Chemodivergent Synthesis of 1,4-Benzo[b]dithiins and 1,4-Benzodithiafulvenes
Douglas B. Paixão, Anita B. Kessler, Fabiano S. Rodembusch, Rafael Stieler, Daniel S. Rampon, Paulo H. Schneider

TL;DR
This paper describes a new method to make two types of sulfur-containing organic compounds using different solvents.
Contribution
A chemodivergent synthesis method that produces different sulfur-based compounds based on solvent choice.
Findings
Using DMF produces six-membered 1,4-dithiins.
Using DMSO leads to ring contraction forming 1,4-dithiafulvenes.
The solvent affects the S2–/S3• – equilibrium and base availability, controlling the product.
Abstract
We report a chemodivergent synthesis of 1,4-benzo[b]dithiins and 1,4-benzodithiafulvenes from 2-iodoaryl alkynyl sulfides using a NaSH·xH2O/KOH system. In DMF, the reaction affords six-membered 1,4-dithiins, whereas in DMSO, these intermediates undergo a base-promoted ring contraction to the corresponding 1,4-dithiafulvenes. Photophysical and mass spectrometry studies support the idea that this chemodivergence arises from the interplay between S2–/S3 • – equilibrium and base availability in each solvent, which governs whether the 1,4-dithiin framework is preserved or undergoes ring contraction to 1,4-benzodithiafulvenes.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
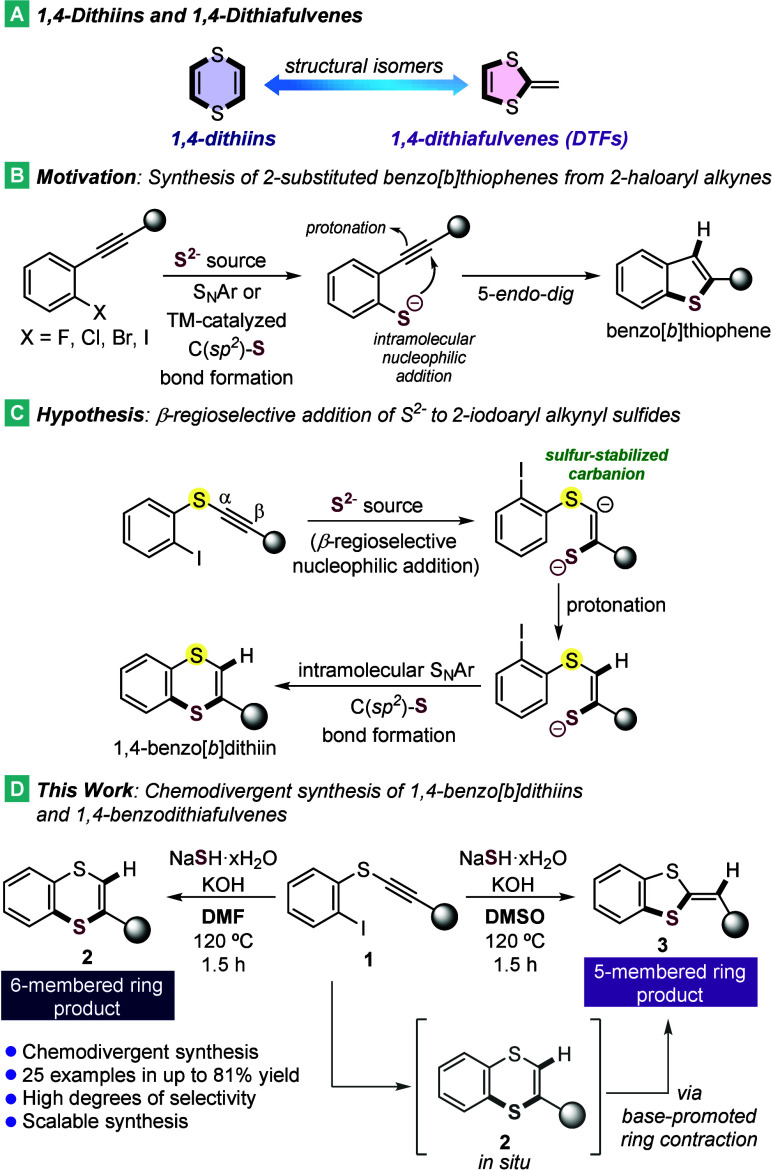 Figure 1
Figure 1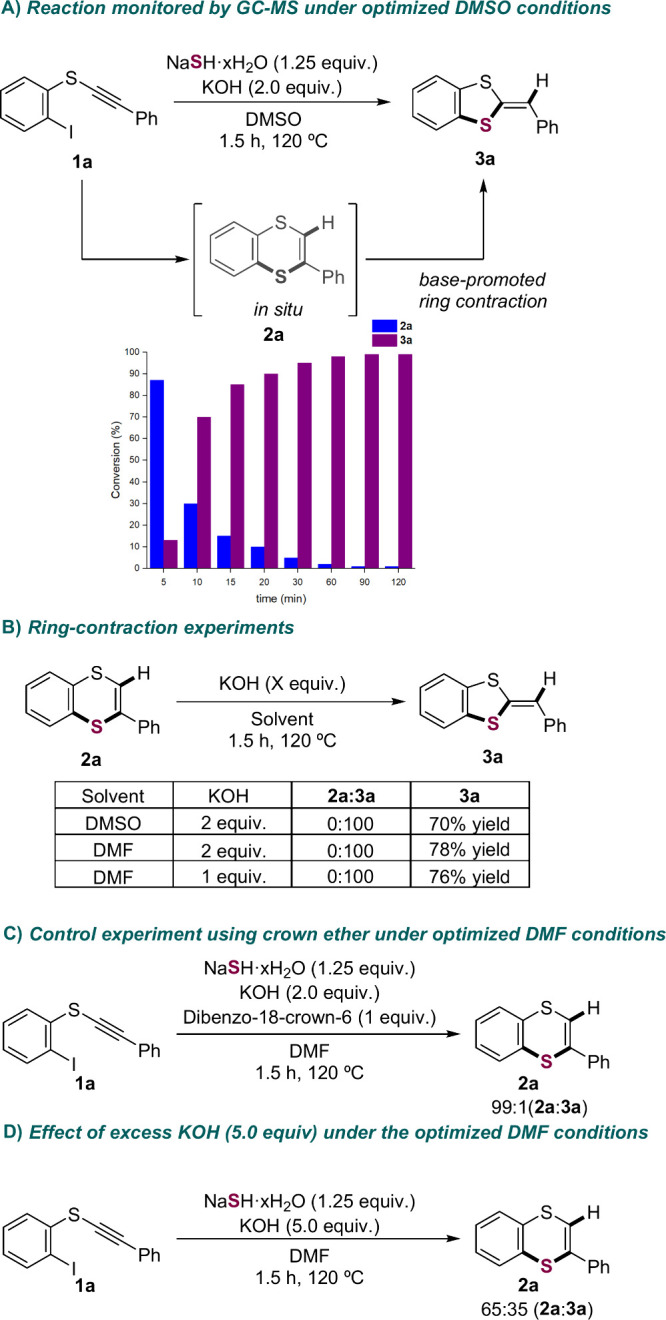 Figure 2
Figure 2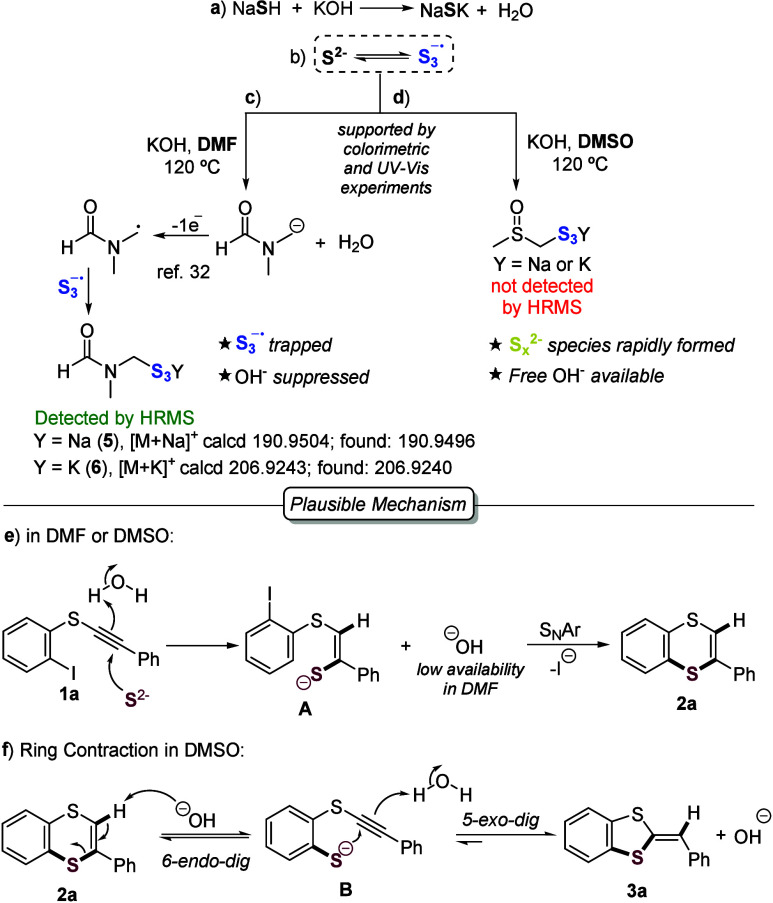 Figure 3
Figure 3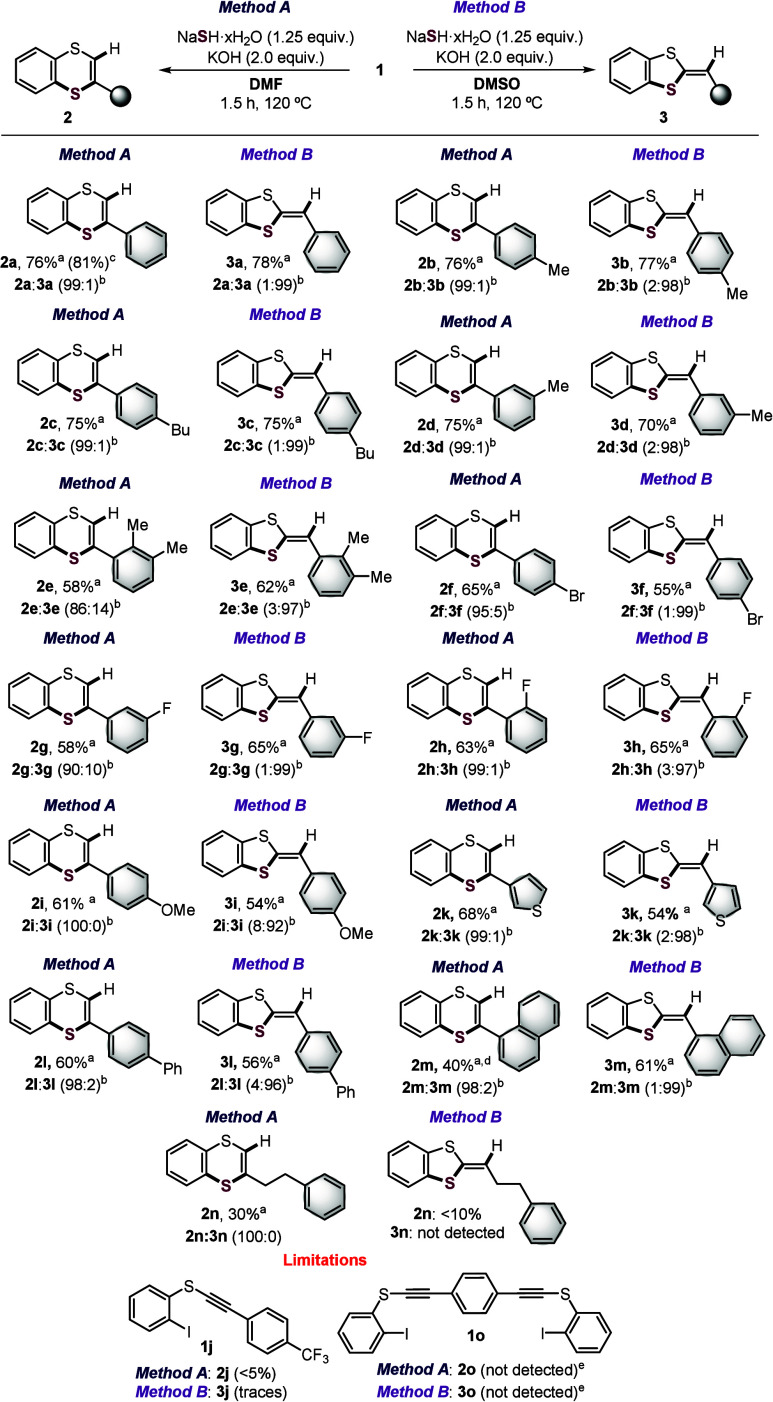 Figure 4
Figure 4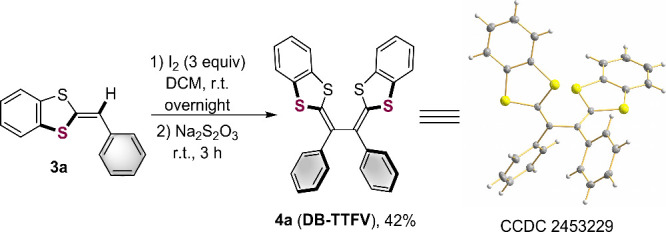 Figure 5
Figure 5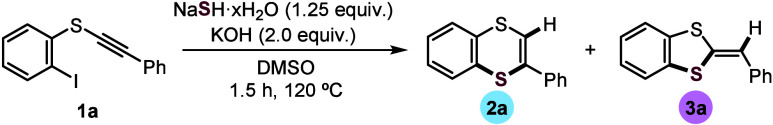 Figure 6
Figure 6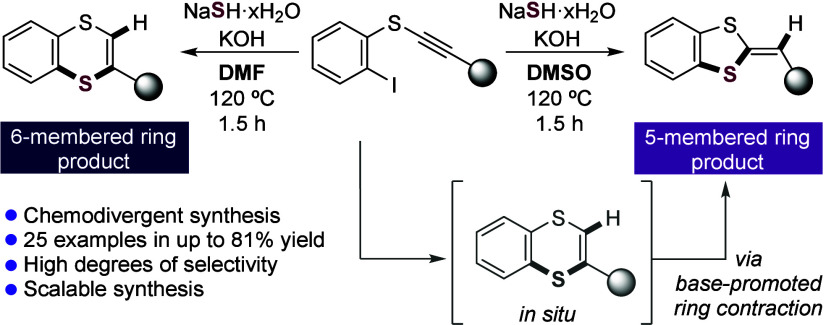 Figure 7
Figure 7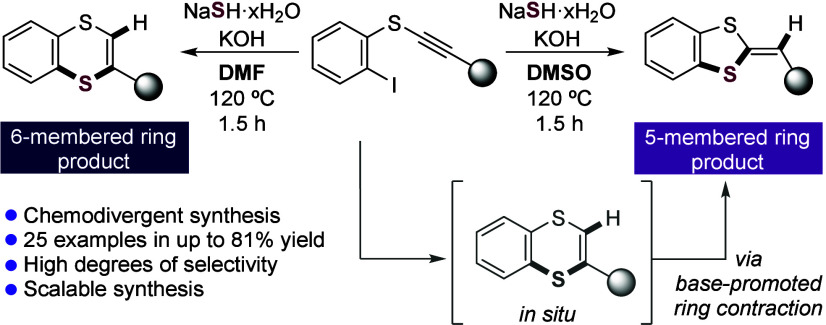 Figure 8
Figure 8- —Coordena??o de Aperfei?oamento de Pessoal de N?vel Superior10.13039/501100002322
- —Conselho Nacional de Desenvolvimento Cient?fico e Tecnol?gico10.13039/501100003593
- —Conselho Nacional de Desenvolvimento Cient?fico e Tecnol?gico10.13039/501100003593
- —Funda??o de Amparo ? Pesquisa do Estado do Rio Grande do Sul10.13039/501100004263
- —Universidade Federal do Paran?10.13039/501100008223
