Harnessing artificial intelligence for genomic variant prediction: advances, challenges, and future directions
Indah Pakpahan, Mentari Sihombing, Haohan Liu, Mengyao Wang, Zheng Su, Mingyan Fang

TL;DR
This paper reviews how artificial intelligence is improving the prediction of genetic variants, highlighting progress, challenges, and future strategies for better accuracy in disease research and personalized medicine.
Contribution
The paper introduces strategies for improving variant prediction through explainable AI and multi-omics integration, emphasizing inclusivity and interpretability.
Findings
Traditional systems are being replaced by machine learning and deep learning in variant prediction.
Explainable AI and inclusive genomic databases are needed to address variant uncertainty and data heterogeneity.
Optimized workflows and multi-omics integration can enhance clinical and research variant interpretation.
Abstract
Accurate genetic variant interpretation is crucial for disease research and the development of targeted therapies. Artificial intelligence is transforming this field by integrating computational methodologies across structural biology, evolutionary analysis, and multimodal genomic data. This review examines the evolution from traditional rule-based systems and statistical models to contemporary machine learning, deep learning, and protein language models, while addressing critical challenges in variant classification. Key obstacles include data heterogeneity, interpretability, and the persistence of variants of uncertain significance, emphasizing the critical need for explainable artificial intelligence frameworks and more inclusive genomic databases to improve predictive accuracy across diverse populations. Based on the assessment of current variant impact predictors, we propose…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
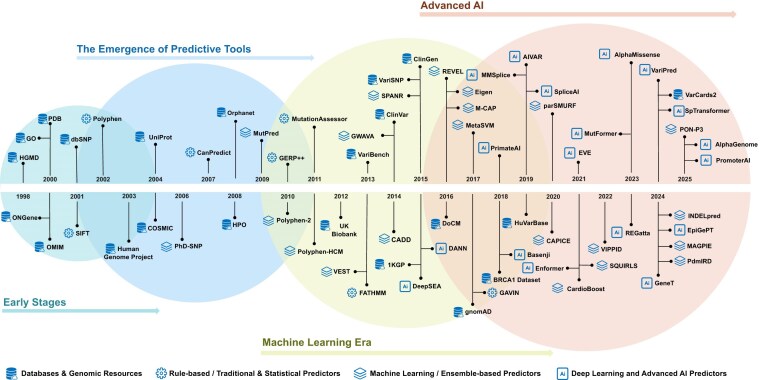 Figure 1
Figure 1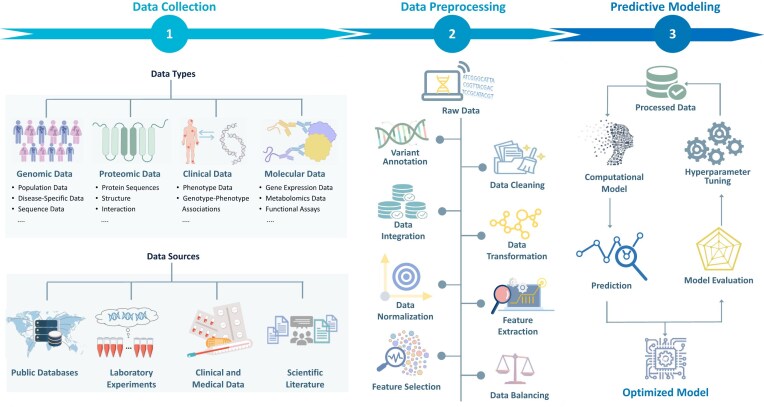 Figure 2
Figure 2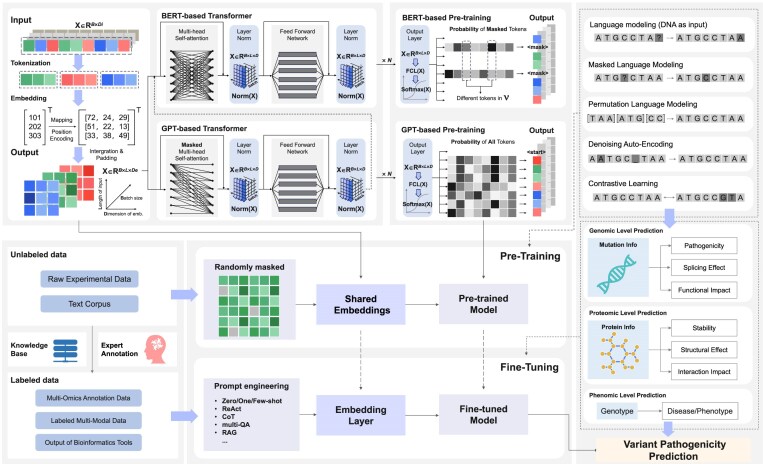 Figure 3
Figure 3| No. | Name | Description | Data types | Cross-link | Entries (as of September 2025) | Last updates | Website | Reference |
|---|---|---|---|---|---|---|---|---|
| 1 | 1KGP | Large-scale project to create a comprehensive resource on human genetic variation | Population genomic data | Uses refID from dbSNP; variants included in gnomAD | >88 million variants (84.7 million SNPs, 3.6 million indels, 60,000 SVs) | 2024–11-18 |
| [ |
| 2 |
| Focused dataset on SNVs in the | Gene-specific genomic data | Variants cross-validated with ClinVar and HGMD | 3,893 SNVs | 2018–08-20 |
| [ |
| 3 | ClinGen | Clinical genomics resource defining the clinical relevance of genes and variants | Clinical genomics resource | Provides gene-disease evidence to ClinVar; uses HPO and OMIM | 3,256 genes, 11,062 variants | 2025–09-18 |
| [ |
| 4 | ClinVar | Public archive of reports of the relationships among human variations and phenotypes. | Clinical genomic data | Integrates rsID from dbSNP; links to ClinGen, OMIM, HPO | 5,640,148 records | 2025–08-24 |
| [ |
| 5 | COSMIC | Catalogue of Somatic Mutations in Cancer | Somatic genomic data | Overlaps with DoCM and ClinVar | 25,014,261 variants | 2025–05-21 |
| [ |
| 6 | dbSNP | Database of Single Nucleotide Polymorphisms and other variants | Genomic variant registry | Referenced by ClinVar and gnomAD via rsID | 1,206,053,617 unique rs | 2025–01-15 |
| [ |
| 7 | DoCM | Manually curated database of clinically relevant mutations. | Clinically curated genomic data | Overlaps with COSMIC and ClinVar | 3,818 variants | 2024–10-15 |
| [ |
| 8 | FAVOR | Aggregated variant and indel functional annotations from multiple databases. | Functional annotation data | Indexed by genomic coordinates and rsID; integrates multi-source functional annotations | 8,892,915,237 variants (8,812,917,339 SNVs and 79,997,898 indels) | 2025–02-05 |
| [ |
| 9 | gnomAD | Aggregated and harmonized human exome and genome sequencing data | Population genomic data | Uses rsID from dbSNP, referenced in ClinVar | 730,947 exomes and 76,215 whole genomes | 2024–04-19 |
| [ |
| 10 | GO | Structured vocabulary for gene product functions and processes | Ontology | Used in UniProt | 39,906 terms, 9.41 million annotations, 1.60 million gene products, 5,497 species | 2025–07-22 |
| [ |
| 11 | HGMD | Comprehensive collection of germline mutations in human genes | Clinically curated genomic data | Often cross-validated with ClinVar and OMIM | 549,178 mutations | 2025–07-07 |
| [ |
| 12 | HPO | Ontology for describing human phenotypic abnormalities | Phenotype ontology | Used by OMIM and ClinGen; mapped to GO | 18,000 terms and >156,000 annotations | 2024–04-19 |
| [ |
| 13 | HuVarBase | Annotated human variation database | Curated genomic variant data | Merged from COSMIC, ClinVar, 1000 Genomes | 774,863 variants from 18,318 proteins (702,048 disease causing and 72,815 neutral variants) | 2018–06-01 |
| [ |
