Enantiopure Pyridinium Bisretinoids of Ocular Lipofuscin with Hexahydrobenzofuran Structure: Total Synthesis and Structure-Dependent Aggregated Morphology
Brais Vidal, Rafael Rodríguez, Angeles Peña-Gallego, Rosana Álvarez, Claudio Martínez, Ángel R. de Lera

TL;DR
Scientists synthesized specific forms of a compound found in eye cells and studied how they form structures.
Contribution
A new enantiopure synthesis method for pyridinium bisretinoids with hexahydrobenzofuran structures is developed.
Findings
Enantiopure pyridinium bisretinoids were synthesized using a key HWE condensation reaction.
Aggregation behavior of the synthesized compounds was observed via nanoprecipitation.
Spherical aggregates formed by (5′R,8′R)-L-trans-hexahydrobenzofuran-A2E were confirmed using TEM.
Abstract
Oxidized photoproducts of pyridinium bisretinoid A2E, including the mono- and bishexahydrobenzofurans, which have been isolated from lipofuscin in the retinal pigment epithelium (RPE) cells of human eyes, have been synthesized in enantiopure form using as key step a Horner-Wadsworth-Emmons (HWE) condensation reaction of pyridinecarbaldehydes and enantiopure cyclohexene oxide pentadienylphosphonates. The synthesis of the trienylcyclohexene oxide branch on the shorter arm (S) of the pyridine ring was followed by a diastereoselective rearrangement to the hexahydrobenzofurandienyl substituent under acidic conditions. In contrast, the construction of the polyenic long arm (L) of the pyridine ring by HWE condensation evolved to the formation of diastereomeric hexahydrobenzofurantrienyl substituents in an unselective rearrangement. The alternative and more straightforward bidirectional HWE…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1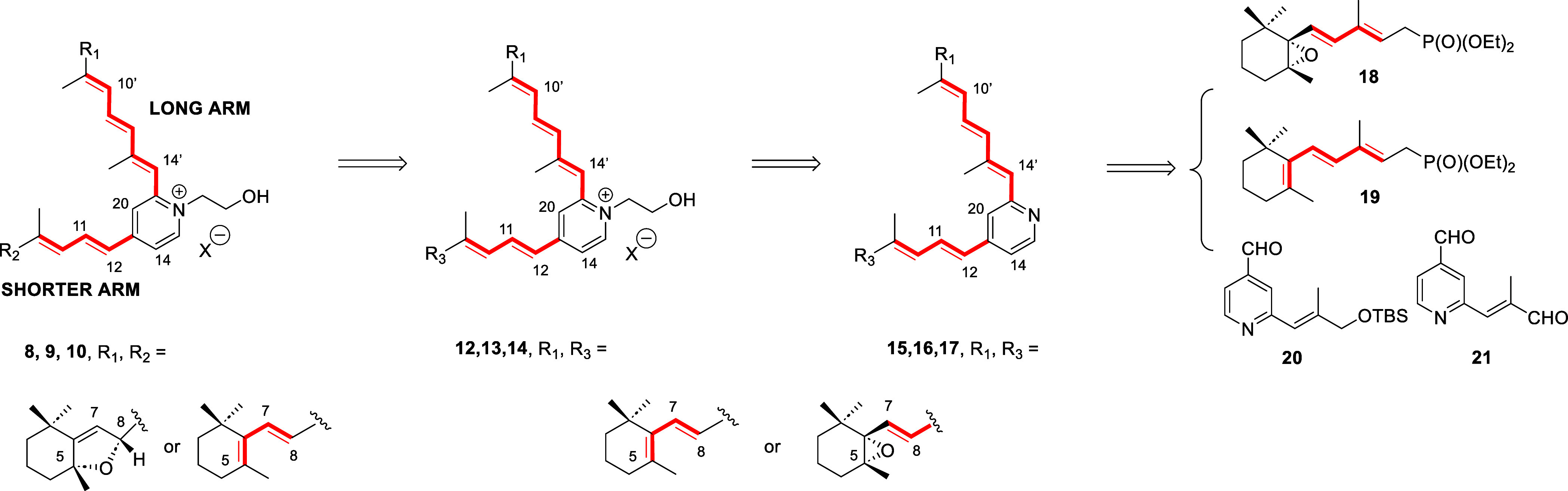 1
1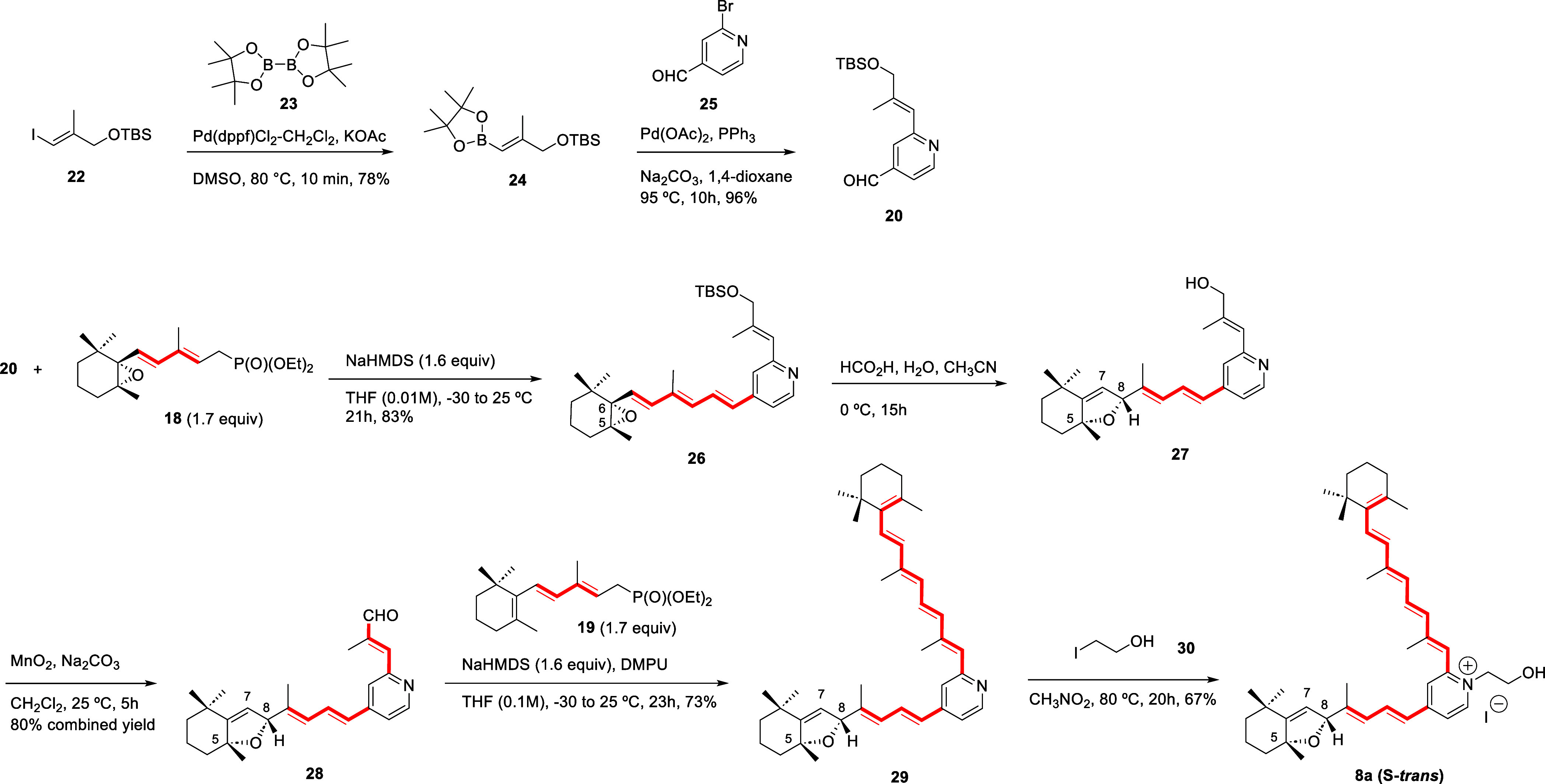 2
2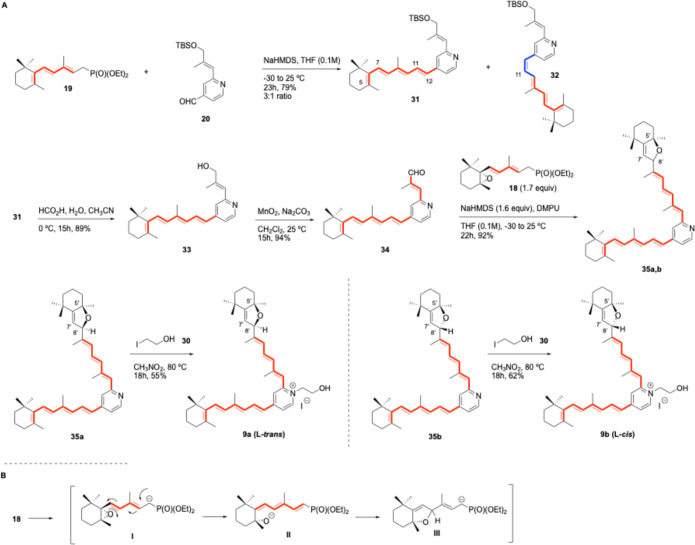 3
3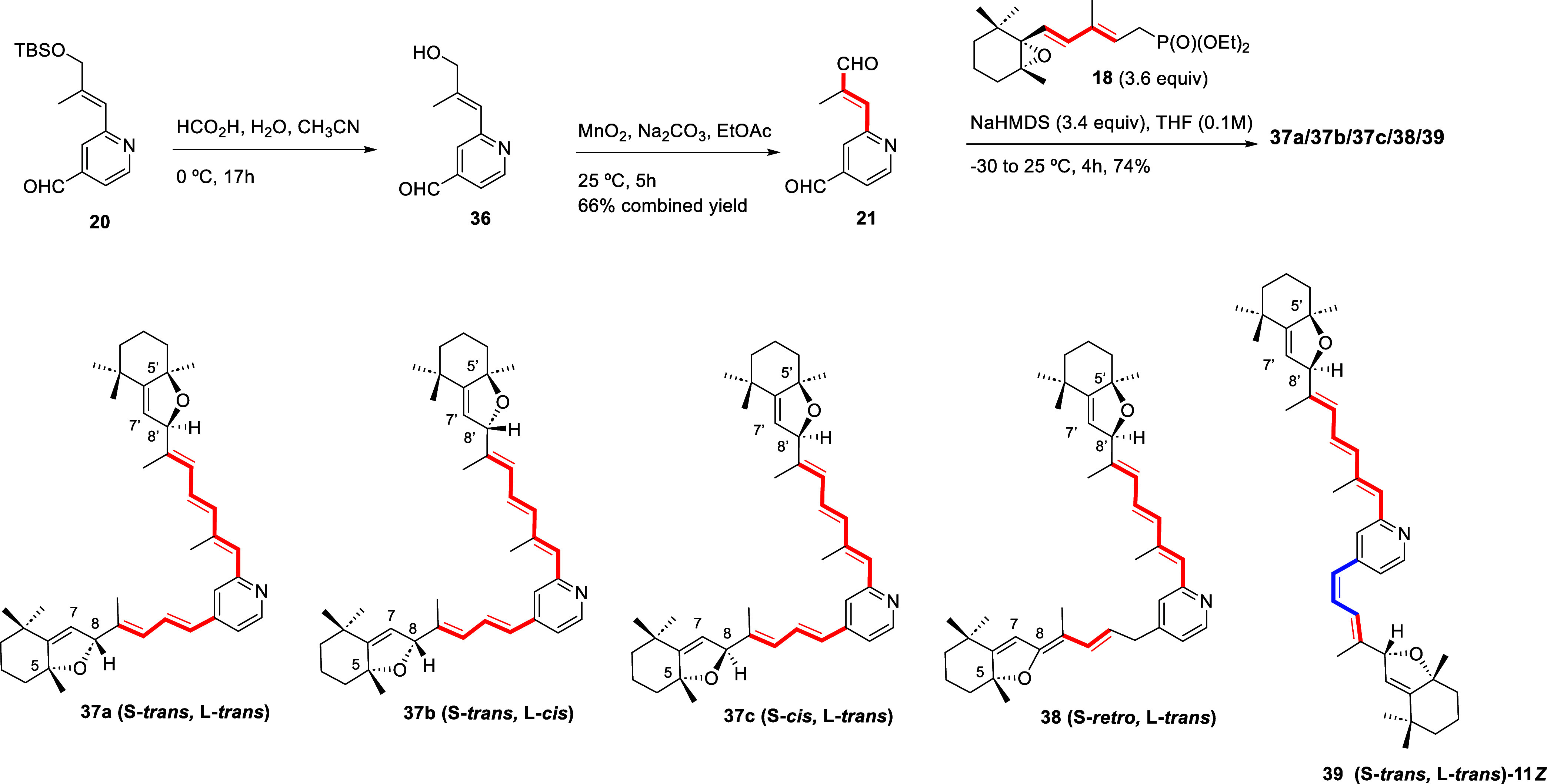 4
4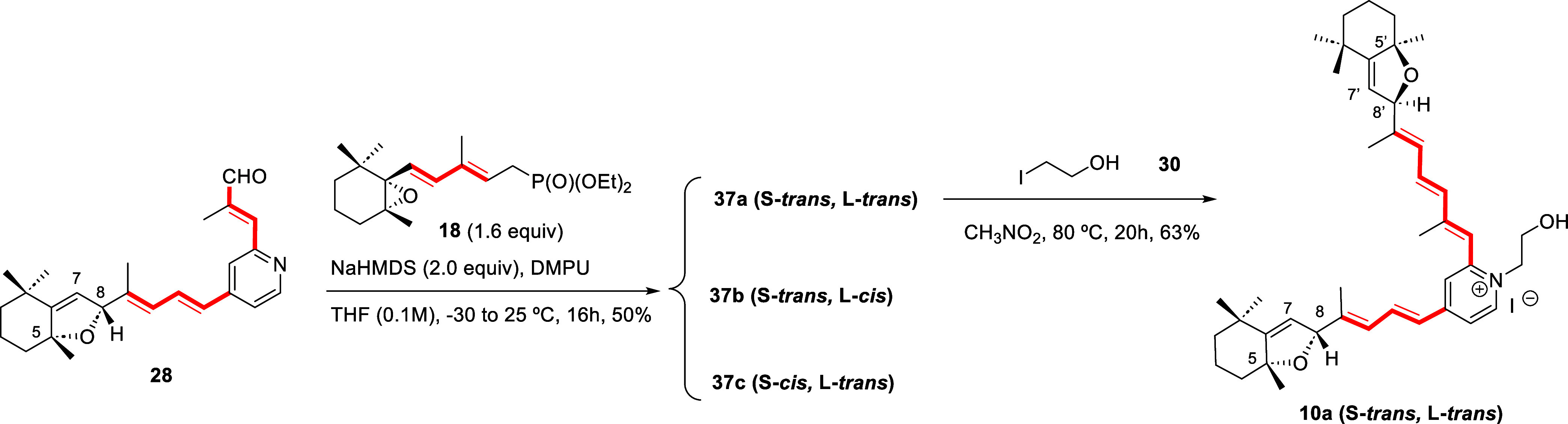 5
5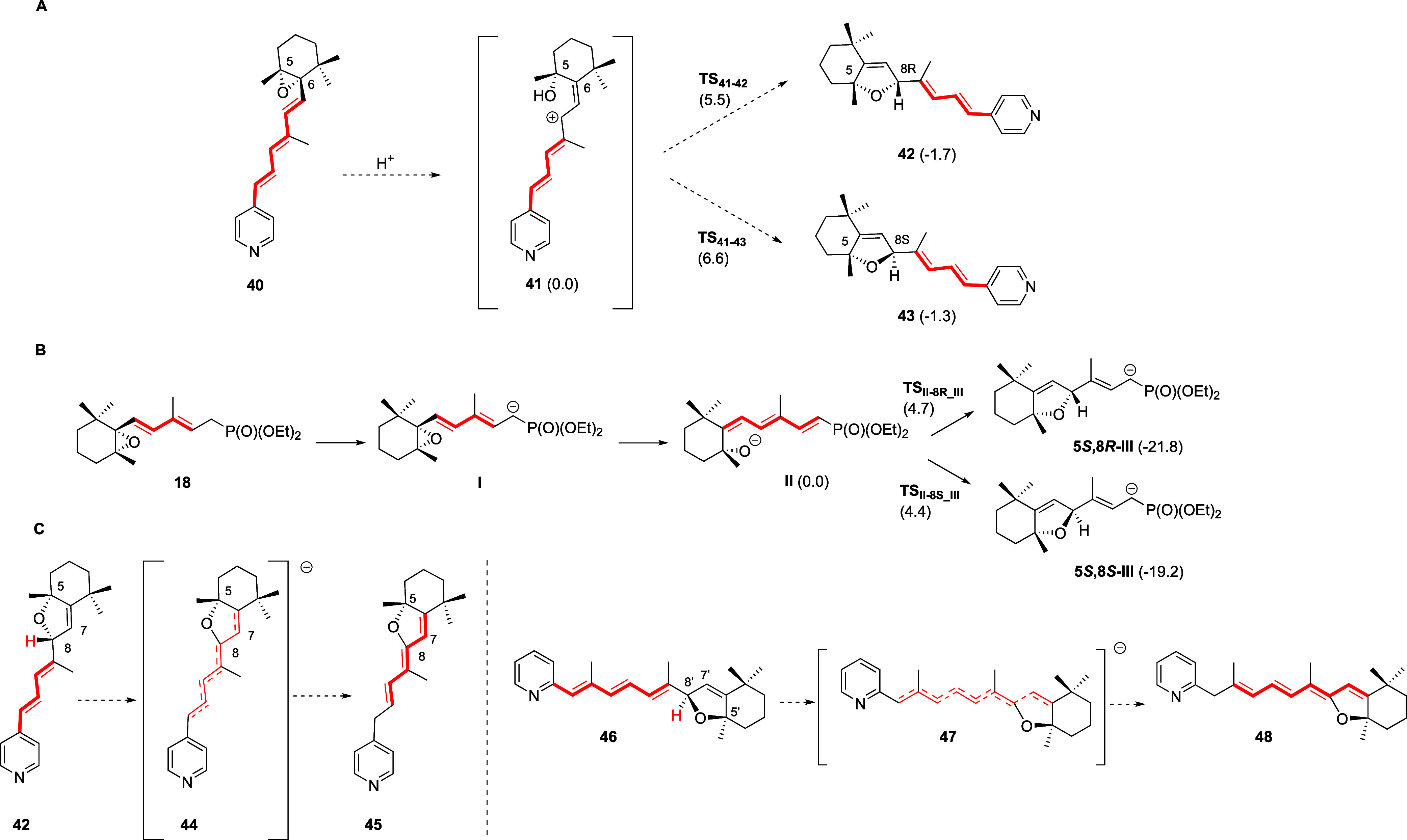 6
6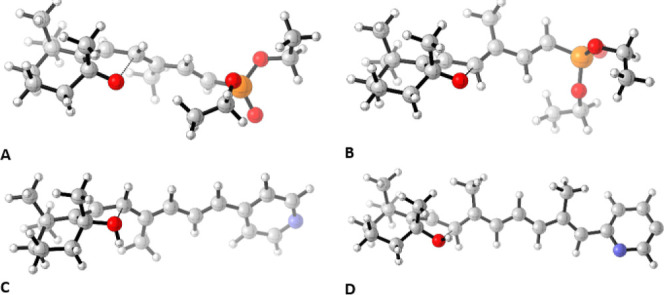 2
2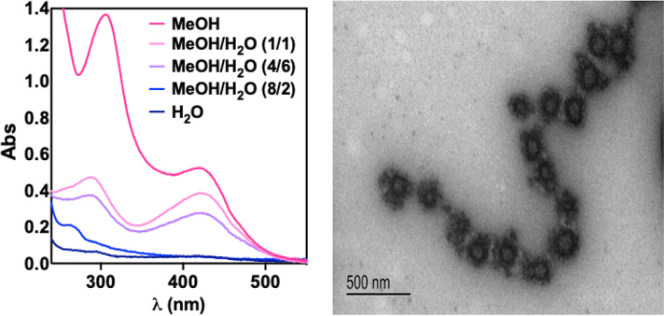 3
3- —Xunta de Galicia10.13039/501100010801
- —Xunta de Galicia10.13039/501100010801
- —Xunta de Galicia10.13039/501100010801
- —Agencia Estatal de Investigaci?n10.13039/501100011033
- —Agencia Estatal de Investigaci?n10.13039/501100011033
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRetinoids in leukemia and cellular processes · Multicomponent Synthesis of Heterocycles · Retinal Diseases and Treatments
Introduction
The absorption of light in rod and cone photoreceptors by visual pigments, which contain a protonated Schiff base derived from the condensation of 11-cis-retinal (1, FigureA) and a lysine group (Lys296) of the protein opsin, is the first event on the complex visual process in vertebrates. ?−? ? The visual cycle? proceeds with the isomerization of the chromophore to the all-trans geometry.? After hydrolysis, all-trans-retinal (2, FigureA) is converted back to 1 in the photoreceptor cells through reduction to all-trans-retinol (3, FigureA) by the action of NADPH-dependent retinol dehydrogenases (RDH8, RDH11, and RDH12), isomerization of the latter via the esters (4, FigureA) to 11-cis-retinol (5, FigureA) promoted by retinal pigment epithelium (RPE)-specific 65 kDa protein (RPE65) isomerohydrolase, and oxidation by retinol dehydrogenase. ?−? ?