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSulfur-Based Synthesis Techniques · Organic and Molecular Conductors Research · Synthesis and Reactivity of Sulfur-Containing Compounds
1,4-Dithiins are six-membered, sulfur-rich heterocycles that have emerged as key building blocks in organic materials due to their reversible redox properties and structural flexibility.? Aromatic-fused 1,4-dithiins can form radical cations and undergo switchable conformational changes, making them valuable for applications in photocatalysis,? supramolecular and host–guest chemistry,? chiroptical systems,? and redox-active materials.? Similarly, 1,4-dithiafulvenes (DTFs), structural isomers of 1,4-dithiins composed of a five-membered 1,3-dithiole ring and an exocyclic CC bond (SchemeA), are recognized as “half units” of tetrathiafulvalenes (TTFs),? combining electron-donating and -withdrawing properties driven by zwitterionic resonance.? Their incorporation into π-extended systems or oxidative dimerization into tetrathiafulvalene vinylogues (TTFVs) has enabled conjugated organic materials,? switchable processes,? and chemosensors.?
The synthesis of benzo-fused analogues of these sulfur-rich heterocycles remains limited and challenging. Early examples of 1,4-benzo[b]dithiins involved the reaction of 1,8-diketones with Lawesson’s reagent, ring expansion of 1,3-dithiolanes, or the use of in situ generated benzodithiete with alkynes.? More recently, Miyake and co-workers employed ethynylbenziodoxolone (EBX) reagents and thiols to access three 1,4-benzo[b]dithiin derivatives,? while Xie’s group reported a Cu-catalyzed diarylthiolation of ynones.? In contrast, 1,4-benzodithiafulvenes are predominantly synthesized by carbonyl olefination,? with only a few reports involving dehalogenation reactions? or base-promoted ring contraction of 1,4-benzodithiins.?
This led us to hypothesize that the unique reactivity of alkynyl sulfides? could provide an efficient platform for synthesizing 1,4-benzo[b]dithiins. Inspired by the well-established synthesis of benzo[b]thiophenes from 2-haloaryl alkynes and S^2–^ sources (SchemeB),? we considered 2-iodoaryl alkynyl sulfides as suitable substrates for a mechanistically “inverse” pathway (SchemeC). The iodine substituent was designed to prevent undesired S_N_Ar-type initiation, commonly associated with cyclizations of 2-bromo-,? 2-chloro-,? or 2-fluoroaryl alkynes? in the synthesis of benzo[b]thiophenes under transition-metal-free conditions. Thus, we proposed to proceed via a chemo- and β-regioselective addition of S^2–^ species to the CC triple bond, generating a vinylic carbanion intermediate potentially stabilized by the adjacent sulfur atom.? Subsequent protonation, followed by an intramolecular S_N_Ar reaction, afforded the six-membered 1,4-dithiin ring.
During our studies on the synthesis of six-membered 1,4-benzo[b]dithiins (2) from 2-iodoaryl alkynyl sulfides (1) using a NaSH·xH_2_O/KOH system in DMSO, we consistently observed the formation of the five-membered product, 1,4-benzodithiafulvenes (3). This outcome prompted us to explore whether the formation of either 2 or 3 could be controlled by adjusting the reaction conditions. Notably, complete chemodivergence was achieved by changing the solvent from DMSO to DMF, motivating a detailed investigation of the origin of this selectivity (SchemeD).