| 14 | OMIM | Comprehensive catalog of human genes and genetic phenotypes | Gene–phenotype data | Includes HPO annotations; links to ClinVar and UniProt | 27,938 entries (26,394 autosomal, 1,407 X linked, 64 Y linked, 73 mitochondrial) | 2025–09-17 |
| [ |
| 15 | ONGene | Curated database of human oncogenes | Cancer gene annotation data | – | 803 oncogenes (698 protein-coding genes + 105 non-coding) | 2016–12-26 |
| [ |
| 16 | Orphanet | Database for rare diseases and orphan drugs | Rare disease ontology | Shares HPO and OMIM terms | 9,785 clinical entities, 6,528 rare disorders, 8,296 disease gene relationships, 115,611 phenotypic annotations, 16,418 epidemiological data, 689 orphan drugs 8,648, expert centers | 2025–06-24 |
| [ |
| 17 | PDB | Repository for 3D structural data of biological macromolecules | Protein structural data | Linked from UniProt entries | 242,296 structures, 1,068,577 computed structure models | 2025–09-17 |
| [ |
| 18 | UK Biobank | Large-scale biomedical database with genetic and health information | Population-scale genotype–phenotype data | Overlaps with dbSNP | 90 million variants | 2024–08-13 |
| [ |
| 19 | UniProt/UniProtKB | Comprehensive protein sequence and annotation resource | Protein sequence and functional annotation data | Cross-links to PDB, GO, dbSNP, ClinGen, Orphanet, | 253,206,171 entries (UniProtKB/Swiss-Prot: 572,970 entries and UniProtKB/TrEMBL: 252,633,201 entries) | 2025–06-17 |
| [ |
| 20 | VarCards2 | Updated version of VarCards, human variant annotation and interpretation | Variant annotation and interpretation data | Some extracted from gnomAD, ClinVar, COSMIC, dbSNP | 368,820,266 indels, 2,773,555 CNVs | 2023–10-21 |
| [ |
| 21 | VariBench | Benchmark database for variation datasets in bioinformatics | Benchmark variant datasets | Some extracted from dbSNP, OMIM | >90 million variants | 2023–05-12 |
| [ |
| 22 | VariSNP | A benchmark database suite comprising variation datasets that can be used for developing and testing the performance of variant effect prediction | Benchmark variant data | Selected from dbSNP | 30,571,777 variants | 2017–02-16 |
| [ |
| No. | Algorithm | Description | VIPs | References |
|---|---|---|---|---|
| 1 | Naive Bayes | A probabilistic graphical model that represents a set of variables and their conditional dependencies | PolyPhen-2, PolyPhen-HCM, CanPredict, SPANR | [ |
| 2 | Support vector machines | A supervised learning model that analyzes data for classification and regression analysis | CADD, PhD-SNP, MetaSVM | [ |
| 3 | Random forest | An ensemble learning method that operates by constructing multiple decision trees | VEST, MutPred, SQUIRLS, GWAVA, VIPPID, PdmIRD, REVEL, parSMURF | [ |
| 4 | Gradient boosting machines | An ML technique for regression and classification problems that builds a model in a stage-wise fashion. | MAGPIE, CAPICE, INDELpred, M-CAP, PON-P3, CardioBoost | [ |
| 5 | Neural networks | A set of algorithms modeled after the human brain, designed to recognize patterns | DANN, PrimateAI, DeepSEA, Basenji, PromoterAI, SpliceAI, MMSplice, AIVAR | [ |
| 6 | Transformer | A DL model that uses self-attention mechanisms to process sequential data, capturing long-range dependencies and relationships in the data. | AlphaMissense, VariPred, Enformer, AlphaGenome, EpiGePT, MutFormer, GeneT, SpTransformer | [ |
| No. | Methodological paradigm | VIPs | Variant type(s) | Reported performance (as published) | References |
|---|---|---|---|---|---|
| 1 | Rule-based | SIFT | Missense (amino acid-altering) variants | ROC-AUC: 0.80–0.82 | [ |
| PolyPhen | Missense (amino acid-altering) variants | ROC-AUC: ≈0.83 | [ | ||
| 2 | Statistical | MutationAssessor | Missense (amino acid-altering) variants | ROC-AUC: ≈0.86 | [ |
| FATHMM | Coding and non-coding variants | Accuracy: ≈0.86 | [ | ||
| 3 | Machine learning | CADD | Coding and non-coding variants | ROC-AUC: 0.90–0.93 | [ |
| REVEL | Missense (amino acid-altering) variants | ROC-AUC: 0.90–0.91 | [ | ||
| 4 | Deep learning | SpliceAI | Splicing-altering variants | PR-AUC: ≈0.90 | [ |
| DeepSEA | Non-coding regulatory variants | ROC-AUC: ≈0.96 | [ | ||
| 5 | Transformer/pLM | AlphaMissense | Missense (amino acid-altering) variants | ROC-AUC: ≈0.94 | [ |
| MutFormer | Missense (amino acid-altering) variants | ROC-AUC: 0.92–0.97 | [ |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Rare Diseases · Genetic Associations and Epidemiology · Bioinformatics and Genomic Networks
Background
High-throughput sequencing and the Human Genome Project have enabled comprehensive catalogs of human genetic variation, yet distinguishing pathogenic from benign variants remains a central bottleneck in research and clinical genetics [1–3]. Over the last decade, variant impact predictors (VIPs) have progressed from rule-based heuristics to statistical models, machine learning (ML), deep learning (DL), and, most recently, transformer-based large language models that integrate evolutionary, structural, and multi-omics signals [4, 5].