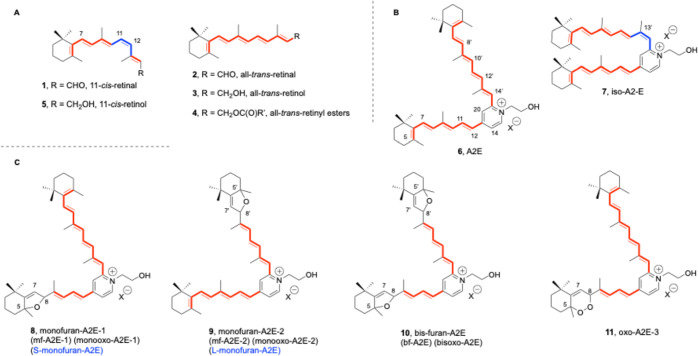
A). Retinoids implicated in the visual cycle. (B). Pyridinium bis-retinoids (6,7). (C). Oxidized derivatives (8–11) of A2E (6).
An excess of all-trans-retinal (2) in human eyes due to anomalies of the retinoid metabolic routes? perturbs lipid metabolism and produces a buildup of pro-inflammatory lipid-containing granules of fluorophores and protein debris called lipofuscin that accumulate in RPE phagolysosomes.? Pyridinium ions substituted with cyclohexenyl-containing unsaturated fragments (tetraenes and pentaenes; the most common are shown in FigureB) derived from all-trans-retinal (2) are members of the structurally complex mixture of lipofuscin components.? The most abundant of these fluorophores, namely A2PE, is generated through condensation of two molecules of all*-trans*-retinal (2) with dipalmitoyl-l-β-phosphatidylethanolamine.? Further enzymatic hydrolysis of the bis-retinoid fluorophore precursors ?,?,? in the photoreceptor outer segment membrane generates as major product N-retinylidene-N-retinylethanolamine or A2E (6, FigureB). ?,?,?,? A ca. 4:1 photostationary equilibrium mixture of A2E (6) and its C13’ = C14′ cis isomer, namely iso-A2E (7, FigureB) has been isolated from human RPE, ?,? and shown to be present upon photochemical irradiation of eye extracts. ?,?,? A2E was proposed to also function as a sensitizer on the photochemical generation of singlet oxygen from triplet oxygen. ?,?
Understanding the etiology of eye diseases and their effects on the visual cycle, as well as the genetic defects of the enzymes responsible for these processes, is crucial to suggest therapeutic options. ?,?−? ? In this regard, ABCR is a high molecular weight glycoprotein found in photoreceptor red outer segment disk membranes (foveal and peripheral cone) that functions as a photoreceptor-specific ATP-binding cassette transporter. Mutations in the gene encoding ABCR have been implicated in eye diseases such as Stargard’s macular dystrophy. ?,?
Oxidized photoproducts of A2E (6) ?,? were first detected in the organic soluble portion of human retinal lipofuscin. ?,? As a model of the photooxidation occurring in the eyes, they were also detected in the organic extracts of bovine RPE cells derived from calf eyes fed with A2E (6) following irradiation of the samples under ambient conditions. ?,?,? They could also be generated when solutions of A2E-laden RPE in abcr^–/–^ mice were exposed to blue light. ?,?
Although the small quantities of these compounds precluded further structural studies, larger amounts where obtained upon treating solutions of synthetic A2E (6)? in MeOH with 2 equiv of meta-chloroperoxybenzoic acid (MCPBA) for 12 h at ambient temperature in the dark.? ^1^H NMR and UV spectroscopic characterization of the main components of the reaction mixture confirmed the presence of the 5,8,5′,8′-bishexahydrobenzofuran 10 (FigureC) (first named 5,8,5′,8′-bis-furanoid oxide, or bis-furan-A2E or bf-A2E? or bis-oxo-A2E?), the 5,8- and 5′,8′-hexahydrobenzofurans 8 and 9 (FigureC)? at the shorter (S) or long (L) arms of A2E (also called monofuran-A2E, mf-A2E-1 and mf-A2E-2, respectively, or monooxo-A2E), ?,?,? and the bisoxygenated 5,8-monoperoxy-A2E 11 (first named oxo-A2E-1) (FigureC). ?,?
The hexahydrobenzofurans were proposed to be generated upon rearrangement under acidic conditions of the corresponding cyclohexene oxides, which would be preferentially formed, as occurs with the bisoxygenated analogue 11 (FigureC), by oxidation at the most electron-rich endocyclic double bond (C5 = C6 and/or C5′ = C6′ as indicated in FigureC). Since lipofuscin accumulates within the RPE cells in lysosomes, which are more acidic than the surrounding cytoplasm,? it was anticipated? that these hexahydrobenzofurans would be generated from the rearrangement of the first formed cyclohexene oxides promoted by small amounts of acid.?
Despite the limited and incomplete spectroscopic characterization of these (bis)hexahydrobenzofurans (FigureC), ?,? and their comparison with the racemates obtained from the oxidation of A2E (6) with MCPBA, no diastereo- and enantioselective synthesis of these compounds has been reported, and therefore we targeted them as an extension of our work on enantiopure hexahydrobenzofuran-containing polyenes. ?,?
Given the stereochemical complexity of the target compounds with six/seven stereogenic double bonds and the presence of two stereocenters in the monohexahydrobenzofurans-A2E (8,9) and four stereocenters in the bishexahydrobenzofuran-A2E (10), a total of 4 and 16 stereoisomers sharing the all-trans geometry of the double bonds are possible for these compounds. Being the absolute and relative configurations of the stereocenters unknown due do the minute amounts isolated from human retinal lipofuscin, ?−? ? we selected a single enantiomer of the putative biogenetic precursors 5,6-cyclohexene oxides 12–14 (Scheme) and directed our efforts to explore the generation of the second stereocenter, as previously demonstrated for carotenoids containing that motif. ?−? ? ? Based on these precedents, we considered feasible that the 5,8-(bis)hexahydrobenzofurans 8, 9 and 10 would be generated by the acid-promoted rearrangement of the corresponding enantiopure A2E-related 5,6-cyclohexene oxides 12–14 and the latter by pyridine alkylation of precursors 15–17. The longer and shorter conjugated arms of these bis-unsaturated pyridines would result from the stepwise condensation reaction? of pyridinecarbaldehyde 20 with cyclohexene oxide dienylphosphonates 18 and 19 as complementary partners to reach 15 and 16, or by the double condensation reaction of 21 with 18 to construct 17 (Scheme). Enantiopure 18 has been previously prepared? using a sequence that included as key step an enantioselective Sharpless asymmetric epoxidation of a precursor allylic alcohol.?