Despite multiple reactive sites, 2-Iodoaryl alkynyl sulfides (1) have been explored almost exclusively as precursors to substrates used in cycloaddition reactions.? The reported syntheses of these compounds are usually limited in scope and require harsh conditions; therefore, we adopted an alternative strategy via oxidative C(sp)–S coupling of 2-aminophenyl disulfides with terminal alkynes, followed by diazotization/iodination (see section 2 of the Supporting Information for details).? Our optimization studies started with NaSH·xH_2_O (1.25 equiv),? KOH (2.0 equiv), and 1a (1.0 equiv) as model substrates (Table). The five-membered product 3a was obtained in 78% yield after 1.5 h at 120 °C in DMSO, with only negligible amounts of six-membered product 2a detected by GC–MS (Table, entry 1). Using NaOH or higher amounts of KOH (3.0 equiv) maintained high selectivity for 3a without yield improvement (Table, entries 2 and 3). Interestingly, lower amounts of KOH (0.5 equiv) shifted from 1:99 to 89:11 (2a/3a), and 2a was isolated in 43% yield (Table, entry 4). In the absence of a base, complete selectivity toward 2a was observed, which was obtained in 40% yield (Table, entry 5). These results suggest that the reaction initially forms the six-membered product 2a, which subsequently undergoes a base-promoted ring contraction to afford the five-membered product 3a.? They also demonstrate that the base plays a crucial role not only in promoting ring contraction but also in enhancing the overall reaction efficiency. To improve the yield of 2a while suppressing ring contraction, milder bases were evaluated (Table, entries 6–8). Among them, Cs_2_CO_3_ (1.0 equiv) provided an optimal balance of reactivity and selectivity, fully inhibiting ring contraction and affording 2a in 62% yield (Table, entry 8). Alternative sulfur sources, including K_2_S_ x _,? EtOCS_2_K,? and S,? or increased amounts of NaSH·xH_2_O, selectively afforded 3a but without yield improvement (Table, entries 9–12). Lowering the temperature to 80 °C reduced both efficiency and selectivity (48:52, 2a/3a), indicating a strong temperature dependence of the ring contraction (Table, entry 13). Remarkably, performing the reaction in DMF with the same base loading employed in DMSO almost completely suppressed the formation of 3a, affording 2a in 76% yield with 99:1 selectivity (2a/3a) (Table, entry 14).
To investigate the mechanism and solvent-dependent selectivity, we performed a series of controlled experiments (Scheme). The optimized reaction conditions for the synthesis of 3a (Table, entry 1) were monitored by GC–MS, confirming the initial formation of 2a and its gradual conversion into 3a, with a 1:99 (2a/3a) ratio observed after 1.5 h (SchemeA). The base-promoted ring contraction was validated by treating 2a with KOH in DMSO at 120 °C, furnishing 3a in 70% yield (SchemeB). Notably, this transformation also occurred in DMF, even at lower KOH loading (SchemeB), ruling out the possibility that the selective formation of 2a in DMF (Table, entry 14) arises from a superbase effect unique to DMSO.? This conclusion was further supported by an experiment using a crown ether to enhance basicity,? which still failed to promote the formation of 3a in DMF (99:1 2a/3a; SchemeC). However, using a large excess of KOH (5.0 equiv) significantly increased the formation of 3a (65:35 2a/3a), indicating that hydroxide availability is likely limited under the optimized DMF conditions, thereby disfavoring the ring contraction pathway.