Despite rapid methodological advances, the field faces a paradox: while computational methods have improved significantly, their clinical utility remains limited. ML and DL approaches have demonstrably improved pathogenicity prediction for coding variants, and sequence-based models have advanced interpretation of splicing and regulatory variants. However, several critical challenges persist. First, a substantial proportion of variants remain classified as variants of uncertain significance (VUS), reducing their diagnostic impact. Second, reference datasets such as Genome Aggregation Database (gnomAD) [6] and ClinVar [7] exhibit ancestry imbalances that affect generalizability across populations. Third, DL and transformer-based models often operate as “black boxes,” complicating their alignment with American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) interpretive guidelines. Finally, experimental validation lags behind computational predictions, creating a gap between algorithmic output and clinical translation.
This review addresses these challenges through a comprehensive, method-focused synthesis. We map the technological evolution of VIPs across computational paradigms, characterize the supporting database ecosystem, and outline practical workflows for data preprocessing, model development, and evaluation. Rather than proposing new algorithms, we assess current artificial intelligence (AI) capabilities: identifying where approaches are robust, where caution is warranted, and what translational gaps must be addressed for clinical implementation. We provide actionable recommendations for model selection, ancestry-aware algorithm design, multi-omics integration, and validation frameworks to improve the reliability, transparency, and equity of variant interpretation in diverse populations.
Methodologically, this review adopts a narrative, method-focused search of PubMed, Google Scholar, and Scopus combining terms such as variant pathogenicity, in silico prediction, deep learning, transformer/foundation model, splicing prediction, non-coding variants, functional assays, and database/tool names. This primary search was supplemented by manual cross-referencing of seminal reviews and benchmarking repositories to ensure a comprehensive coverage of diverse computational frameworks. We prioritized peer-reviewed method papers with clear training/validation descriptions, widely used tools across coding and non-coding tasks, comparative evaluations, and resources on functional screening, fairness, and explainable AI (XAI).
Curated database infrastructure supporting variant interpretation
The accurate interpretation of genetic variants relies on a stratified data ecosystem integrating genomic, clinical, and functional evidence, in which each layer offers a distinct analytical perspective while remaining interconnected to enable comprehensive pathogenicity assessment.
Population-scale genomic initiatives provide baseline frequency profiling, establishing context for variant rarity assessment. Resources including the Single Nucleotide Polymorphism Database (dbSNP) [8], the 1000 Genomes Project (1KGP) [9], gnomAD [6], and UK Biobank [10] provide allele frequency distributions and mutational constraint profiles across diverse ancestries, enabling distinction between rare pathogenic variants and common benign polymorphisms, especially in underrepresented populations [11]. Functional Annotation of Variants—Online Resource (FAVOR) [12] complements these resources by integrating functional annotations to assess variants lacking clinical evidence.
Pathogenicity evidence emerges from curated clinical and disease-specific repositories. ClinVar [7], Human Gene Mutation Database (HGMD) [13], Human Variants Database (HuVarBase) [14], and ClinGen [15] aggregate experimentally or clinically validated genotype–phenotype associations, forming an empirical foundation for supervised learning approaches in variant classification.
Standardized disease ontologies help transform clinical observations into computational frameworks. Online Mendelian Inheritance in Man (OMIM) [16], Orphanet [17], and Human Phenotype Ontology (HPO) [18] connect genetic alterations to specific disease mechanisms and biological pathways. Additionally, Gene Ontology (GO) [19] complements phenotype-focused resources by clarifying functional impacts at molecular and cellular levels.
Domain-specific repositories provide further refinement of variant interpretation in specialized contexts. Oncology–focused databases, such as Catalogue of Somatic Mutations in Cancer (COSMIC) [20], Database of Curated Mutations (DoCM) [21], and ONGene [22], aggregate tumor-specific mutations. Gene-centric functional assays (e.g., a curated BRCA1 functional dataset [23]) provide confidence labels for many clinically relevant genes.
Beyond sequence and phenotype resources, protein architecture databases including Universal Protein Resource (UniProt) [24] and Protein Data Bank (PDB) [25] provide the three-dimensional framework essential for understanding variant consequences at the molecular level, particularly useful for structure–function relationship modeling.
Finally, specialized benchmarking and validation resources, including the Benchmark Database for Variations (VariBench) [26], VariSNP [27], and VarCards2 [28], support model evaluation and comparison of emerging AI predictors by providing standardized test sets and performance metrics.