Retrosynthetic Analysis of the Stepwise and Bidirectional HWE Reaction for the Total Synthesis of Enantiopure A2E-Oxidized Derivatives Containing Hexahydrobenzofuran Rings (8–10)
Results and Discussion
Synthetic Studies
As target for methodological development, the mono-oxidized A2E in the shorter arm (namely, 8, FigureC) was first selected. Based on our prior experience on hexahydrobenzofuran containing carotenoids,? the Horner-Wadsworth-Emmons (HWE) condensation reaction ?−? ? ? ? ? was considered reliable to construct the polyene fragments on each arm of the disubstituted pyridine skeleton.
The required 2-alkenylisonicotinaldehyde 20 was generated in 96% yield by Suzuki–Miyaura cross-coupling reaction ?−? ? of commercial 2-bromoisonicotinaldehyde 25 with alkenylboronate 24 promoted by Pd(OAc)2 and PPh_3_ with Na_2_CO_3_ as base in dioxane at 95 °C (Scheme). Alkenylboronate 24 was previously formed in 78% yield upon heating, at 80 °C in DMSO solution, protected (E)-3-iodo-2-methylpropenol 22 with bis(pinacolato)diboron 23 under catalysis of Pd(dppf)Cl_2_·CH_2_Cl_2_ in the presence of KOAc.?
Bidirectional HWE Condensation Reaction for the Total Synthesis of Diastereomer 8a
For the HWE condensation ?−? ? ? ? ? of aldehyde 20 and C_15_ phosphonate 18
?−? ? ?,? an excess (1.7 mol equivalents) of the anion generated using NaHMDS (1.6 mol equivalents) as base provided the trienylcyclohexene oxide fragment on the shorter arm of 26 with complete stereoselectivity as E isomer in 83% yield (Scheme). Under the classical phosphonate anion generation conditions (KOtBu, THF, rt) used previously for the synthesis of related carotenoids, ?,?,? a 3:1 trans/cis isomer ratio was obtained (see Supporting Information).
Deprotection of the allylic alcohol under acidic conditions? was accompanied by the rearrangement of the C5,C8-alkenylcyclohexene oxide fragment to the hexahydrobenzofuran of R configuration at C8 (27, Scheme) as major stereoisomer (5:1 dr) in excellent yields.? Oxidation of the allylic alcohol 27 using MnO_2_ afforded the C2-methylpropenal-substituted pyridine 28 in 80% combined yield, which was subjected to the second HWE condensation. ?−? ? ? ? ? This step required optimization, since the use of NaHMDS as a base under the same conditions used for 26 (Scheme) afforded compound 29 incorporating the additional unsaturated fragment, but in low yields (27% at most). Fortunately, addition of DMPU to the reaction media increased the yield to 73% and afforded stereoselectively the thermodynamically favored? all-trans isomer of the pentaene branch present in 29. After HPLC purification (Chiralpak IA column, 25 × 1 cm; heptane/CH_2_Cl_2_/EtOH/Et_2_NH 70:30:1:0.1 v/v/v/v), full characterization confirmed the structure of the bis-polyenic pyridine 29.
As discussed in the case of carotenoids with bis-hexahydrobenzofuran skeletons,? structural confirmation of the C8 configuration in 29 and synthetic precursors (Scheme) rested on the chemical shift values for H7 and H8 of the hexahydrobenzofuran ring,? which appear in the δ ≈ 5.0–5.3 ppm region of the ^1^H NMR spectra. To compare the data with those previously reported for bishexahydrobenzofuran-A2E (vide infra),? the ^1^H NMR spectra were acquired in C_6_D_6_, and the data correlated with those of related carotenoids. In these more conjugated (bis)hexahydrobenzofuran-containing carotenoids it has been shown that the chemical shift differences of H7 and H8 for the trans diastereomers of 8R relative configuration are very small (Δδ_H7–H8_ ≈ 0.02 ppm) and the signal for H7 appears as a broad singlet. In contrast, those for the 8S diastereomer show larger chemical shift differences (Δδ_H7–H8_ ≈ 0.15–0.22 ppm) and noticeable coupling constants (J H7–H8 > 1.4 Hz). ?−? ? Based on these precedents, the ^1^H NMR data for the major disubstituted pyridine 29 (and also for pyridinium salt 8a, vide infra) showed full consistency with those of the 8R stereoisomer, given the small chemical shift differences noted for H7 and H8 (δ values of 5.22 and 5.15 ppm, respectively, in CD_2_Cl_2_ solutions).? The R configuration of the newly formed C8 stereocenter in 27 was further supported by the NOE correlations observed between the methyl group at C5 and C8-H.
Lastly, alkylation of the pyridine was carried out under similar conditions to those reported for A2E (6), ?,? namely heating 29 with 2-iodoethanol (30) in nitromethane but at 80 °C instead of 100 °C for 20h, and (5R,8R)-S-trans-hexahydrobenzofuran-A2E (8a, Scheme) was obtained in 67% yield. The spectroscopic data of 8a followed the same trends described above for the disubstituted pyridines and confirmed the formation of the hydroxyethylpyridinium ion,? thus complementing the limited characterization data reported for the racemate. ?,?
Inspired on the previous results with 8, a similar approach but with the reversed order of the reacting phosphonates, was next adopted for the synthesis of enantiopure 9 (Scheme) containing a trienylhexahydrobenzofuran motif in the longer pentaenyl arm of A2E (6). The first HWE condensation reaction ?−? ? ? ? ? of the anion of trienylphosphonate 19 generated using NaHMDS at −30 °C with C2-substituted pyridine-2-carbaldehyde 20 took place in this case with a 3:1 E/Z diastereoselectivity of the newly formed olefin on the tetraene fragment. ?−? ? The geometric isomers 31 and 32 could be easily separated by column chromatography. Following the deprotection of the allylic alcohol of 31 (HCO_2_H, 89% yield), oxidation of 33 with MnO_2_ and Na_2_CO_3_ afforded enal 34 in 94% yield (SchemeA).
(A). Iterative HWE Reaction for the Total Synthesis of the Diastereomers of 9: (B). Proposed Reaction Pathway for Formation of the Anion of Cyclohexene Oxide Dienylphosphonate 18 and Rearrangement of Reactive Anionic Species I to III
The second HWE condensation reaction of 34 with the anion of the enantiopure cyclohexene oxide pentadienylphosphonate 18
?−? ? ?,? generated using NaHMDS as a base and DMPU, under the same conditions described for the formation of 26 (Scheme), afforded the thermodynamically favored? all-trans isomer of the newly formed unsaturated branch in excellent yield (92%). However, the NMR data analysis confirmed that the hexahydrobenzofuran skeleton was already present, and in contrast to the epoxytrienyl chain at C4, the rearrangement of the cyclohexene oxide fragment on the epoxytetraenyl chain at C2 of the pyridine ring had taken place under the basic reaction conditions. Moreover, a 50:50 mixture of diastereomers at the new stereocenter at C8 with polyene fragments of all-trans geometry was isolated (SchemeA).?
