting example of how genomic elasticity of an organism gives it a multifaceted potential to thrive and survive in the gut ecosystem. This further reinforces the value of the integration of genome-derived data coupled with experimental phenotyping to unleash the functional characteristics of the individual microbes within communities.
Conclusion
This study presents a unique CASGM strategy for the targeted isolation and characterization of key interaction dynamics as well as the resistome of core anaerobic bacteria of the healthy human gut. By utilizing this targeted single, optimized CASGM approach, we were able to culture a major proportion of the core anaerobic gut species, where some of those were cultured for the first time. Through the combinatorial approach of cross-interaction, AMR profiling, and WGS-resolved functional insights, we uncovered interesting insights into interdependencies and competition among these core strains as well as their ecological flexibilities. Moreover, we observed that some strains, such as B. thetaiotaomicron and B. angulatum, are capable of either inhibiting or promoting the growth of certain strains depending on the pairing. Such findings go against the traditional binary view of microbes as being purely commensal or pathogenic and suggest that microbial behavior is highly context-dependent. Furthermore, WGS analysis provided useful insights into the potential genomic basis of the observed phenotype. Taken together, our findings provide valuable data and enhance our current understanding of how core anaerobes interact with each other, survive, and adapt within the gut environment. Moreover, our work also lays the critical basis for a more rational design of synthetic microbial communities as well as precision-based microbiome aimed at targeted gut health rehabilitation, both with accuracy and sensitivity.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sender R, Fuchs S, Milo R. 2016. Revised estimates for the number of human and bacteria cells in the body. P Lo S Biol 14:e 1002533. doi:10.1371/journal.pbio.100253327541692 PMC 4991899 · doi ↗ · pubmed ↗
- 2Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. 2005. Diversity of the human intestinal microbial flora. Science 308:1635–1638. doi:10.1126/science.111059115831718 PMC 1395357 · doi ↗ · pubmed ↗
- 3Lloyd-Price J, Abu-Ali G, Huttenhower C. 2016. The healthy human microbiome. Genome Med 8:51. doi:10.1186/s 13073-016-0307-y 27122046 PMC 4848870 · doi ↗ · pubmed ↗
- 4Maioli TU, Borras-Nogues E, Torres L, Barbosa SC, Martins VD, Langella P, Azevedo VA, Chatel J-M. 2021. Possible benefits of Faecalibacterium prausnitzii for obesity-associated gut disorders. Front Pharmacol 12:740636. doi:10.3389/fphar.2021.74063634925006 PMC 8677946 · doi ↗ · pubmed ↗
- 5Tamanai-Shacoori Z, Smida I, Bousarghin L, Loreal O, Meuric V, Fong SB, Bonnaure-Mallet M, Jolivet-Gougeon A. 2017. Roseburia spp.: a marker of health? Future Microbiol 12:157–170. doi:10.2217/fmb-2016-013028139139 · doi ↗ · pubmed ↗
- 6Hou K, Wu Z-X, Chen X-Y, Wang J-Q, Zhang D, Xiao C, Zhu D, Koya JB, Wei L, Li J, Chen Z-S. 2022. Microbiota in health and diseases. Signal Transduct Target Ther 7:135. doi:10.1038/s 41392-022-00974-435461318 PMC 9034083 · doi ↗ · pubmed ↗
- 7Hu C, Shen H. 2024. Microbes in health and disease: human gut microbiota. Appl Sci (Basel) 14:11354. doi:10.3390/app 142311354 · doi ↗
- 8Armour CR, Nayfach S, Pollard KS, Sharpton TJ. 2019. A metagenomic meta-analysis reveals functional signatures of health and disease in the human gut microbiome. m Systems 4:10. doi:10.1128/m Systems.00332-18 · doi ↗