Across this data ecosystem (Table 1) spanning evolutionary, biochemical, structural, and regulatory domains lies the foundation for advanced AI architectures that can effectively model the complex relationships underlying variant pathogenicity [29]. Access to these resources is governed by institutional guidelines and data use agreements ensuring appropriate use of de-identified data.
The technological trajectory of variant pathogenicity assessment
The development of VIPs has evolved through five distinct yet overlapping paradigms, each addressing limitations of previous approaches while expanding analytical capabilities (Fig. 1). This progression mirrors broader technological trends within computational biology, and has evolved from initial rule-based heuristics toward advanced transformer architectures. Each step has facilitated progressively refined insights into genomic variation (Supplementary Table 1).
Evolution of in silico tools for predicting genetic variant pathogenicity. This figure illustrates the temporal progression of computational approaches for genetic variant pathogenicity prediction. The evolutionary trajectory is categorized into four phases: (1) early stages, characterized by rudimentary rule-based algorithms; (2) the emergence of predictive tools, marking the transition to more sophisticated statistical frameworks; (3) machine learning era, defined by the integration of supervised and unsupervised learning methodologies; and (4) advanced AI, representing contemporary approaches that leverage deep learning architectures and multi-modal data integration.
Phase 1: Rule-based biological heuristics
Early predictive tools emerged from foundational biological insights. Pioneering rule-based predictors such as Sorting Intolerant From Tolerant (SIFT) [30] and Polymorphism Phenotyping (PolyPhen) [31] relied on empirical knowledge and evolutionary principles to evaluate variant pathogenicity. These tools primarily relied on sequence conservation and amino acid physicochemical properties to distinguish between benign and deleterious variants. Gene-Aware Variant INterpretation (GAVIN) [32] applied predefined classification logic to refine pathogenicity predictions within gene-specific contexts. Their strength lay in their high interpretability, as the underlying biological rules were explicit and transparent.
However, this rule-based approach inherently limited their scalability and ability to capture the complex, nonlinear patterns emerging from rapidly expanding genomic datasets. Though computationally efficient, these methods typically operated within limited genomic contexts and largely overlooked non-coding or regulatory regions such as promoters or splice sites [33]. Despite these constraints, early-phase predictors remain valuable for preliminary assessments and continue to be incorporated into comprehensive prediction frameworks [34], particularly where interpretability is prioritized over prediction complexity.
Phase 2: Statistical modeling and probabilistic frameworks
As genomic databases expanded in both size and diversity, statistical methods emerged to enhance the prediction accuracy through probabilistic modeling. Tools such as MutationAssessor [35] utilized evolutionary conservation patterns within protein families, while Functional Analysis Through Hidden Markov Models (FATHMM) [36] integrated evolutionary conservation scores into sequence-based probabilistic models to estimate variant pathogenicity. Eigen [37], an unsupervised spectral method, prioritized variants by analyzing annotation correlations and constructing a weighted score across both coding and non-coding genomic regions. In parallel, Genomic Evolutionary Rate Profiling++ (GERP++) [38] quantifies evolutionary constraint using a maximum likelihood model to calculate rejected substitutions, and is widely used as an annotation feature in downstream predictive frameworks.
While these tools have advanced the ability to contextualize genomic variation, predictive methods still face substantial challenges when analyzing rare or novel variants due to insufficient representation in existing reference datasets. Additionally, their heavy reliance on high-quality reference annotations limited their effectiveness in classifying clinically important yet poorly characterized VUS [39], prompting further developments toward data-driven ML methods.
Phase 3: Traditional machine learning and ensemble approaches
ML algorithms transformed variant prediction by capturing complex, nonlinear patterns in multidimensional genomic datasets, enabling integration of diverse biological features [40]. Approaches in this era can be broadly categorized into:
Classical machine learning classifiers: Naive Bayes classifiers were applied in tools like PolyPhen-2 [41], which integrates sequence and structure-based features to predict the effects of amino acid substitutions. This algorithm was also effectively deployed in disease-specific contexts, such as Polymorphism Phenotyping for Hypertrophic Cardiomyopathy (PolyPhen-HCM) [42], CanPredict [43], and Splicing-based Analysis of Variants (SPANR) [44]. Support vector machines (SVMs) and random forests (RFs) underpin tools such as Combined Annotation Dependent Depletion (CADD) [33], Predictor of human Deleterious Single Nucleotide Polymorphisms (PhD-SNP) [45], Variant Effect Scoring Tool (VEST) [46], Meta-analytic Support Vector Machine (MetaSVM) [47], MutPred [48], incorporating diverse features such as evolutionary conservation, protein structure, and gene-level annotations within unified predictive frameworks. SQUIRLS [49] focuses on splice-altering variants, while genome-wide annotation of variants (GWAVA) [50] extends this approach to non-coding regions, prioritizing functional regulatory variants across diverse biological contexts.
The RF algorithm was similarly adapted for specialized tasks, powering tools like the Variant Impact Predictor for Primary Immunodeficiency Diseases (VIPPID) [51] for immunodeficiencies and Prediction of Deleterious Missense Mutation for Inherited Retinal Diseases (PdmIRD) [52] for retinal diseases. For instance, SVMs identify optimal hyperplanes to separate classes, while RFs build multiple decision trees and aggregate their results, offering robustness and handling high-dimensional data effectively.
Gradient boosting approaches: More recent implementations employ gradient boosting machines (GBMs) to enhance classification performance. Multimodal Annotation Generated Pathogenic Impact Evaluator (MAGPIE) [5], Consequence-Agnostic Pathogenicity Interpretation of Clinical Exome variations (CAPICE) [53], INDELpred [54], Mendelian Clinically Applicable Pathogenicity (M-CAP) [55], and PON-P3 [56] exemplify this approach by integrating multi-source annotations via gradient-boosted decision trees. The strength of GBMs for leveraging complex feature sets also made them ideal for building specialized predictors like CardioBoost [57] for cardiac genetics. GBMs operate on the principle of iterative improvement: they sequentially build models, with each new model attempting to correct the errors of its predecessor.