The finding is in agreement with the previously proposed ?,? reactivity of phosphonate anion I, which is stabilized through conjugation, and evolves by ring-opening of the 5,6-epoxide and subsequent ring-closure by conjugate addition of the generated alkoxide to the trienylphosphonate intermediate II (SchemeB) to afford the reacting 5,8-dihydrofurandienylphosphonate anion III, ?,? which would then be engaged in the condensation with alkenal 34 to provide 35 (SchemeA). ?,?
In contrast to the experimental results on the rearrangement of 26 (Scheme) and to the general case of 5,6-cyclohexene oxides to 5,8-hexahydrobenzofurans promoted by formic acid in carotenoid structures,? the formation of the hexahydrobenzofuran 35 did not show selectivity under basic conditions. To justify the diastereomeric ratio on the rearrangement of the trienylcyclohexene oxides to the hexahydrobenzofuran isomers under acidic conditions (see Scheme) and after formation of the phosphonate anion (SchemeB), these alternative mechanisms were computed by DFT using the I to III (SchemeB) model system (vide infra).
Diastereomers 35a and 35b were separated by HPLC under the same conditions and spectroscopically characterized. Each compound was converted (SchemeA) as described above into the corresponding pyridinium salts, namely (5′R,8′R)-L-trans- and (5′R,8′S)-L-cis-hexahydrobenzofuran-A2E (9a and 9b, respectively).
^1^H NMR data of 9a and 9b showed full consistency with those of the 8′R and 8′S epimers, since as indicated for the smaller branch hexahydrobenzofuran 8a (Scheme), the Δδ_H7′‑H8′_ for the formed diastereomer L-trans 9a is very small (around 0.07 ppm in this case) and the signal for H7′ appears as a broad singlet, whereas chemical shift values for H7′ and H8′ on the L-cis epimer 9b were different (δ ≈ 5.35 and 5.10 ppm, respectively) and a measurable coupling constant was clearly noted in the latter signal (J H7′‑H8′ = 1.9 Hz). ?−? ? ? In the case of the 8′R diastereomer 9a the configuration of the newly formed C8′ stereocenter was further confirmed by the NOE effect observed between the methyl group at C5′ and the hydrogen at C8′ (Scheme). ?,?
Lastly, bidirectional condensation of the conjugated anion of phosphonate 18 (NaHMDS, THF, −30 °C) with dialdehyde 21,? which was obtained from 20 after deprotection of the silyl ether with formic acid and oxidation of the allylic alcohol 36 (66% combined yield, Scheme), using the described Barbier conditions (addition of 3.6 equiv of 18, from −30 to 0 °C) took place in comparable overall yields (74%) to those of the monocondensation process of 20 (Scheme). Surprisingly, the reaction time required for completion to obtain 37 (Scheme) was shorter (4 h), when compared to the case of the monocondensation (23 h). Moreover, although the main products were the hexahydrobenzofuran diastereomers at the conjugated chains (37), additional compounds were present in the more complex product mixture.
Bidirectional HWE Reaction for the Total Synthesis of Diastereomers 37a–c, and Structures of Secondary Products 38 and 39
HPLC purification as described above allowed to isolate compounds 37 a /37 b /37 c /38/39 (Scheme) in a 1.0:0.5:0.4:0.25:0.12 relative ratio. The structure of the major components could be confirmed by comparison of their ^1^H NMR and UV data with those discussed above for the hexahydrobenzofurans (29 and 35). Using the same analysis of the chemical shift and coupling constant values for H7 and H8 in their ^1^H NMR spectra, the major bishexahydrobenzofuran (37a) was identified as the diastereomer with L-trans, S-trans relative configuration, accounting for the presence of NMR signals at δ values of 5.22, 5.21, 5.15, and 5.13 ppm, all of them as singlet, which were further assigned using NOE analysis to H7, H7́, H8, and H8′, respectively.? Diastereomers at C8 and C8′ (37b and 37c, respectively) were also obtained, and could be identified as those with the S-trans, L-cis (J H7′‑H8′ ≈ 1.9 Hz) and S-cis, L-trans (J H7–H8 ≈ 2.0 Hz) relative configurations, respectively, based on the coupling constant values. NOE correlations further confirmed the geometries of the conjugated skeletons and the relative and absolute configurations of the dihydrofurans fused to the cyclohexanes in these stereoisomers.
Furthermore, two minor condensation products (38 and 39, Scheme) were characterized, both of which showed structural differences on the smaller branch relative to 37. Spectroscopic analysis was consistent with 38, which we named S-retro, L-trans, being the product corresponding to the presumed rearrangement of the unsaturated fragment of either 37a or 37c (given the lack of the former H8 signals) across the C8-C12 region with formation of the alkylidenedihydrofuran structure.
The structural proposal for 39, the stereoisomer of 37a with 11-cis geometry, was based on comparison of the NMR data with those of 37a and further analysis of the coupling constants values of the C11 = C12 double bond (overlapping of signals in CD_2_Cl_2_; J H11–H12 ≈ 11.6 Hz) and the presence of singlets assigned to H7, H7′, H8, and H8′ in the ^1^H NMR spectra. NOE correlations were fully consistent with the proposed geometries.
To improve the synthesis of the major diastereomer (37a), we took advantage of the diastereoselective formation of 27 from 26 under acidic conditions (Scheme) and subjected enal 28 to HWE condensation reaction with excess phosphonate 18. Although the stereochemical outcome of the newly generated stereocenter was like that described for 35 (SchemeA), no further components were found in the reaction mixture, and bishexahydrobenzofurans 37a and 37b were isolated in similar yields, together with minor amounts of 37c (Scheme).
Stepwise HWE Reaction for the Total Synthesis of Diastereomers 37, and Alkylation of the Major Diastereomer 37a to Pyridinium Salt 10a
Diastereomer 37a was then treated with iodoethanol 30 as described before ?,? to afford (5R,8*R,*5′R,8′R)-S-trans-L-trans-bishexahydrobenzofuran-A2E (10a) in comparable yields to those of the hexahydrobenzofurans (Scheme). The ^1^H NMR data for this compound matched those reported for the racemate in this diastereomeric form,? and the complete ^13^C NMR characterization data (previously reported? only for the C5C6C7C8 dihydrofuran-containing unit) confirmed the formation of the hexahydrobenzofuran skeleton on each polyenyl branch.