Considering that HS^–^/S^2–^ species can disproportionate to S_3_ ^•^ ^–^ in both DMF and DMSO (SchemeA and B), ?,? we carried out colorimetric and UV–vis experiments to evaluate the influence of KOH on the S^2–^/S_3_ ^•^ ^–^ equilibrium in these solvents. The presence of sulfur species in solution was evidenced by characteristic color changes and spectroscopic analyses (see sections 5 and 6 of the Supporting Information for details). These studies revealed that S_3_ ^•^ ^–^ exhibits a significantly longer lifetime in DMF, consistent with its reported higher stability in this solvent.? Our results further indicate that S_3_ ^•^ ^–^ is gradually trapped by a DMF-derived radical generated via solvent deprotonation followed by single-electron oxidation? (SchemeC), as suggested by photophysical analyses and HRMS detection of adducts 5 and 6. This trapping process consumes hydroxide, thereby reducing its availability and suppressing the ring contraction of 2a in DMF. In contrast, in DMSO, the basic medium rapidly converts sulfur radicals into S_ x _ ^2–^ species? and no trapping of S_3_ ^•^ ^–^ was observed, indicating that free hydroxide remains available to promote the ring contraction of 2a (SchemeD).
Based on these results, along with control experiments employing TEMPO that ruled out a radical pathway (see section 8 of the Supporting Information for details), a plausible mechanism is proposed (SchemeE). In both solvents, the reaction is initiated with a β-regioselective addition of S^2–^ to the CC triple bond of 1a, followed by protonation to give intermediate A. This intermediate then undergoes an intramolecular S_N_Ar reaction to afford six-membered product 2a. In DMSO, hydroxide ions promote the ring opening of in situ generated 2a by abstracting vinylic hydrogen adjacent to sulfur,? forming intermediate B. This intermediate preferentially undergoes a 5-exo-dig cyclization rather than a 6-endo-dig process, driven by the greater thermodynamic stability of the 1,3-dithiole ring and the lower acidity of dithiafulvenes,? leading to the formation of the five-membered product 3a (SchemeF).
We next investigated the scope and limitations of the reactions (Scheme, methods A and B). The scalability was demonstrated using 1.0 mmol of 1a under method A, yielding 2a in 81% with 99:1 selectivity (2a/3a) (see the Supporting Information for details). Initial scope studies showed that both methods proceeded smoothly with substrates bearing alkyl groups, such as methyl and butyl, at various positions on the benzene ring, affording the corresponding six- and five-membered products in good yields and excellent selectivity (2b–2d and 3b–3d). A 2,3-dimethyl substitution pattern was also well-tolerated, indicating compatibility with steric hindrance (2e and 3e).
Methods A and B were similarly effective and selective for substrates bearing weak electron-withdrawing groups, such as bromo and fluoro substituents, at different positions (2f–2h and 3f–3h). Importantly, the bromo substituent in 2f and 3f offers a valuable handle for further functionalizations via cross-coupling reactions,? while the successful cyclizations using 2-fluorinated substrate 1h (2h and 3h) highlight the chemoselectivity of the addition of S^2–^ species to the CC triple bond, with no evidence of competing benzo[b]thiophene formation.? Additionally, the protocols also proved effective for 4-methoxy-substituted substrate 1i (2i and 3i), which contains a key functional group for developing push–pull dithiafulvene derivatives,? affording moderate to good yields with exclusive formation of 2i (100:0) and high selectivity for 3i (8:92 2i/3i).
The scope was successfully expanded to heteroaromatic and π-extended systems, such as thienyl, biphenyl, and naphtyl groups, which are important motifs in the design of conjugated materials (2k–2m and 3k–3m). Interestingly, an inseparable low-selectivity mixture (48:52 2m/3m) was obtained for 2m under optimized method A, whereas method B furnished the five-membered product 3m in 61% yield with excellent selectivity (1:99 2m/3m). Gratifyingly, 2m could be accessed with high selectivity at a lower reaction temperature (90 °C, 98:2 2m/3m), which suppressed ring opening but resulted in a modest yield.