While these models demonstrate superior accuracy, they require high-quality, well-curated training datasets; when data are limited, sparse, or imbalanced, challenges such as overfitting and limited generalizability can compromise their clinical utility [58].
Ensemble prediction systems: To address the inherent limitations of individual algorithms, ensemble methodologies aggregate outputs from multiple predictors, thereby enhancing robustness and reliability [59]. Tools such as the Rare Exome Variant Ensemble Learner (REVEL) [60] and Parallel SMote Undersampled Random Forest (parSMURF) [61] exemplify this strategy by combining complementary models to mitigate individual weaknesses while amplifying collective strengths. Although ensemble methods introduce additional computational costs and diminish transparency, these frameworks have become integral to clinical genetics pipelines where reliability is paramount. Despite improved accuracy, ML-based methods remain sensitive to training data and often lack interpretability, limiting their use as standalone clinical tools.
Phase 4: Deep learning approaches
DL has further advanced variant interpretation by exploiting neural networks to model nonlinear relationships in large, multidimensional genomic datasets [62]. DL models can learn hierarchical feature representations directly from raw data, reducing the reliance on manual feature engineering. Deleterious annotation of genetic variants using neural networks (DANN) [63], employs a deep neural network trained on the same functional annotations as CADD to score both coding and non-coding variants. PrimateAI [64] leverages evolutionary signatures across primates to enhance prediction accuracy. This capacity for learning from raw sequence data has improved the prediction of non-coding and regulatory variants. Models like DeepSEA [65] and Basenji [66] demonstrated that chromatin accessibility and gene expression could be predicted directly from DNA sequence, capturing both proximal and distal effects. PromoterAI [67] focused on predicting the impact of promoter variants, further refining DL-based modeling of regulatory sequences.
In splicing prediction, DL brought a significant leap in accuracy. SpliceAI [68] utilized convolutional neural networks to capture long-range dependencies in pre-mRNA sequences, often outperforming previous models, though at the cost of interpretability. MMSplice [69] attempted to address this interpretability gap through more modular or explainable designs. While these approaches demonstrate substantial sensitivity, challenges persist regarding their “black box” nature and intensive computational requirements [70].
Phase 5: Transformer architectures
The most recent technological leap has been catalyzed by adapting transformer architectures and language modeling principles to genomic and protein sequences. Originally developed for natural language processing, transformer models have become powerful tools for predicting functional impacts of genetic variants [71]. The core innovation lies in the self-attention mechanism, which allows the model to weigh the importance of different parts of the input sequence when processing each element [72]. This enables the capture of long-range dependencies and global context within biological sequences, a key aspect for understanding how mutations in one part of a protein can affect distant functional regions [73]. The application of transformers has evolved into two powerful, complementary paradigms:
Protein language models (pLMs): Pre-trained on vast corpora of evolutionary sequences, pLMs learn fundamental principles of protein structure and function. Models like ESM-1b [74] excel at capturing subtle sequence constraints, distinguishing isoform-specific pathogenic variants with high accuracy (area under the receiver operating characteristic curve [ROC-AUC]: 0.905 on ClinVar, 0.897 on HGMD/gnomAD). This approach is exemplified by tools like Variant impact Predictor (VariPred) [75], which leverages these learned representations to predict variant effects, often outperforming traditional structure-dependent methods. The strength of pLMs is further demonstrated by their versatility in tasks such as masked residue prediction (82% accuracy) [76] and sequence conservation analysis (Matthews Correlation Coefficient (MCC) = 0.596) [77]. Regulatory genome transformers: These models are designed to interpret the non-coding genome by learning the regulatory code directly from DNA sequence. Enformer [78] set a new standard by using a transformer architecture to achieve leading performance in predicting chromatin accessibility and gene expression profiles from sequence context, capturing effects of distal enhancers. This paradigm is extended by tools like AlphaGenome [79], which integrates >100 regulatory features in a multitask framework. EpiGePT [80] focuses on precise prediction of context-specific epigenomic signals, achieving high performance (Pearson r = 0.710; auROC = 0.949).
The potential of this era is revealed in models that fuse these approaches or leverage their insights for specific clinical tasks. AlphaMissense [71] by DeepMind represents a seminal work, fusing structural insights from AlphaFold [81] with the pattern recognition of language models to generate a massive, highly accurate map of missense variant pathogenicity. Similarly, MutFormer [82] integrates self-attention with convolutional layers for missense analysis, and Genetic Transformer (GeneT) [83] achieves high recall rates (99% in synthetic data, 97.85% in clinical cohorts) for identifying causative variants. This fusion also enables more specialized applications, such as SpTransformer [84], which incorporates tissue specificity into splicing prediction.
Together, these findings suggest that transformer architectures can achieve improved performance across diverse variant interpretation tasks, including coding, non-coding, and splicing variants. In benchmark comparisons, these models often outperform earlier DL approaches, though performance gains vary considerably with task definition, training data, and evaluation frameworks. These advances reflect a transition from feature-engineered methods to models that learn context-aware representations directly from biological sequences, potentially improving predictive capacity while introducing challenges in scalability and interpretability. Representative algorithms and VIPs across these technological phases are summarized in Table 2, with detailed tool characteristics in Supplementary Table 1.
To provide illustrative quantitative context, Table 3 presents performance metrics for representative VIPs spanning major methodological phases, as reported in original publications. These values derive from heterogeneous evaluation settings and are not directly comparable.
Integrative tools for variant annotation
Comprehensive annotation pipelines consolidate outputs from diverse databases and predictive algorithms to streamline variant interpretation. Ensembl Variant Effect Predictor (VEP) [86] annotates variants with gene-level and regulatory features, supporting flexible plug-in integration of tools such as SIFT, CADD, and SpliceAI. ANNOVAR [87] enables gene-based, region-based, and filter-based annotation in unified workflows, incorporating population frequency data (e.g., gnomAD) and multiple pathogenicity scores.