In retrospect, since ^1^H NMR spectra in CD_3_OD for the synthetic hexahydrobenzofurans generated by MCPBA oxidation of A2E (6) showed signals at δ ≈ 5.15 ppm, δ ≈ 5.19 ppm, δ ≈ 5.22 ppm, and δ ≈ 5.26 ppm,? which were assigned using HSQC to the dienyl (δ ≈ 5.15 ppm and δ ≈ 5.22 ppm) and the trienyl (δ ≈ 5.19 ppm and δ ≈ 5.26 ppm) branches, the diastereoselective synthesis along this work confirmed that the major diastereomer obtained in these experiments? was the S-trans-L-trans-bishexahydrobenzofuran-A2E, presumably promoted by the acidic conditions used for HPLC purification. In natural settings, the rearrangement of the 5,6-cyclohexene oxide-A2E to the 5,8-hexahydrobenzofuran-A2E structural isomer could most likely occur under acidic conditions, in full consistency with the acidic medium of RPE lysosomes.
Computational Studies
To justify the diastereoselective formation of hexahydrobenzofuran structures from cyclohexene oxide precursors under acidic conditions (26 to 27, Scheme), and the lack of stereoselectivity under basic conditions (21 to 37a–c) accompanied by the generation of alkylidene hexahydrobenzofuran rearrangement product 38 (Scheme), we carried out DFT studies on model systems 40, 42 and 46 (Scheme) featuring when required one of the unsaturated side chains as substituent at C2 or C4 of the pyridine ring (see S. I. for details). In addition, we also addressed the lack of stereoselectivity on the rearrangement of the anion of cyclohexene oxide dienylphosphonate 18 (SchemeB) to the alkylidene hexahydrobenzofuran intermediate (I to III, SchemeB).
(A) Rearrangement of Model System 40 Under Acidic Conditions by Epoxide Ring Opening to 41 and Ring Closure to Diastereomers 42 and 43 (ΔG Values in kcal/mol are Shown in Brackets). (B) Rearrangement of Cyclohexene Oxide Dienylphosphonate 18 to Diastereomeric Hexahydrobenzofuran Alkenylphosphonates 5S,8R-III and 5S,8S-III Under Basic Conditions (ΔG Values in kcal/mol are Shown in Brackets). (C) Rearrangement of Model Systems 44 and 47 Under Basic Conditions Followed by Protonation to 46 and 49, Respectively.
A. The rearrangement of pyridine-C4-trienylcyclohexene oxide model system 40 promoted by acid, which requires the ring opening of the oxirane to afford a carbocation intermediate and its trapping with the tertiary alcohol (40 to 42 or 43, SchemeA), was computed using the Gaussian 16 suite of programs.? All calculations were performed at the wB97XD level,? with def2svp as basis function? and PCM as solvation model (in THF or CH_3_CN).?
For the acid-mediated rearrangement, the proposed carbenium ion intermediate 41 was shown by NBO analysis? to be mostly located at C8 but enjoy stabilization by resonance given its bis-allylic structure (Figure). The additions to either face of the carbenium ion are exergonic, and the formation of diastereomeric hexahydrobenzofurans 42 and 43 proceeds through transition states TS_41–42_ and TS_41–43_ with computed activation energies of 5.5 and 6.6 kcal/mol, respectively (see Supporting Information). Therefore, the predicted diastereomeric ratio is consistent with that experimentally observed (ca. 5:1 R/S).
Top. Transition states TS41–42 (A, C8–O distance 2.41 Å) and TS41–43 (B, C8–O distance 2.48 Å, CC5‑Me-CC9‑Me 5.06 Å) for the 5,6-cyclohexene oxide to 5,8-hexahydrobenzofuran rearrangement on the corresponding epoxydienylphosphonates promoted by base. Bottom. Transition states TSII‑8RIII (C, C8–O distance 2.12 Å) and TSII‑8SIII (D, C8–O distance 2.07 Å, CC5‑Me-CC9‑Me 3.88 Å) for the rearrangement of 5,6-cyclohexene oxide trienylpyridine at C4 and 5,6-cyclohexene oxide tetraenylpyridine at C2, to the corresponding 5,8-hexahydrobenzofurans promoted by acid, computed at the wB97XD/def2svp (PCM, THF) level.
B. We also addressed by DFT the lack of face selection, and therefore the lack of stereoselectivity on the rearrangement of the anion of phosphonate 18 in THF solution. As indicated in SchemeB, after ring-opening of the 5,6-epoxide, the ring-closure by conjugate addition of the generated alkoxide to C_8_ of the trienylphosphonate intermediate II (SchemeB) along the two available orientations generates the reacting diastereomeric 5,8-hexahydrobenzofuran alkenylphosphonate anions (5R,8R)-III and (5R,8S)-III. ?,?
A conformationally stabilizing interaction between the heteroatom and H_8_ (H_8_···O distance of ca. 1.89 (Å) was characterized for alkoxide II, despite the 1,3-allylic interactions with the methyl groups at C1 and C5 (SchemeB). NBO analysis? confirmed the alkoxytrienylphosphonate structure for intermediate II (SchemeB). The greater nucleophilicity of the alkoxide on II, as confirmed by the f(−) Fukui index, ?,? led to the formation of diastereomers (5R,8R)-III and (5R,8S)-III on highly exergonic processes by conjugate addition through transition states TS_II‑8RIII_ and TS_II‑8SIII_ (Figure), which have similar computed activation energies of 4.7 and 4.4 kcal/mol, respectively (Table S2). The activation energy values predict a 1:1.6 (5R,8R)-35b/(5R,8S)-35b product diastereomeric ratio, in accordance with the experimental findings of this process under kinetic control (Scheme).
C. The mechanism of the selective generation of alkylidenedihydrofuran on the shorter arm in compound 38 (Scheme) was likely due to a sequence of deprotonation at H8 taking place under the basic reaction conditions and further protonation at H12 during workup.
To validate this proposal, we estimated the pK a of the hexahydrobenzofuran bis-allylic hydrogen on model systems 42 and 46, as well as that of phosphonate 18, using the simplest and direct method based on the computational determination of the free energy of the corresponding species AH, A^–^ and H_3_O^+^ by DFT (SchemeC) using M06-2X,? with aug-cc-pVDZ as basis function,? and SMD as solvation model (in water).? Since the outcome of this method depends on the determination of the free energy of the solvated proton, we followed the described procedure? (G̅ _aq_H_3_O^+^ = −273.14 kcal/mol), which is based on the estimation of the average proton solvation energy from a training set of molecules. The free energies for G_aq_AH and G_aq_A^–^ were computed using the same level of theory (see Supporting Information).
The difference on pK a values for the hexahydrobenzofuran hydrogen on model structures 42 (pK a ≈ 19 for H_8_) relative to 46 (pK a ≈ 22 for H_8’_) might justify the ability of the phosphonate anion (pK a ≈ 22), itself formed by deprotonation of 18 with NaHMDS (Scheme), to deprotonate the hexahydrobenzofuran on the shorter arm (pK a ≈ 19).?
The higher acidity of H_8_ on 42 when compared with H_8′_ on 46 could be explained by the greater stabilization of the electron density along the chain at position C4 of the pyridine ring. Electronic delocalization for both anions (44 and 47) was indirectly estimated by ACID (Anisotropy of the Current Induced Density) analysis,? and the Critical Isosurface Values (CIVs) showed slightly stronger conjugation for 44 relative to 47 (0.056 versus 0.053, Figure S9).