Method A also enabled the synthesis of compound 2n bearing an alkyl substituent with complete selectivity but in only 30% yield, highlighting the importance of an adjacent π system to facilitate the nucleophilic attack to the CC bond. In contrast, method B produced less than 10% 2n and no detectable formation of 3n, suggesting that the ring-opening process is also disfavored in the absence of an aryl substituent. Finally, the p-CF_3_-substituted substrate 1j proved incompatible with both methods (2j, <5% yield; 3j, trace), and attempts to cyclize bifunctionalized alkynyl sulfide 1o resulted in complex mixtures of unidentified products under both conditions.
Considering the relevance of TTFV derivatives and the applications of their benzo-fused analogues in electroluminescent devices,? the synthetic utility of 3a was demonstrated through its oxidative dimerization? to afford dibenzo-fused tetrathiafulvalene vinylogue (DB-TTFV) 4a in 42% yield (Scheme). Structural analysis by X-ray diffraction confirmed that 4a adopts a pseudo-cisoid conformation, which reduces steric repulsion between the phenyl substituents (see the Supporting Information for details).?
We report a chemodivergent protocol for the selective synthesis of 1,4-benzo[b]dithiins and 1,4-benzodithiafulvenes from readily accessible 2-iodoaryl alkynyl sulfides. Six- or five-membered sulfur-rich heterocycles are obtained simply by changing the reaction medium from DMF to DMSO. Preliminary mechanistic studies suggest that differences in hydroxide availability, together with shifts in the S^2–^/S_3_ ^•^ ^–^ equilibrium under basic conditions, play a decisive role in enabling the observed chemodivergence between ring retention and ring contraction. The methodology demonstrates good functional group tolerance and operational simplicity, affording 25 examples in moderate to good yields with excellent selectivity. This study provides a useful platform for accessing structurally diverse 1,4-dithiins and 1,4-dithiafulvenes, offering valuable insights into sulfur-mediated cyclizations.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1a Etkind S. I.Swager T. M.The Properties, Synthesis, and Materials Applications of 1,4-Dithiins and Thianthrenes Synthesis 2022544843486310.1055/s-0042-1751368 · doi ↗
- 2a Meng H.Liu M.-S.Shu W.Organothianthrenium Salts: Synthesis and Utilization Chem. Sci.202213136901370710.1039/D 2SC 04507 A 36544727 PMC 9710214 · doi ↗ · pubmed ↗
- 3a Yuan J.Song Y.Li X.Xie J.Dong S.Zhu K.A Tubular Belt and a Möbius Strip with Dynamic Joints: Synthesis, Structure, and Host–Guest Chemistry Org. Lett.2021239554955810.1021/acs.orglett.1c 0378134870442 · doi ↗ · pubmed ↗
- 4Keck C.Rominger F.Mastalerz M.Synthesis of Chiral Pyrene-Based 1,4-Dithiins Angew. Chem., Int. Ed.202463 e 20231938910.1002/anie.20231938938179861 · doi ↗ · pubmed ↗
- 5a Speer M. E.Kolek M.Jassoy J. J.Heine J.Winter M.Bieker P. M.Esser B.Thianthrene-Functionalized Polynorbornenes as High-Voltage Materials for Organic Cathode-Based Dual-Ion Batteries Chem. Commun.201551152611526410.1039/C 5CC 04932 F 26235336 · doi ↗ · pubmed ↗
- 6a Segura J. L.Martín N.New Concepts in Tetrathiafulvalene Chemistry Angew. Chem., Int. Ed.2001401372140910.1002/1521-3773(20010417)40:8<1372::AID-ANIE 1372>3.0.CO;2-I 11317287 · doi ↗ · pubmed ↗
- 7Nielsen M. B.The 1,3-dithiol-2-ide carbanion Org. Biomol. Chem.2021195999600610.1039/D 1OB 00975 C 34190306 · doi ↗ · pubmed ↗
- 8a Bendikov M.Wudl F.Perepichka D. F.Tetrathiafulvalenes, Oligoacenenes, and Their Buckminsterfullerene Derivatives: The Brick and Mortar of Organic Electronics Chem. Rev.20041044891494610.1021/cr 030666 m 15535637 · doi ↗ · pubmed ↗