Web-based platforms further enhance usability and evidence integration. VarSome [88] and MobiDetails [89] aggregate clinical annotations, in silico predictions, and allele frequencies via interactive interfaces apply ACMG-based classification framework. InterVar [90] emphasizes rule-based implementation of ACMG guidelines, offering reproducible, guideline-concordant classification support. Exomiser [91] combines variant pathogenicity scores with phenotype data (HPO terms) to prioritize candidate variants, especially in rare disease diagnosis.
These integrative tools reduce the burden of manual curation by consolidating diverse resources into unified workflows, enabling efficient filtering and evidence synthesis for both research and clinical applications. However, while these pipelines effectively consolidate evidence from multiple sources, the complexity of variant interpretation requires a systematic approach to optimize their implementation. The selection of appropriate predictors and the integration of their outputs into coherent clinical decisions necessitates a structured workflow framework.
Variant interpretation workflow and evaluation considerations
To address these challenges systematically, the variant interpretation process can be conceptualized as a structured pipeline that links raw genomic data to actionable clinical predictions (Fig. 2). This systematic approach provides a comprehensive framework for improved variant classification accuracy and supports the development of more equitable genomic medicine applications.
Integrated pipeline for variant pathogenicity prediction. This figure delineates a systematic framework for developing variant pathogenicity prediction models. The pipeline harnesses multi-modal data inputs (genomic, proteomic, molecular, and clinical) acquired from public repositories, experimental assays, clinical documentation, and literature curation. Critical preprocessing steps include variant annotation, normalization, cross-platform integration, and feature engineering to enhance signal integrity. The analytical workflow culminates in a machine learning implementation with rigorous hyperparameter optimization and comprehensive performance assessment to ensure robust predictive capacity across diverse genetic contexts.
The framework encompasses three interconnected phases that collectively transform raw genomic data into clinically actionable insights. The initial phase centers on comprehensive data acquisition, integrating population-level variant frequencies, disease-specific variants repositories, high-throughput molecular characterization profiles, and curated clinical annotations. This foundation transitions into an advanced preprocessing phase by systematically integrating heterogeneous data sources, including evolutionary conservation, protein structural, and regulatory element characterizations. Subsequent steps include normalization, feature extraction, and dimensionality reduction to optimize the computational feature space for downstream analytical applications [92].
The analytical phase leverages sophisticated predictive modeling approaches, employing rigorous hyperparameter optimization strategies to achieve optimal discriminative performance [93]. This computational framework generates probabilistic assessments that require systematic validation through robust benchmarking protocols. Evaluation encompasses multiple complementary metrics: sensitivity and specificity for detection of pathogenic versus benign variants, precision metrics that characterize predictive accuracy, and ROC-AUC, which provides threshold-independent assessment of discriminative performance [94]. Comparative analyses against benchmark datasets, such as ClinVar or consortium-established validation cohorts, enable cross-method evaluations while ensuring generalizability through cross-validation [95].
The optimization of these computational models requires adherence to stringent development protocols that emphasize data integrity through integration of high-quality variant annotations from diverse repositories such as gnomAD and ClinVar. Advanced feature engineering requires integration of multi-omics data layers spanning genetic, proteomic, regulatory, and clinical domains [96]. Robust model development paradigms demand thorough cross-validation, systematic hyperparameter tuning, and validation against independent external datasets to ensure reproducibility and clinical applicability [97].
Challenges limiting clinical translation
Despite these methodological advances across both general and specialized AI approaches, several interconnected challenges persist in limiting the clinical deployment of VIPs. These challenges span data limitations, dataset biases, polygenic and complex traits, and model interpretability, each of which would benefit from targeted solutions to bridge the gap between computational prediction and clinical application.
Data limitations and functional validation bottlenecks
VUS continue to represent a large fraction of clinical findings, particularly for rare and novel or non-coding variants [98]. AI-based models address this challenge by leveraging evolutionary conservation, regulatory annotations, and protein structural features to infer potential pathogenicity and prioritize variants lacking database evidence or functional validation, although significant VUS challenges remain. While computational methods such as matrix factorization [99] and active learning [100] provide incremental gains, they often remain dependent on sparse or biased training data.
Recent years have seen growing reliance on high-throughput functional assays such as CRISPR-Cas9 screens and multiplexed assays of variant effects (MAVEs), which can directly measure variant function at scale [101]. These approaches are beginning to be incorporated into clinical frameworks, e.g., MAVE-derived scores have been incorporated into ClinGen rules for BRCA1, TP53, and PTEN [102]. Yet, widespread clinical use remains limited by assay cost, turnaround time, and availability outside specialized centers.
Dataset biases and generalizability issues
Beyond general data limitations, demographic imbalances in genomic databases primarily skewed toward individuals of European ancestry constrain model performance across diverse populations [103]. Such sampling biases may reduce the reliability of variant interpretation in underrepresented groups [104]. Training datasets for AI models are typically de-identified and accessed through controlled mechanisms governed by data use agreements and ethical guidelines. While targeted sequencing and federated learning are emerging, coverage remains incomplete for many populations, limiting equitable variant interpretation [105].
Modeling polygenic and complex traits
Current single-variant VIPs like AlphaMissense are optimized for high-penetrance Mendelian variants, which differs substantially from diverges from the genetic architecture of complex, polygenic diseases. While these models excel at identifying variants disrupting protein stability or evolutionary conservation, they struggle to capture the cumulative contribution of numerous small-effect variants underlying conditions such as hypertension, hyperlipidemia, and type 2 diabetes [106].
Furthermore, current predictors operate largely in isolation, neglecting the “omnigenic” reality in which disease susceptibility is influenced by epistatic interactions and regulatory networks rather than single-locus disruptions. Consequently, binary classification of variants as “pathogenic” or “benign” lacks the granularity required for complex trait prediction, where risk is continuous and context-dependent [107].