Aggregation Studies
The aggregation trend in biological media of these pyridinium bisretinoid hexahydrobenzofurans? prompted us to study this behavior by UV–vis spectroscopy. Given the insolubility of all the compounds tested in water, we turned our attention to the use of a potential cosolvent fully miscible with water. This strategy has been effectively employed to aggregate not only a variety of small molecules but also of macromolecules. ?−? ? Methanol was the solvent of choice, since it solubilizes these pyridinium salts and is fully miscible with water. As shown in Figure, solutions of (5′R,8′R)-L-trans-hexahydrobenzofuran-A2E (9a) in CH_3_OH exhibited UV–vis spectra with absorption maxima at 292 and 425 nm. Upon gradually increasing the volume fraction of H_2_O, a concomitant hypochromic effect was observed, which is indicative of the aggregation of the molecules (see Figure S2–S6 for similar experiments performed with the analogues, and Figure S8 for UV spectra).
Left. UV–vis spectra of (5′R,8′R)-L-trans-hexahydrobenzofuran-A2E (9a) with different volume fractions of H2O in MeOH (c T = 75 μM). Right. TEM image showing the formation of spherical aggregates of 9a (c T = 75 μM, 20/80 v/v MeOH/H2O).
Further evidence of the aggregation upon increasing the water volume fraction was obtained by ^1^H NMR experiments (Figure S7). In CD_3_OD, 9a showed a well-defined NMR spectrum. However, progressive increase of D_2_O volume fractions led to a dramatical sharp decrease in NMR signal intensity, ultimately resulting in near-complete disappearance of signals in 20:80 (v/v) CD_3_OD/D_2_O solvent mixtures. This behavior is indicative of effective nanoprecipitation.
Finally, the morphology of the aggregates was studied by transmission electron microscopy (TEM). TEM images of (5′R,8′R)-L-trans-hexahydrobenzofuran-A2E (9a) showed the formation of spherical aggregates with diameters of ca. 175 nm (See Figure for 9a, Figures S2–S5 for the analogues, and Figure S6 for the estimation of the size of the aggregates).
The formation of spherical aggregates of 9a might justify some of the deleterious effects of these compounds, since oxidized derivatives of A2E were also detected in the eye cups of mice with null mutations in Abca4/Abcr, the gene responsible for recessive Stargardt disease, which is the most common inherited macular dystrophy. ?,? Moreover, they were found to be more abundant in albino vs pigmented abcr ^ –/– ^ mice exposed to increasing ambient light,? and were shown to also induce DNA damage,? suggesting that these effects might also take place in vivo.?
Conclusions
We have synthesized and fully characterized enantiopure hexahydrobenzofurans derived from the oxidation of all-trans-retinal condensation product A2E in the human retinal pigment epithelium (RPE). These conjugates have been shown to accumulate in human eyes with age,? due to their slow or nonexistent breakdown by RPE cells. The Horner-Wadsworth-Emmons (HWE) condensation reaction conditions for olefination on the shorter arm (S) preserved the trienyl 5,6-cyclohexene oxide, which underwent stereoselective rearrangement to the hexahydrobenzofuran under acidic deprotection conditions of an allylic alcohol protected as silyl ether. The same HWE reaction for construction of the long arm (L) of the skeleton induced an unselective rearrangement which led to diastereomeric hexahydrobenzofurans on L or to bishexahydrobenzofurans starting from the dienylhexahydrobenzofuran on S. Computational studies justified the contrasting behavior of the unsaturation on each branch of the pyridine ring under acidic or basic conditions. The morphology of these diastereo- and enantiopure A2E-derived hexahydrobenzofurans was studied by TEM and shown for (5′R,8′R)-L-trans-hexahydrobenzofuran-A2E (9a) to form spherical aggregates upon nanoprecipitation using methanol/water solvent mixtures. Since A2E-derived hexahydrobenzofurans have also been detected in aged human donor RPE, and therefore contribute to the formation of lipofuscin, the toxic fluorescent material that accumulates in the retina, these synthetic spherical aggregates might be useful as models to study their formation and deposition on human eyes.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Zhang J.Choi E. H.Tworak A.Salom D.Leinonen H.Sander C. L.Hoang T. V.Handa J. T.Blackshaw S.Palczewska G.Kiser P. D.Palczewski K.Photic generation of 11-cis-retinal in bovine retinal pigment epithelium J. Biol. Chem.2019294191371915410.1074/jbc.RA 119.01116931694912 PMC 6916499 · doi ↗ · pubmed ↗
- 2Palczewski K.Kiser P. D.Shedding new light on the generation of the visual chromophore Proc. Natl. Acad. Sci. U.S.A.2020117196291963810.1073/pnas.200821111732759209 PMC 7443880 · doi ↗ · pubmed ↗
- 3Chen S.Getter T.Salom D.Wu D.Quetschlich D.Chorev D. S.Palczewski K.Robinson C. V.Capturing a rhodopsin receptor signalling cascade across a native membrane Nature 202260438439010.1038/s 41586-022-04547-x 35388214 PMC 9007743 · doi ↗ · pubmed ↗
- 4Gruhl T.Weinert T.Rodrigues M. J.Milne C. J.Ortolani G.Nass K.Nango E.Sen S.Johnson P. J. M.Cirelli C.Furrer A.Mous S.Skopintsev P.James D.Dworkowski F.Båth P.Kekilli D.Ozerov D.Tanaka R.Glover H.Bacellar C.Brünle S.Casadei C. M.Diethelm A. D.Gashi D.Gotthard G.Guixà-González R.Joti Y.Kabanova V.Knopp G.Lesca E.Ma P.Martiel I.Mühle J.Owada S.Pamula F.Sarabi D.Tejero O.Tsai C.-J.Varma N.Wach A.Boutet S.Tono K.Nogly P.Deupi X.Iwata S.Neutze R.Standfuss J.Schertler G.Panneels V.Ultrafast structural changes direct the first molecular events of visi · doi ↗ · pubmed ↗
- 5Hong J. D.Salom D.Kochman M. A.Kubas A.Kiser P. D.Palczewski K.Chromophore hydrolysis and release from photoactivated rhodopsin in native membranes Proc. Natl. Acad. Sci. U.S.A.2022119 e 221391111910.1073/pnas.221391111936322748 PMC 9659404 · doi ↗ · pubmed ↗
- 6Yakovleva M. A.Radchenko A. S.Feldman T. B.Kostyukov A. A.Arbukhanova P. M.Borzenok S. A.Kuzmin V. A.Ostrovsky M. A.Fluorescence characteristics of lipofuscin fluorophores from human retinal pigment epithelium Photochem. Photobiol. Sci.20201992093010.1039/c 9pp 00406 h 32441276 · doi ↗ · pubmed ↗
- 7Mata N. L.Weng J.Travis G. H.Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration Proc. Natl. Acad. Sci. U.S.A.2000977154715910.1073/pnas.13011049710852960 PMC 16515 · doi ↗ · pubmed ↗
- 8Kim H. J.Sparrow J. R.Bisretinoid phospholipid and vitamin A aldehyde: shining light J. Lipid Res.20216210004210.1194/jlr.TR 12000074232371567 PMC 7933493 · doi ↗ · pubmed ↗