Model interpretability versus predictive power
Even when data issues are mitigated, the interpretability of advanced AI models remains an important barrier to clinical adoption. ML models like evolutionary model of variant effect (EVE) [108] have demonstrated high performance in variant prediction but are often too complex to explain and their opacity poses challenges for clinical trust, regulatory approval, and usability [109, 110]. Recent efforts in XAI strategies [111] and interpretable classifiers like Artificial Intelligent Variant Classifier (AIVAR) [85] seek to bridge this gap by providing transparent and biologically meaningful rationales for prediction. XAI frameworks help distinguish robust predictions from algorithmic artifacts, but AI outputs are best interpreted as probabilistic prioritization requiring validation with clinical and experimental evidence.
Clinical role in AI-based variant interpretation
AI-based VIPs are designed to support clinical decision-making within established diagnostic workflows rather than operate as standalone classifiers. Empirical evaluations show VIPs function most effectively when augmenting expert review, with clinicians retaining final classification decisions, particularly for VUS [112, 113].
In practice, clinicians integrate AI predictions with phenotypic information, family history, segregation validation, and functional evidence to guide variant classification. This workflow underscores that successful clinical adoption is strongly influenced by how AI outputs are presented: interpretable scores, confidence estimates, and evidence aligned with ACMG/AMP guidelines are essential. Ultimately, clinical utility depends as much on usability, explainability, and seamless workflow integration as on algorithmic performance.
Toward more interpretable and clinically actionable predictions
Emerging trends ranging from multi–omics fusion to foundation models have the potential to advance variant interpretation along five strategic axes. These axes collectively span molecular, computational, functional, and population-level dimensions of clinical genomics.
Phenotype-conditioned variant interpretation
Phenotype-conditioned approaches link genetic alterations directly to clinical manifestations, addressing allelic heterogeneity. Multi-task architectures such as the Variant-to-Phenotype framework [114] integrate protein interactomes, structural descriptors, and evolutionary constraints to predict pathogenicity conditioned on HPO categories. By capturing genotype–phenotype associations, these approaches accelerate rare disease diagnosis and identify therapeutic targets, enhancing variant scoring into a clinically useful tool that can inform patient-specific pathology.
Multi-omics evidence integration
Integrating genomic, transcriptomic, proteomic, and epigenetic data [115] improves statistical power for associations with low-frequency variants [116]. Effective integration typically involves selectively, prioritizing omics layers most informative for specific variant types [117, 118]. Transcriptomic and splicing data are critical for interpreting non-coding and splice-site variants, particularly for resolving VUS [119, 120], whereas proteomic and structural information is more informative for missense variants [121]. Tissue-specific expression needs to be considered, as variant effects can be context-dependent.
AI-based approaches integrate multi-omics through feature-level aggregation, model-level fusion, or post hoc synthesis according to ACMG/AMP guidelines. Despite their potential, these approaches remain constrained by data heterogeneity, tissue specificity, and incomplete population coverage. Selective prioritization of high-value omics layers and shared databases offer a promising strategy for scalable clinical adoption.
Foundation models and few–shot adaptation
Transformer-based language models enable fine-tuning with minimal labeled data, addressing challenges posed by rare variants. Pre-trained on large, unlabeled datasets, these models can be efficiently adapted to novel variant types using parameter-efficient fine-tuning [122] and prompt engineering [123], potentially making high-performance variant prediction accessible for understudied conditions. Recent benchmarking of DNA foundation models demonstrates their capacity for zero-shot variant effect prediction, with large, multi-species architectures demonstrating strong discriminative power in capturing both local and extended contextual effects [124].
These transformer-based approaches can be conceptually unified through a shared pipeline that transforms biological sequences into clinical predictions (Fig. 3). Biological sequences undergo tokenization and embedding generation, converting raw genomic data into computational representations. Contextual encoding via multi-head self-attention processes these embeddings, where unsupervised pre-training objectives, including masked language modeling, permutation language modeling, and contrastive learning [125], enable the models to capture meaningful sequence patterns without labeled data. Task-specific adaptation occurs through specialized prediction heads (task-specific output layers) coupled with either frozen or fine-tuned backbones. Prompt-engineering strategies enable efficient knowledge transfer when limited labeled datasets are limited, and predictions can be further interpreted or calibrated using functional validation and systems-level analyses to support clinical interpretation.
Transformer-based framework for variant pathogenicity prediction. This figure illustrates a transformer-based pipeline for predicting genetic variant pathogenicity. Input sequences (X∈RB×L×D, where X represents the input tensor, R denotes real numbers, B = batch size, L = sequence length, and D = embedding dimension) are processed through three key stages: (1) tokenization and embedding, (2) contextual encoding via multi-head self-attention, and (3) classification. The model undergoes pre-training on multi-omics datasets followed by pathogenicity-specific fine-tuning. Prompt engineering facilitates performance with limited labeled data. Other key components include LayerNorm to mitigate vanishing or exploding gradients, fully connected layers for effective feature extraction, and Softmax activation for generating robust probability distributions. Together, the overall architecture and training strategies enable the model to leverage latent biological mechanisms for accurate variant classification.
Scalable functional validation and systems-level context
Scaling functional validation beyond specialized centers is increasingly feasible through collaborative resources. Resources such as the Atlas of Variant Effects [126] and MaveDB [127] provide standardized repositories of multiplexed assay data directly incorporated into ACMG/AMP classification frameworks. At the same time, integration of functional readouts with systems biology analyses, including pathway and interaction networks, may help contextualize variant effects at multiple biological levels [128]. Functional evidence is being more systematically integrated into precision medicine workflows, rather than an ad hoc supplement, and pairing these insights with diverse population data supports robust cumulative-risk interpretation while accounting for population-specific variation.
Equity-aware modeling across variant and polygenic risk
Systematically diversifying genomic databases addresses biases from population underrepresentation [103]. Collaborative initiatives like the Human Heredity and Health in Africa consortium [129] provide models for ethical, scientifically robust data collection among underserved populations. Ethical AI practices, such as fairness assessments, transparent model documentation, and inclusive stakeholder engagement, help mitigate algorithmic biases.
Algorithmic strategies for polygenic risk score transferability
Equity challenges become particularly pronounced when extending variant-level prediction frameworks to polygenic risk modeling. While generating diverse population-scale data is essential for addressing these challenges, several computational strategies address ancestry bias using existing resources. Transfer learning adjusts models for population-specific linkage disequilibrium and variants shift using existing summary statistics [130, 131], while domain adaptation constructs ancestry-invariant representations through adversarial learning [132, 133]. Data augmentation via generative models supplements scarce data with synthetic genomes [134, 135].
For polygenic risk prediction, ensemble methods integrate multiple polygenic risk score construction strategies: supervised approaches like CT-SLEB [136] and multi-ancestry polygenic risk scores based on ensemble of penalized regression models [137] combine clumping-thresholding, empirical Bayes, and penalized regression using target population genome-wide association study data, while unsupervised frameworks such as Unsupervised Ensembles [138] aggregate pre-trained models based on prediction concordance without requiring phenotype data from target populations, which enables robust performance even in underrepresented populations by circumventing sparse phenotype availability.
Transitioning to polygenic modeling
For complex diseases, single-variant predictions may have limited utility. Emerging frameworks are moving toward systems-level polygenic modeling that integrates cumulative genetic architecture. Ensemble methods like polygenic risk predictions integrating common and rare variants [139] combine polygenic background with rare variant burden, enhancing predictive accuracy across ancestries. Graph neural networks like PRS-Net [140] model genes and pathways as networks, capturing epistatic dependencies [141] and potentially enhancing both interpretability and clinical actionability. Benchmark studies have shown that combining rare and common variant modeling improves cross-ancestry predictive performance, providing stronger clinical evidence for polygenic risk application. Realizing these advances will require continued efforts in both algorithmic innovation and data diversification to promote equitable benefits across populations.
Implications and limitations
Recent advances in AI-based variant prediction have shown substantial progress. DL approaches, particularly transformer architectures and pLMs, have improved sequence context capture and biological signal integration across diverse variant interpretation tasks.
However, important limitations constrain clinical translation of these findings. Reported performance gains are context-dependent, derived from specific benchmarks and curated datasets, and do not establish universal superiority of any single method. Differences in data composition, labeling practices, and evaluation protocols limit direct cross-method comparisons. Despite improved predictive accuracy, many models require further validation or interpretability improvements for standalone clinical use, and challenges including ancestry bias and limited experimental validation persist.
These limitations underscore that AI-based VIPs function primarily as decision-support tools that complement expert judgment within established clinical workflows rather than as definitive classifiers. Continued progress requires standardized benchmarking across diverse populations, transparent reporting of model scope and limitations, development of interpretable architectures, and systematic experimental validation of computational predictions.
Conclusions
In summary, the integration of diverse datasets, transparent predictive algorithms, and consistent validation practices will be critical for advancing AI-driven variant interpretation into routine clinical use. The rapid advancement of computational tools has significantly enhanced our ability to predict genetic variant pathogenicity, offering scalability and accuracy. However, several challenges remain, particularly around accurately classifying VUS, and ensuring fairness and transparency of predictive models. Continued improvement will require effectively integrating multi-omics information, expanding international cooperation to diversify genomic datasets, and systematically linking computational predictions to robust experimental validations. With these concerted efforts, next-generation computational tools can help realize the promise of personalized medicine, guiding clinicians and researchers toward deeper mechanistic insights and improved patient care.
Supplementary Material
giag004_Supplemental_File
giag004_Authors_Response_To_Reviewer_Comments_original_submission
giag004_GIGA-D-25-00463_original_submission
giag004_GIGA-D-25-00463_Revision_1
giag004_Reviewer_1_Report_original_submissionAmmar Husami -- 11/12/2025
giag004_Reviewer_1_Report_Revision_1Ammar Husami -- 1/5/2026
giag004_Reviewer_2_Report_original_submissionWeiyang Li -- 12/8/2025
giag004_Reviewer_2_Report_Revision_1Weiyang Li -- 1/6/2026
giag004_Reviewer_3_Report_original_submissionJinming Han -- 12/17/2025
giag004_Reviewer_3_Report_Revision_1Jinming Han -- 1/5/2026
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Aworunse OS, Adeniji O, Oyesola OL, et al. Genomic interventions in medicine. Bioinf Biol Insights. 2018;12:1177932218816100. 10.1177/1177932218816100.PMC 628730730546257 · doi ↗ · pubmed ↗
- 2Rego SM, Snyder MP. High throughput sequencing and assessing disease risk. Cold Spring Harb Perspect Med. 2019;9:a 026849. 10.1101/cshperspect.a 026849.29959131 PMC 6314070 · doi ↗ · pubmed ↗
- 3Spielmann M, Kircher M. Computational and experimental methods for classifying variants of unknown clinical significance. Cold Spring Harb Mol Case Stud. 2022;8:a 006196. 10.1101/mcs.a 006196.35483875 PMC 9059783 · doi ↗ · pubmed ↗
- 4Boulaimen Y, Fossi G, Outemzabet L, et al. Integrating large language models for genetic variant classification. ar Xiv 2024; 10.48550/ar Xiv.2411.05055. · doi ↗
- 5Liu Y, Zhang T, You N et al. MAGPIE: accurate pathogenic prediction for multiple variant types using machine learning approach. Genome Med. 2024;16:3. 10.1186/s 13073-023-01274-4.38185709 PMC 10773112 · doi ↗ · pubmed ↗
- 6Karczewski KJ, Weisburd B, Thomas B et al. The Ex AC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017;45:D 840–45. 10.1093/nar/gkw 971.27899611 PMC 5210650 · doi ↗ · pubmed ↗
- 7Landrum MJ, Lee JM, Riley GR, et al. Clin Var: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D 980–85. 10.1093/nar/gkt 1113.24234437 PMC 3965032 · doi ↗ · pubmed ↗
- 8Sherry ST, Ward M-H, Kholodov M et al. db SNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11. 10.1093/nar/29.1.308.11125122 PMC 29783 · doi ↗ · pubmed ↗